
The Role of BDNF in Experimental and Clinical Traumatic Brain Injury


Abstract
:1. Introduction
1.1. Brain Derived Neurotrophic Factor (BDNF)
BDNF Val66met Polymorphism
2. BDNF Polymorphism in Human Studies
2.1. BDNF Polymorphism and Outcome after Mild and Moderate TBI
2.2. BDNF Polymorphism and Outcome in Severe TBI
2.3. BDNF Levels in CSF and Plasma after TBI
3. Experimental TBI
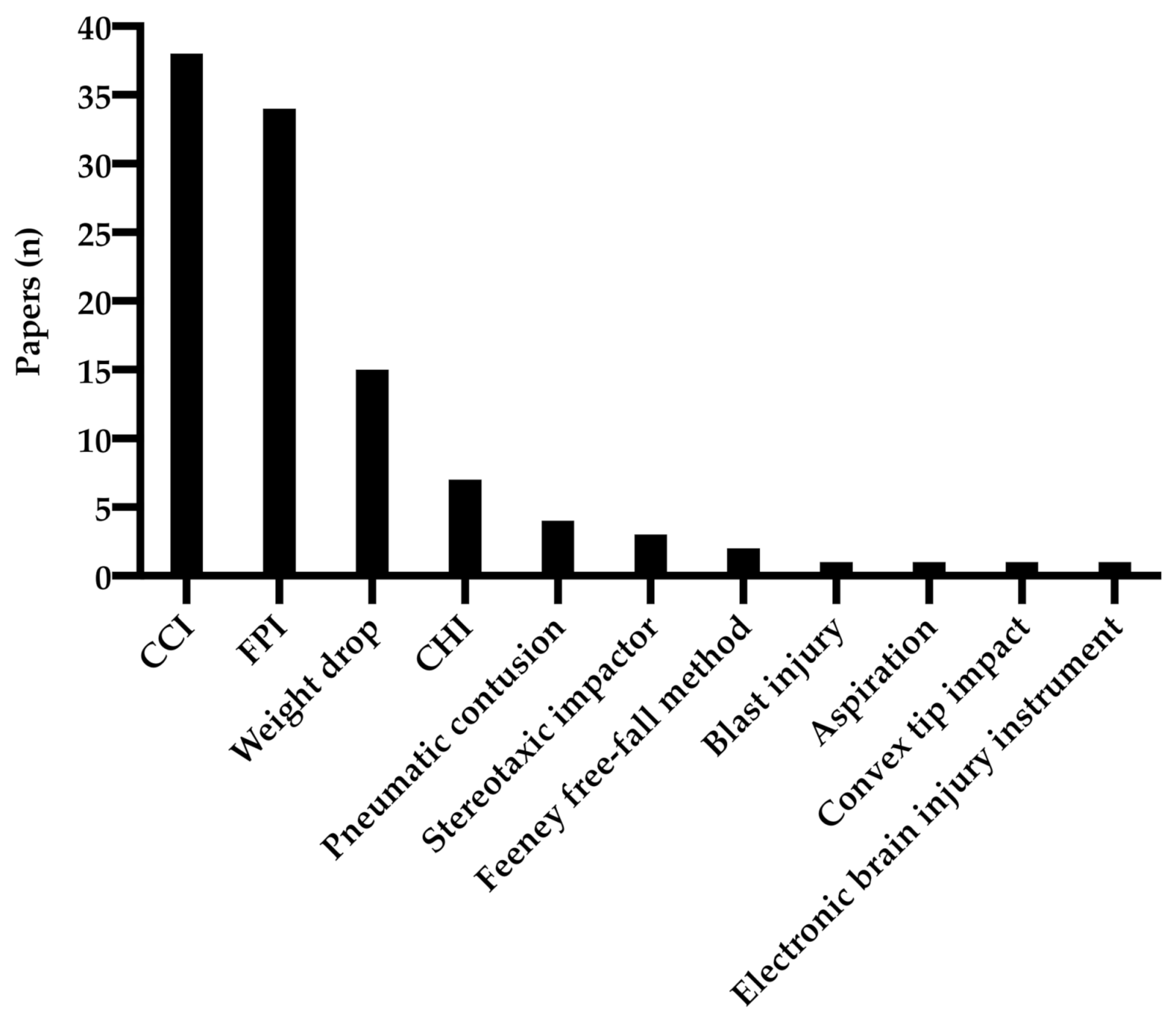
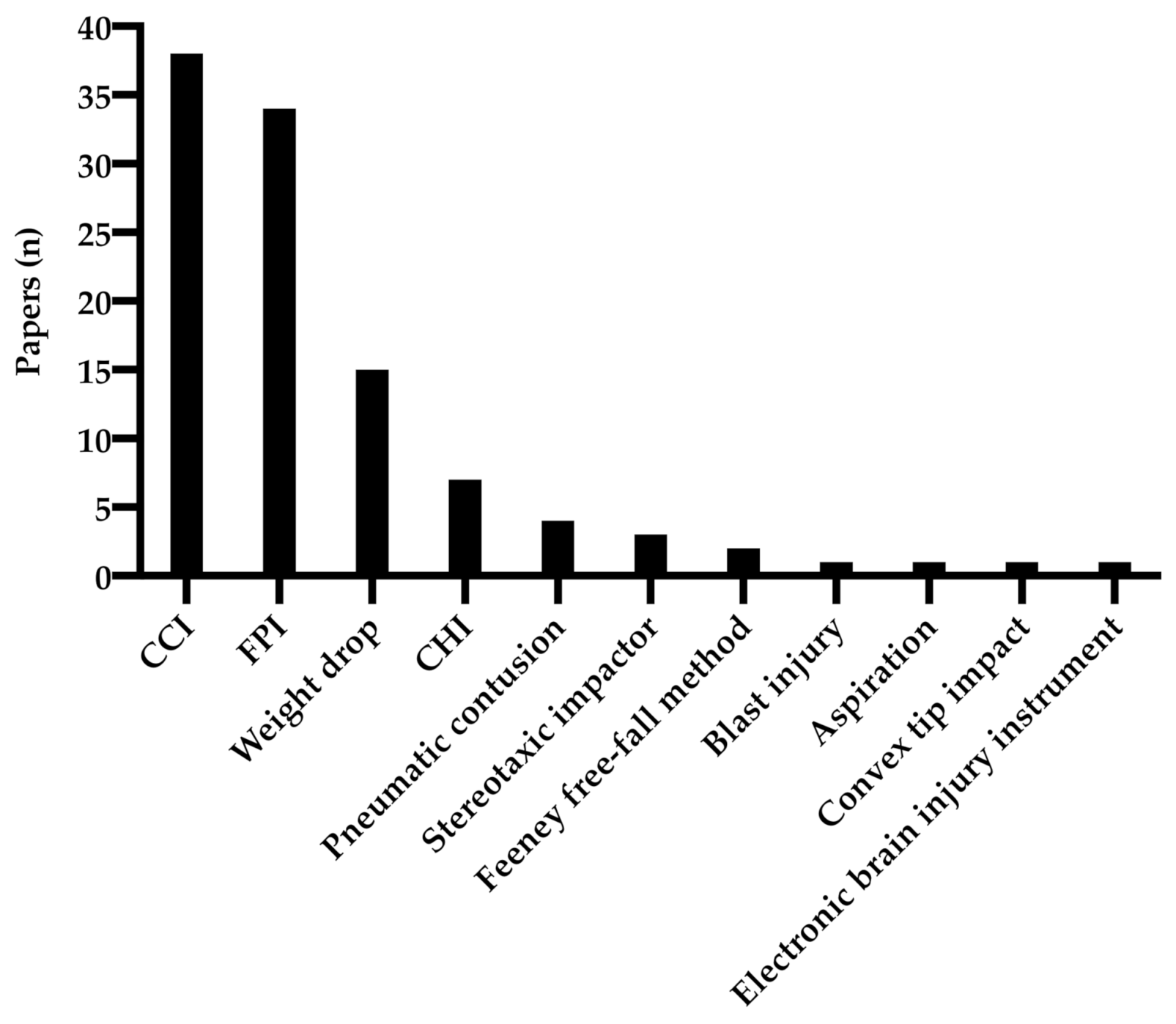
3.1. Experimental Animal Models
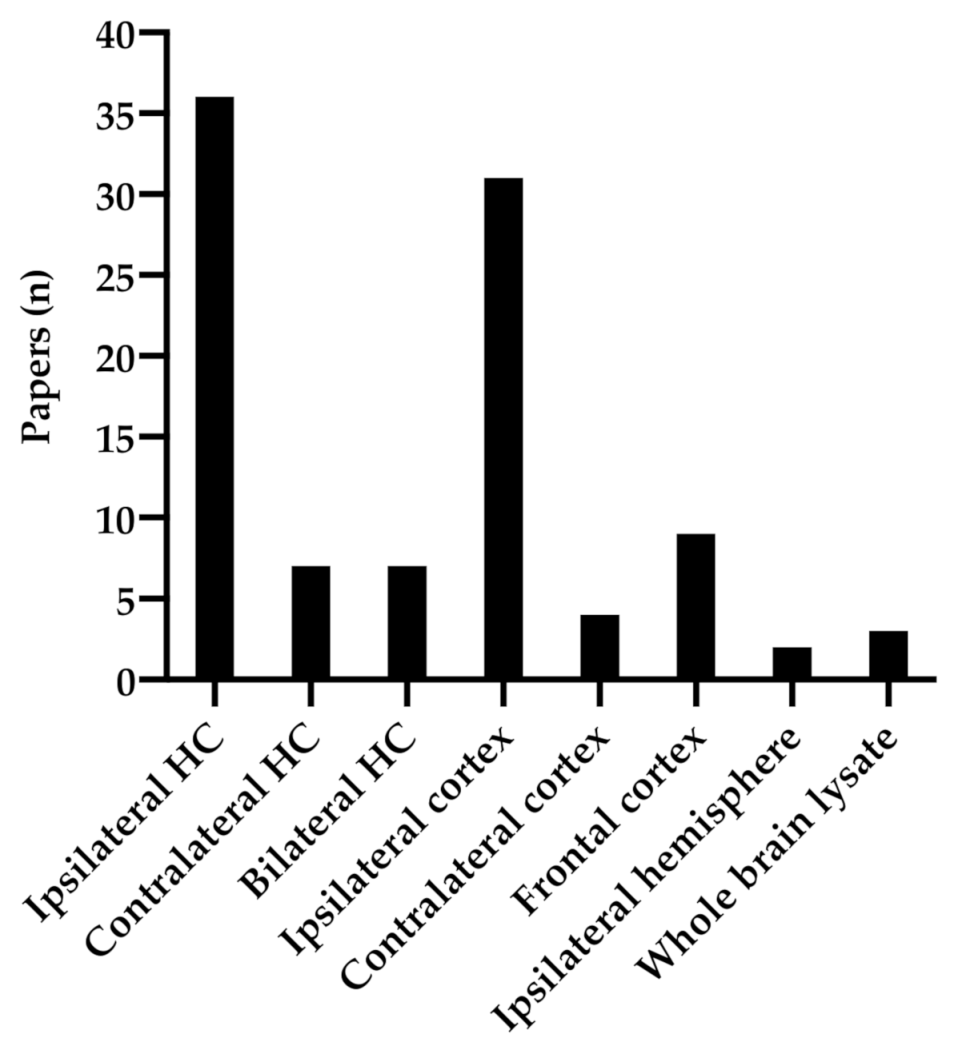
3.2. Anatomical Regions of BDNF Analysis
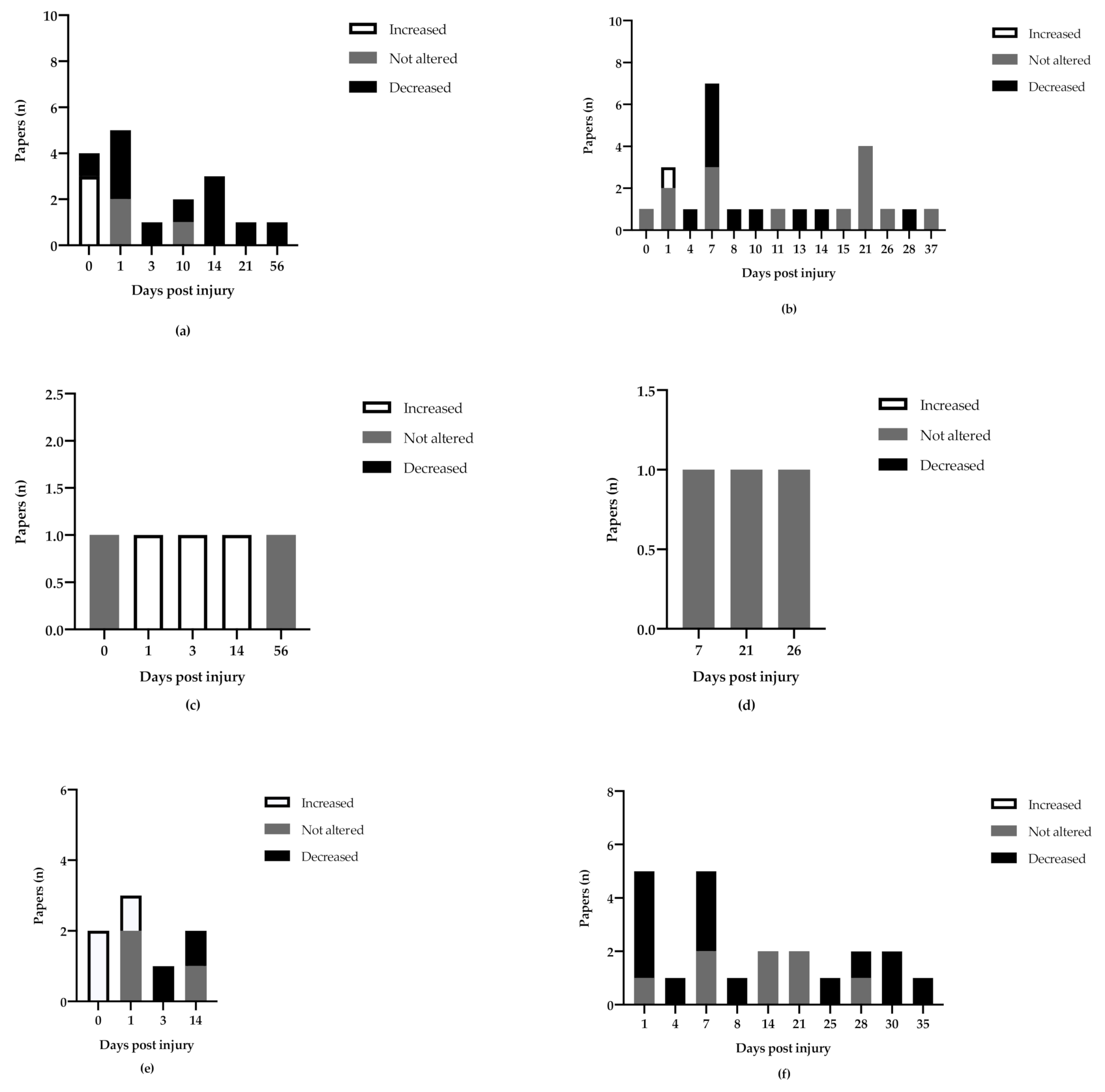
3.3. Assessment of BDNF Expression
3.3.1. Hippocampus
3.3.2. Cortex
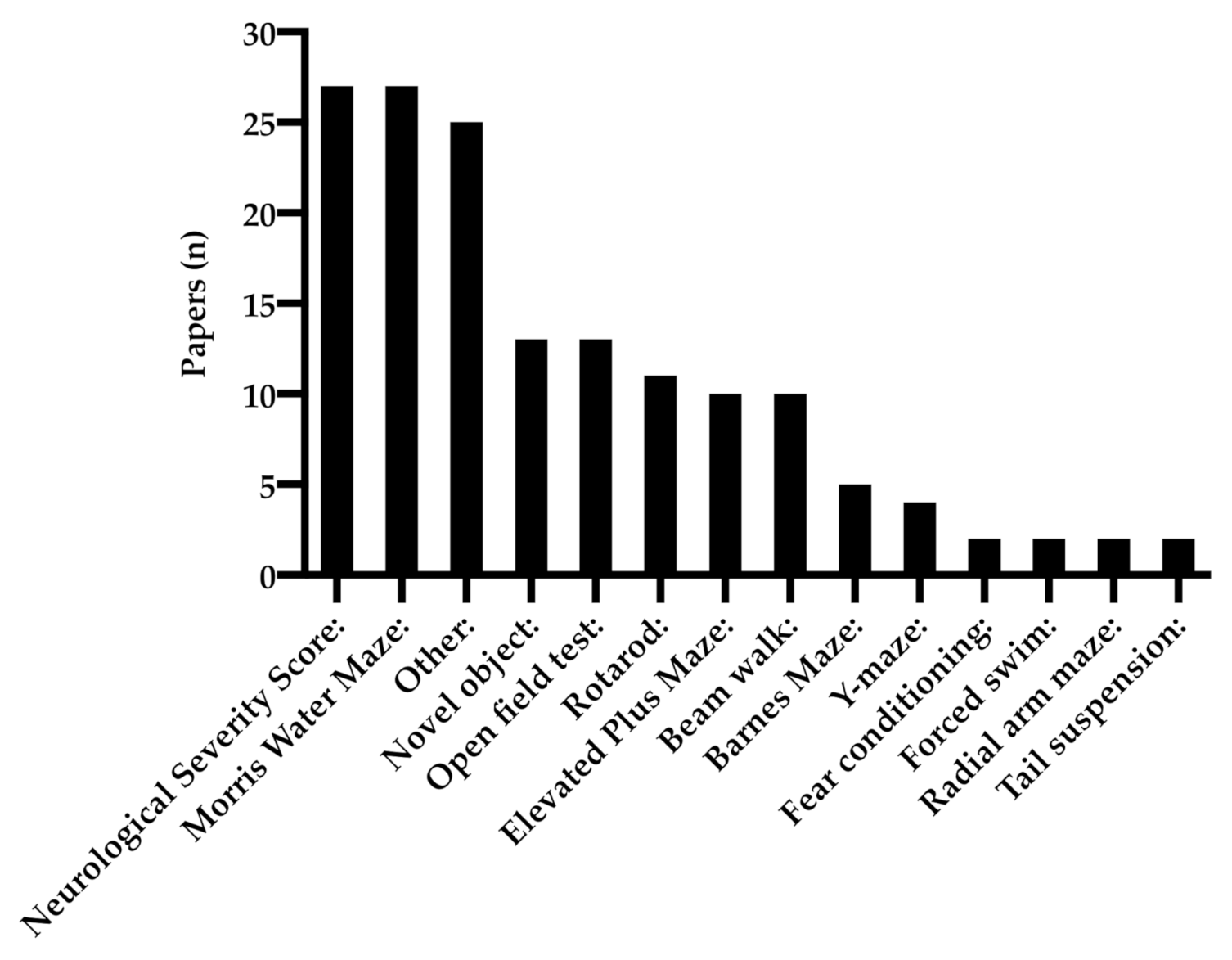
3.4. Behavioral Tests
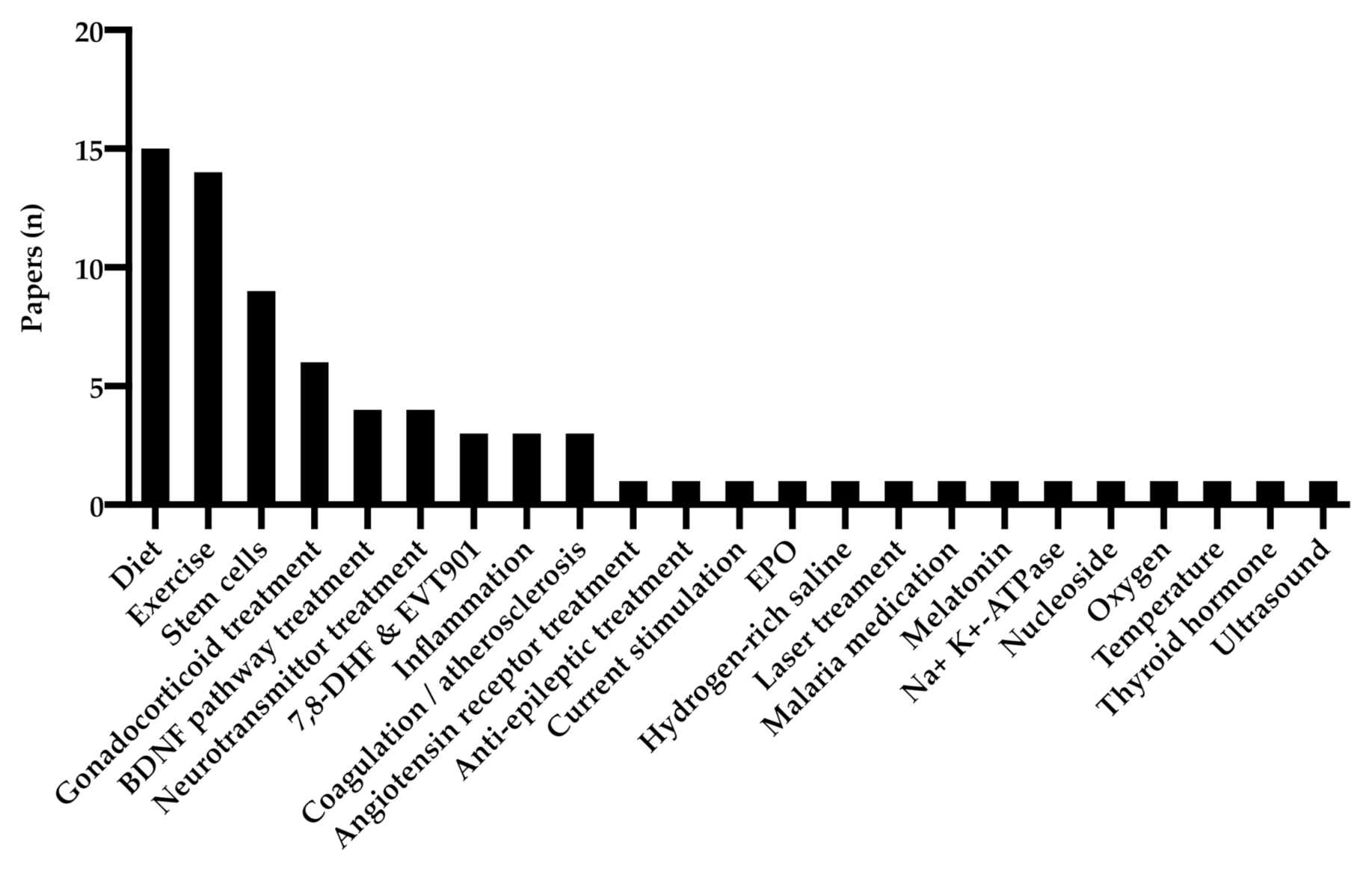
3.5. Treatment of TBI
3.5.1. Exercise
3.5.2. Diet
3.5.3. Stem Cell Treatment
3.5.4. BDNF Pathway Treatment
3.5.5. 7,8-DHF & EVT901
3.6. BDNF in Transgenic Animals
4. Discussion
4.1. Human Induced Pluripotent Stem Cell-Models in TBI Research
4.2. Potential Advantages with iPSC-Models
4.3. Studying the Impact of BDNF Val66met Polymorphism
4.4. Considerations Regarding Translation to Humans
4.5. Treatment of TBI and Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total N | TBI | Age | Timing | BDNF SNP | Outcome Assessed | Findings | |
|---|---|---|---|---|---|---|---|
| BDNF Global Outcome | |||||||
| (Failla et al. 2016) [33] | 203 and 10 controls | Severe TBI | 16–74 y | 0–7 d + 8–365 d | Rs6265 and rs7124442 (c-carrier) | CSF and serum BDNF levels. Mortaility GOS-scores | BDNF-GRS + serum BDNF predictive of mortality |
| (Failla et al. 2015) [18] | 315 | Severe TBI | 16–75 y | 0–7 d + 8–365 d | Rs6265 and rs7124442 (c-carrier) | Mortality GOS BDNF-GRS | BDNF met-carrier higher mortality first year and long term in younger individuals (<45 y), lower mortality >45 y. |
| (Munoz et al. 2017) [19] | 117 | Severe TBI | 16–73 y | 6 months | Rs6265 and rs7124442 (c-carrier) | Mortality GOS CSF cortisol CSF/serum BDNF BDNF-GRS | BDNF-GRS predict mortality CSF levels of BDNF in association with CSF cortisol correlate to mortality |
| BDNF cognition | |||||||
| (Bagnato et al. 2012) [39] | 53 | Vegetative | 15–55 y | 1–12 m | Rs6265 2 groups val/val + ohters | Levels of cognitive function 1–8 | No difference in LCF (1–8) |
| (Barbey et al. 2014) [40] | 156 | Penetrating, frontal lobe lesion | 30–40 y | Rs6265 2 groups val/val + ohters | WAIS | val/val 6–8 lower IQ score | |
| (Krueger et al. 2011) [16] | 168 and 47 controls | Penetrating, frontal lobe lesion | 52–70 y | Ca 40 y | Rs 6265 2 groups val/val + others | D-KEFS cognitive and executive function | Val/val post TBI lower score |
| (McAllister et al. 2012) [17] | 75 and 38 controls | Mild/moderate TBI | 33.1 y mean | 1 month | Rs6265 (3 groups) Rs11030102 Rs11030107 Rs12273363 | CVLT CPT Gordon WAIS + WRAT for baseline. | Met/met slower RT, no difference between TBI and controls |
| (Merritt et al. 2020) [28] | 75 and 63 military controls | Mild TBI | 22–53 y | Mean 66.7 months | Rs6265 (2 groups) | WRAT D-KEFS CVLT-II RCFT WAIS | Met-carriers better memory and executive function post TBI. Reverse for controls. |
| (Narayanan et al. 2016) [27] | 48 | Mild TBI | 18–53 y | 10 h post GCS 15 and 6 months | Rs6265 (2 groups) Rs1048218 Rs1048220 Rs1048221 Rs8192466 Rs139352447 | S-NAB | Met-carriers worse memory at 6 months |
| (Rostami et al. 2011) [20] | 109 and 38 controls | Focal, penetrating TBI | Phase II = 10–15 y Phase III = 30–35 y | Rs1519480 Rs7124442 rs6265 rs7934165 Rs11030121 Rs12273363 Rs908867 | Army Force Qualification Test WAIS | C/C of Rs1519480 (and Rs7124442 = worse performance phase II, unchanged phase III | |
| BDNF psychological factors | |||||||
| (Dretsch et al. 2016) [24] | 231 of 458 soldiers | Mild TBI | Within 30 days of return | Rs6265 (3 groups) (APOE, DRD2) | Pre- and post deployment: CNS-VS Neurobehavioral Symptom Inventory | Met-homozygote higher incidence of mTBI Met-homozygote higher incidence of PTSD | |
| (Gabrys et al. 2019) [120] | 219 | Mild TBI | Life time | Rs6265 (2 groups) | WCST Subtypes of rumination BDI (Beck Depression Inventory) | Met-carriers with history of mTBI had a higher frequency of brooding, depressive symtoms and less cognitive flexibility. More pronounced difference among female participants. | |
| (Lanctt et al. 2010) [38] | 90 | Mild/moderate TBI | Mean 39.9 y | Unknown | Rs6265 5HT1A/2A TPH2 MTHFR | HAMD | Val-homozygote better responders to Citalopram |
| (Wang et al. 2018) [36] | 192 (103 in the follow up) | Mild TBI | 20–83 y | 1 week and 6 weeks | Rs6265 (C/T) | BAI BDI | Male, T-carriers had more anxiety and depression |
| BDNF other | |||||||
| (Hunter et al. 2019) [32] | 312 + 110 controls | Heading (subconcussive brain injury) | 18–55 y | Baseline and 24 months follow up | Rs6265 (2 groups) | Heading self report MR DTI | Met-carriers had less remyeliniation if q4 (>1460 headings) |
| (Larson-Dupuis et al. 2015) [25] | 54 and 51 controls | Concussion | Mean 20.8 y (3–85 y) | Mean 27 months | Rs6265 (3 groups) | Sniffin’ Sticks Inventory Test PCS-scale | Val-homozygote with history of mTBI had worse olfactory function |
| (Hayes et al. 2018) [21] | 110 and 55 controls | Mild TBI | 19–58 y | N/A | BDNF Rs1157659 Rs 6265) | Hippocampal volume on MRI fMRI—functional connecticity | Rs1157659 minor allele homozygote had reduced functional connectivity in defualt mode network and smaller hippocampal volume |
| BDNF Animal Studies | N = | Animal | Trauma Model | BDNF Analysis | Site of Analysis | Date of Analysis | BDNF Expression | Functional Tests |
|---|---|---|---|---|---|---|---|---|
| (Edut et al. 2014) [121] | 9–13/group | Mouse | Weight drop | Western blot | Cortex, striatum | 1 h, 24 h, 72 h, 7days and 30 days | Not altered by trauma, not altered by treatment at any timepoints | Novel object recognition, Y-maze |
| (Sen et al. 2017) [83] | 8–10/group | Mouse | CCI | Western blot | Site of impact | 25 days | Decreased by trauma, increased by treatment at all timepoints | Morris Water Maze |
| (Li et al. 2017) [122] | 20/group | Rat | FPI | Western blot and ELISA | "tissue homogenates" | 6 h, 12 h, 48 h, 96 h and 168 h | No sham. Increased by treatment at all timepoints | Beam walking test, Morris Water Maze, and modified Neurological Severity Score |
| (Rostami et al. 2014) [9] | 3–4/group | Rat | Penetrating injury | In situ hybridization and Microarray | Hippocampus | 1, 3, 14 and 56 days | Contralateral DG and CA3: Significant increase at day 1, 3, and 14 but not at day 56. Ipsilateral/contralateral ratio of BDNF mRNA did not differ from levels in sham until 8 weeks following injury | Not performed |
| (Kobori et al. 2002) [70] | 10/group | Mouse | CCI | RNA Microarray and real-time PCR | Cortex | 2 h, 6 h, 24 h, 3 days and 14 days | Increased at 2 h, 6 h, 24 h, decreased at 3 days, 14 days. | Not performed |
| (Wang et al. 2018) [123] | 12/group, 6/group for western | Mouse | Blunt weight drop | Western blot | Homogenized hippocampal tissues | 21 days | Decreased by trauma, increased by treatment at all timepoints | Morris Water Maze, step-down test |
| (Umschweif et al. 2014) [124] | 6–9/group | Mouse | Closed head injury | Western blot | Cortex and subcortical regions | 24 h | No sham. Increased by treatment. | Neurological Severity Score and Novel Object Recognition |
| (Ji et al. 2017) [77] | 7/group | Mouse | Blunt weight drop | Western blot | Cortex | 7 days | Decreased by trauma, increased by treatment | Neurological Severity Score, rotarod, object recognition, Y-maze |
| (Umschweif et al., 2014) [125] | 6/grp | Mouse | CHI | Western blot | Frontal segments of injured hemisphere | 6, 24 and 72 h | Increased by treatment | Neurological severity score and novel object recognition Test |
| (Feng et al. 2017) [81] | 5/group | Rat | Blunt weight drop | Western blot and immunochemistry | Cortex | 14 days | Not altered by trauma, increased by treatment | Modified Neurological Severity Score, rotarod |
| (Wu et al. 2011) [61] | 5/group | Rat | Fluid percussion injury | Western blot | Hippocampus | Not specified | Decreased by trauma, increased by treatment | Beam walking test |
| (Rich et al. 2010) [94] | 3/group | Rat | Blunt weight drop | Western blot | Site of injury, hippocampus | 24 h | No sham, increased by treatment | Morris Water Maze |
| (Lee et al. 2012) [126] | 4/group | Rat | Blunt weight drop | In situ hybridization and immunostaining | Hippocampus | 0, 2, 4, 12 and 24 h | Increased by trauma (peaked at 12 h), increased by treatment at all timepoints | Not performed |
| (Gu et al. 2014) [127] | 7 for Western blot | Mouse | Convex tip impact | Western blot | Hippocampus | 20 days | Decreased by trauma, increased by treatment | Morris Water Maze |
| (Griesbach et al. 2009) [48] | 6/group | Rat | CCI | Real-time PCR | Hippocampus | 10 days | Decreased by trauma in ipsilateral hippocampus, no differences in contralateral hippocampus between injured and sham | Morris Water Maze |
| (Agrawal et al. 2014) [99] | 3/group | Rat | FPI | Immunoblotting | Cortex | 7 days | Decreased by trauma, increased by treatment | Elevated plus maze |
| (Wu et al. 2006) [53] | 8/group | Rat | FPI | ELISA | Hippocampus | Not specified | Decreased by trauma, increased by treatment | Morris water maze |
| (Griesbach et al. 2012) [63] | 11–23/group | Rat | FPI | Western blot and ELISA | Hippocampus | 11 days | Not altered by trauma, not altered by treatment. Although increased by treatment in non-injured group | Not performed |
| (Zhao et al. 2017) [82] | 5/group | Mouse | CCI | Western blot | Pericontusion tissue | 21 days | Not altered by trauma, increased by treatment | Adhesive dot removal test, corner test, home cage behavioral test, cylinder test, forced swim test, open field test |
| (Yoon et al. 2016) [128] | 12/group | Rat | FPI | Immunohistochemistry | Cortex and hippocampus | 14 and 21 days | No sham. Increased by treatment in ipsilateral hippocampus and cortex | Rotarod, Barnes Maze |
| (Deng et al. 2018) [102] | 10/group | Rat | Electronic brain injury instrument | Immunohistochemistry | Peri-injured area | 28 days | No sham, increased by treatment | Morris water maze |
| (Liraz-Zaltsman et al. 2018) [129] | 3/group | Mouse | Closed head injury | Immunohistochemistry | Cortex and hippocampus | 42 days | No sham, increased by treatment | Neurological severity score, Novel object recognition, Y-maze, Barnes maze, staircase test |
| (Gatson et al. 2012) [130] | 6 for control, 8 for sham, 10 for placebo, and 9 for estrone | Rat | Benchmark Stereotaxic Impactor | Immunohistochemistry | Cortex | 72 h | No sham, increased by treatment | Not performed |
| (Wu et al. 2013) [64] | 5–6/group | Rat | FPI | Western blot | Hippocampus | 7 days | Decreased by trauma, increased by treatment | Morris water maze, Barnes maze |
| (Chytrova et al. 2008) [62] | 6/group | Rat | FPI | Immunohistochemistry | Hippocampus | 10 days | Decreased by trauma, increased by treatment | Not performed |
| (Chou et al. 2018) [69] | 10/group | Rat | FPI | Western blot | Cortex and hippocampus | 28 days | Decreased by trauma, increased by treatment | Passive avoidance test and Y-maze |
| (Griesbach et al. 2009) [67] | 8/grp | Rat | FPI | Western blot | Hippocampus | 21 days | Not altered by trauma, increased by treatment | Morris water maze |
| (Crupi et al. 2013) [73] | 5/group | Mouse | CCI | Western blot | Cortex and hippocampus | 24 h | Decreased by trauma, increased by treatment | Swing test and rotarod |
| (Grundy et al. 2000) [131] | 34 for TBI, 13 for sham | Rat | FPI | In situ hybridization | Hippocampus | 4 h | Increased by trauma, further increased by treatment | Not performed |
| (Song et al. 2016) [132] | 20/grp | Mouse | CCI | ELISA | Cortex and hippocampus | 3, 7 and 14 days | Sham not presented, increased by treatment at day 3 ipsilaterally and day 7 contralaterally | Radial Arm Water Maze and rotarod |
| (Dobrachinski et al. 2019) [50] | 5/group | Rat | FPI | Quantitative PCR | Hippocampus | 21 days | Decreased by trauma, increased by treatment | Locomotor behavior, Elevated plus maze, Inhibitory avoidance task, and object recognition test |
| (Matzilevich et al. 2002) [46] | 10/group | Rat | CCI | Microarray, northern blot and immunohistochemistry | Hippocampus | 5 h and 26 h | Not altered at 5 h, increased at 26 h | Not performed |
| (Wang et al. 2014) [44] | 4/group | Rat | FPI | Quantitative real-time PCR | Hippocampus | 3 h and 6 h | Increased by trauma, decreased by treatment in ipsilateral hippocampus at 3 h but not altered in contralateral | Not performed |
| (Hou et al. 2012) [56] | 12/group | Rat | FPI | ELISA | HC | 7 days | Decreased by trauma, increased by treatment | Morris water maze |
| (Algamal et al. 2019) [133] | 15/group | Mouse | Closed head injury | ELISA | HC | 3 months | Not altered by trauma | Contextual and cued fear test, open field, elevated plus maze, forced swim, radial arm water maze and three chamber test |
| (Yang et al. 1996) [47] | 3/group | Rat | CCI | In situ hybridization | Cortex and hippocampus | 1, 3, and 5 h | Increased by trauma | Postural somatomotor reflexes |
| (Mahmood et al. 2004) [100] | 9/group | Rat | Pneumatic piston compression | Immunohistochemistry and ELISA | Periinjured area | 2, 5 and 8 days | No sham. Increased by treatment at day 8 but no significantly altered at day 2 or 5 | Modified neurological severity score |
| (Ko et al. 2018) [134] | 10/grp | Rat | Stereotaxic Impactor | Western blot | Hippocampus | 11 weeks | Increased by trauma, decreased by treatment | Radial 8-arm maze |
| (Mahmood et al. 2009) [135] | 8/group | Rat | CCI | ELISA | Cortex and hippocampus | 3 months | No sham, increased by treatment. | Modified neurological severity score |
| (Mahmood et al. 2006) [101] | 10/grp | Rat | CCI | ELISA | Contralateral and ipsilateral hemispheres | 3 months | No sham. Increased by treatment. | Neurological severity score |
| (Xuan et al. 2015) [136] | 10/group | Mouse | CCI | Immunofluorescence staining | Cortex and hippocampus | 7 and 28 days | Not altered by trauma, increased by treatment | Neurological severity score |
| (Wu et al. 2016) [74] | 12/grp | Rat | CCI | Western blot | Cortex | 24 h | Decreased by trauma, increased by treatment | Neurological severity score |
| (Colak et al. 2012) [43] | 15/grp | Rat | CHI | Quantitative real-time PCR and whole genome microarray | Frontal and parietal brain tissues | 1, 12 and 48 h | Increased at 1h and at 12 h compared to control group, decreased at 48 h. | Not performed |
| (Shah et al. 2006) [137] | 4/group | Mouse | FPI | Real-time PCR | Hippocampus | 1 day | Not altered by trauma | Not performed |
| (Esenaliev et al. 2018) [138] | 3–4/grp | Rat | Blast injury | Quantitative real-time PCR | Cortex and hippocampus | 3 and 7 days | Decreased by trauma at 3 in cortex and at 7 days in the hippocampus. Increased by treatment. | Beam balance test |
| (Xiong et al. 2018) [79] | 5–8/grp | Rat | Blunt weight drop | Immunofluorescent staining and ELISA | Pericontusional regions | 7 days | Decreased by trauma, increased by treatment | Neurological severity score and rotarod |
| (Bhatt et al. 2017) [139] | 6/grp | Rat | Blunt weight drop | ELISA | Whole brain | 28 days | Decreased by trauma, increased by treatment | Open field, elevated plus maze, sucrose consumption test and marble burying test |
| (Chandrasekar et al. 2018) [95] | Not available | Mouse | Blunt weight drop | Real-time PCR | Hippocampus | 1 h and 3 h | Increased by trauma, decreased by treatment | Neurological severity score |
| (Gugliandolo et al. 2018) [75] | 10/grp | Mouse | CCI | Immunohistochemistry and immunofluorescence | Perilesional tissue | 1 day | Decreased by trauma, increased by treatment | Not performed |
| (Shin et al. 2016) [93] | 10/grp | Rat | Stereotaxic Impactor | Western blot | Hippocampus | 41 days | Deceased by trauma, increased by treatment | Step-down avoidance test and radial 8-arm maze test |
| (Sönmez et al. 2015) [140] | 7/grp | Rat | Percussion trauma model in immature rats | Immunohistochemistry | Hippocampus | 4 days | Decreased by trauma, increased by treatment | Elevated plus maze and novel object recognition |
| (Shang et al. 2014) [141] | 8/group | Mouse | CCI | Western blot | Hippocampus | 1, 3, 7, 14 and 21 days | Decreased by trauma, increased by treatment | Open field test, beam walk and Morris water maze |
| (Mao et al. 2015) [65] | 24 for TBI, 20 for sham | Rat | CCI | ELISA | Hippocampus | 14 days | Decreased by trauma, increased by treatment | Morris water maze |
| (Ghadiri et al. 2019) [142] | 14/group | Rat | Weight drop | Western blot | Hippocampus | 15 days | No sham, increased by treatment | Modified neurological severity score and tonic-clonic seizure score |
| (Cekic et al. 2012) [88] | 18/group | Rat | CCI | Western blot | Perilesional tissue | 1, 3 and 7 days | Increased by trauma, decreased by treatment at day 1. Not altered by trauma, decreased by treatment at day 3. Increased by trauma, not altered by treatment at day 7. | Spontaneous locomotor activity and behavioral data |
| (Nagamoto-Combs et al. 2007) [89] | 1/timepoint, (3 total) | Rhesus monkey | Aspiration injury | Immunohistochemistry | Cortex | 1, 6, and 12 months | Non-detectable BDNF-immunoreactivity in sham. Increased BDNF-immunoreactivity at all time points, especially at 6 months. | Modified monkey assessment panel |
| (Ma et al. 2018) [85] | 14/group | Rat | CCI | Western blot and immunohistochemistry | Cortex | 30 days | Decreased by trauma, increased by treatment | Modified neurological severity score and Object Recognition Test |
| (Griesbach et al. 2014) [91] | 29 for TBI, 28 for sham | Rat | FPI | ELISA | Hippocampus | 39 days | Decreased by trauma, increased by treatment | Activity level |
| (Oyesiku et al. 1999) [143] | 6–9/group | Rat | Pneumatic piston compression | Northern blot | Lesion site | 12, 24 and 36 h | Increased by trauma at 12 and 24 h, not altered at 36 h at lesion site and remote site. | Not performed |
| (Gölz et al. 2019) [144] | 10 for TBI, 6 for sham | Mouse | CCI | Western blot | Ipsilesional brain quadrants | 72 h | Not altered by trauma, not altered by treatment | Neurological severity score |
| (Khan et al. 2011) [49] | 13 for control, 30 for sham, 28 or 40 for treated | Rat | CCI | Immunohistochemistry | Whole brain sections | 14 days | Decreased by trauma, increased by treatment | Not performed |
| (Corne et al. 2019) [90] | 6–8/grp | Mouse | CCI | Quantitative real-time PCR | Whole brain sections | 3 months | Decreased by trauma | Fear conditioning and fear extinction, elevated plus maze, open field test, Barnes maze and sensory-motor skills |
| (Wang et al. 2019) [105] | 10/grp | Rat | Blunt weight drop | Immunofluorescence | Perilesional tissue | 10, 14 and 20 days | No sham, increased by treatment | Neurological severity score |
| (Meng et al. 2014) [80] | 4/group | Rat | CCI | Western blot and immunohistochemistry | Cortex | 8 and 35 days | Decreased by trauma, increased by treatment | Morris water maze and modified neurological severity score |
| (Cutler et al. 2006) [145] | 10/grp | Rat | Pneumatic cortical contusion | Western blot | Perilesional tissue | 3 weeks | Decreased by trauma, increased by treatment | Locomotor activity and somatosensory neglect |
| (Tyagi et al. 2014) [54] | 6/grp | Rat | FPI | Immunoblotting | Hippocampus | 7 days | Decreased by trauma, increased by treatment | Elevated plus maze |
| (Ignowski et al. 2018) [96] | 3–4/group | Mouse | CCI | Immunohistochemistry | Whole brain lysates | 72 h | Deceased by trauma, increased by treatment. | Beam walking test, rotarod and Barnes maze |
| (Mychasiuk et al. 2016) [146] | 12/grp | Rat | Modified weight drop mTBI and Lateral impact mTBI | Quantitative real-time PCR | Cortex and hippocampus | 14 days | No significant difference between sham, weight drop or lateral impact. BDNF expression was dependent on examined brain region and animal sex | Time-to-right, open field test, elevated plus maze, novel context mismatch and forced swim test |
| (Shen et al. 2013) [68] | 10 for trauma, 5 for sham | Rat | CCI | Western blot | Hippocampus | 26 days | Not altered by trauma, increased by treatment | Neurologic Deficit Scores and Morris water maze |
| (da Silva Fiorin et al. 2016) [66] | 40/grp | Rat | FPI | Western blot | Hippocampus | 1 and 15 days | Not altered by trauma, increased by treatment at both timepoints. 1, 15 DPI. | Neuroscore and object recognition test |
| (Qi et al. 2018) [104] | 10/grp | Rat | CCI | Quantitative real-time PCR and ELISA | Whole brain | 7, 14, 21 and 28 days | No sham, increased by treatment | Neurological severity score |
| (Xing et al. 2018) [147] | 3/grp | Rat | Modified Marmarou weight drop | Quantitative real-time PCR | Ipsilateral hemisphere | 3 h, 6 h, 1 day, 3 days and 7 days | Not altered by trauma, increased by treatment | Not performed |
| (Wu et al. 2011) [61] | 12/grp | Rat | FPI | Western blot | Hippocampus | 7 days | Decreased by trauma, increased by treatment. | Morris water maze |
| (Griesbach et al. 2004) [78] | 4/grp | Rat | FPI | ELISA | Cortex | 7 days | Not altered by trauma, not altered by treatment | Beam-walk test |
| (Kim et al. 2010) [103] | 6 for ELISA, 2 for immunohistochemistry | Rat | Pneumatic cortical contusion | ELISA | Ipsilateral hemispheres | 2, 8, 15, 29 days | Increased by trauma, increased by treatment at day 2 but not at day 8, 15 or 29 | Modified neurological severity score and rotarod |
| (Griesbach et al. 2007) [59] | 8/grp | Rat | FPI | ELISA | Hippocampus | 7, 21 and 37 days | Not altered by trauma, increased by treatment. Dependent on timing of exercise. | Gross motor impairments |
| (Su et al. 2017) [76] | 6-7/grp | Mouse | CCI | Western blot and immunohistochemistry | Cortex | 1 and 4 days | Decreased by trauma, increased by treatment at day 4 but not day 1. | Not performed |
| (Impellizzeri et al. 2016) [148] | 5/grp | Mouse | Chronic CCI | Immunohistochemistry | Midbrain samples | 30 days | Decreased by trauma. | Open field, Elevated plus maze and Barnes maze |
| (Boone et al. 2012) [45] | 6/grp | Rat | FPI | Quantitative real-time PCR | Ipsilateral hippocampus | 20 hours | Decreased by trauma and sham | Locomotor behavior |
| (Mahmood et al. 2007) [149] | 5/grp | Rat | CCI | ELISA | Ipsilateral hemisphere | 6 and 24 h | Increased by treatment | Morris water maze |
| (Portbury et al. 2017) [98] | 5/grp | Mouse | CCI | Western blot | Ipsilateral and contralateral cortex and hippocampus | 24 h, 72 h, 7 days, 14 days and 28 days | Increased by treatment in the contralateral cortex, but not in the other examined areas | Morris water maze and Y-maze |
| (Chang et al. 2019) [84] | 5/grp | Swine | CCI | Western blot | Cortical sections adjacent to the injury. | 30 days | Decreased by trauma, increased by treatment | neurological severity score |
| (Wu et al. 2010) [55] | 6–8/grp | Rat | FPI | ELISA | ipsilateral hippocampus | 7 days dietary | Decreased by trauma, increased by treatment | Morris water maze |
| (Griesbach et al. 2004) [58] | FPI = 89, sham = 72 | Rat | FPI | ELISA and immunohistochemistry | Ipsilateral and contralateral hippocampus | 7 and 21 days | Not altered by trauma. Not altered by treatment ipsilaterally at day 7, but increased by treatment ipsilaterally at day 21. Decreased by treatment contralaterally at day 7 but increased contralaterally at day 21 | Water maze |
| (Zhao et al. 2015) [71] | 5–8/grp | Mouse | CCI | Quantitative real-time PCR | Ipsilateral cortex | 24 hours | Not altered by trauma, increased by treatment | Beam walking, Morris water maze, tail-suspension, open field test, and novel object recognition test |
| (Cheng et al. 2015) [72] | 6/grp | Rat | Weight drop | Western blot and quantitative real-time PCR | Ipsilateral perilesional cortex and subcortical regions. | 14 days | Increased by treatment | Modified neurological severity score, Morris water maze, and novel object recognition |
| Transgenic animal studies | N = | Animal | Trauma Model | BDNF Analysis | Site of Analysis | Date of Analysis | BDNF Expression | Functional Tests |
| (Giarratana et al. 2019) [110] | 4–6/group | Mouse | Lateral fluid percussion (repeated mild TBI) | Western blot | Cortex and hippocampus | 1 and 21 DPI | Decreased total BDNF in Val66Met in ipsilateral cortex at 21 DPI, increased pro/mature-BDNF in hippocampus at 1 DPI compared to Val66Val. | Rotarod, balance beam, and Morris water maze |
| Gao et al. (2009) [111] | 5–10/group | Mouse | CCI | Western blot and immunohistochemistry | Hippocampus | 1 and 21 DPI | TBI significantly increased the level of BDNF protein in the dentate gyrus, but less in conditional KO mice | Not performed |
| Cheng et al. (2017) [112] | 4/group | Mouse | CCI | Western blot | Cortex | 1 and 21 DPI | TBI increased BDNF protein expression in both contra- and ipsilaterally in WT at 21 days after trauma. In TSP-1 KO BDNF increased only ipsilaterally. | Neurological severity score, Morris water maze, wire grip and corner test |
| BDNF Polymorphism and TBI in Humans | Genes and Polymorphism | Findings | Limitations |
|---|---|---|---|
| (Davidson et al. 2015) [150] | Several genes including BDNF rs6265. | Met-carriers slower reaction time both healthy subjects and post TBI. No association with PTSD. | Few and small studies. Met-carriers one group including both heterozygote and homozygote. |
| (Zeiler et al. 2019) [3] | Several genes, including rs6265 and other polymorphisms of BDNF | Varying results for BDNF rs6265. Some studies show better cognitive function for met-carriers, others better results for val-homozygote. Higher mortality first year for met-carriers. | Small studies with overlapping cohorts. Bias towards positive results. Risk for confounding factors. |
| (Finan et al. 2018) [151] | BDNF rs6265 | The effect of the Met-allele depends on time point after TBI. Early evaluation, negative impact on cognition while protective effect of cognition at a later timing (years). | Confounding factors: gender, age, injury, severity, ethnicity, time point. Interaction with other genetic polymorphisms. |
References
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiler, F.A.; McFadyen, C.; Newcombe, V.F.J.; Synnot, A.; Donoghue, E.L.; Ripatti, S.; Steyerberg, E.W.; Gruen, R.L.; McAllister, T.W.; Rosand, J.; et al. Genetic Influences on Patient-Oriented Outcomes in Traumatic Brain Injury: A Living Systematic Review of Non-Apolipoprotein E Single-Nucleotide Polymorphisms. J. Neurotrauma 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar] [CrossRef]
- Lipsky, R.H.; Marini, A.M. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann. N. Y. Acad. Sci. 2007, 1122, 130–143. [Google Scholar] [CrossRef]
- Poo, M.M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef]
- Klein, R.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Frisen, J.; Verge, V.M.; Fried, K.; Risling, M.; Persson, H.; Trotter, J.; Hokfelt, T.; Lindholm, D. Characterization of glial trkB receptors: Differential response to injury in the central and peripheral nervous systems. Proc. Natl. Acad. Sci. USA 1993, 90, 4971–4975. [Google Scholar] [CrossRef] [Green Version]
- Rostami, E.; Krueger, F.; Plantman, S.; Davidsson, J.; Agoston, D.; Grafman, J.; Risling, M. Alteration in BDNF and its receptors, full-length and truncated TrkB and p75(NTR) following penetrating traumatic brain injury. Brain Res. 2014, 1542, 195–205. [Google Scholar] [CrossRef]
- Dechant, G.; Barde, Y.A. The neurotrophin receptor p75(NTR): Novel functions and implications for diseases of the nervous system. Nat. Neurosci. 2002, 5, 1131–1136. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function and its val/met polymorphism in human memory and hippocampal function and suggest val/met exerts these effects by impacting intracellular trafficking and activity-dependent secretion of BDNF. Cell 2003, 112, 257–269. [Google Scholar]
- Notaras, M.; Hill, R.; van den Buuse, M. The BDNF gene Val66Met polymorphism as a modifier of psychiatric disorder susceptibility: Progress and controversy. Mol. Psychiatry 2015, 20, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, D.P.; Ramanathan, M.; Cox, J.L.; Antulov, R.; Weinstock-Guttman, B.; Bergsland, N.; Benedict, R.H.; Dwyer, M.G.; Minagar, A.; Zivadinov, R. Effect of Met66 allele of the BDNF rs6265 SNP on regional gray matter volumes in patients with multiple sclerosis: A voxel-based morphometry study. Pathophysiology 2011, 18, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Voineskos, A.N.; Lerch, J.P.; Felsky, D.; Shaikh, S.; Rajji, T.K.; Miranda, D.; Lobaugh, N.J.; Mulsant, B.H.; Pollock, B.G.; Kennedy, J.L. The Brain-Derived Neurotrophic Factor Val66Met Polymorphism and Prediction of Neural Risk for Alzheimer Disease. Arch. Gen. Psychiatry 2011, 68, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.E.; Fox, H.; Wright, A.F.; Hayward, C.; Starr, J.M.; Whalley, L.J.; Deary, I.J. The brain-derived neurotrophic factor Val66Met polymorphism is associated with age-related change in reasoning skills. Mol. Psychiatry 2006, 11, 505–513. [Google Scholar] [CrossRef]
- Krueger, F.; Pardini, M.; Huey, E.D.; Raymont, V.; Solomon, J.; Lipsky, R.H.; Hodgkinson, C.A.; Goldman, D.; Grafman, J. The role of the met66 brain-derived neurotrophic factor allele in the recovery of executive functioning after combat-related traumatic brain injury. J. Neurosci. 2011, 31, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, T.W.; Tyler, A.L.; Flashman, L.A.; Rhodes, C.H.; McDonald, B.C.; Saykin, A.J.; Tosteson, T.D.; Tsongalis, G.J.; Moore, J.H. Polymorphisms in the brain-derived neurotrophic factor gene influence memory and processing speed one month after brain injury. J. Neurotrauma 2012, 29, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Failla, M.D.; Kumar, R.G.; Peitzman, A.B.; Conley, Y.P.; Ferrell, R.E.; Wagner, A.K. Variation in the BDNF gene interacts with age to predict mortality in a prospective, longitudinal cohort with severe TBI. Neurorehabilit. Neural Repair 2015, 29, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.J.; Kumar, R.G.; Oh, B.M.; Conley, Y.P.; Wang, Z.; Failla, M.D.; Wagner, A.K. Cerebrospinal fluid cortisol mediates brain-derived neurotrophic factor relationships to mortality after severe TBI: A prospective cohort study. Front. Mol. Neurosci. 2017, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Rostami, E.; Krueger, F.; Zoubak, S.; Monte, O.D.; Raymont, V.; Pardini, M.; Hodgkinson, C.A.; Goldman, D.; Risling, M.; Grafman, J. Bdnf polymorphism predicts general intelligence after penetrating traumatic brain injury. PLoS ONE 2011, 6, e27389. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.P.; Reagan, A.; Logue, M.W.; Hayes, S.M.; Sadeh, N.; Miller, D.R.; Verfaellie, M.; Wolf, E.J.; McGlinchey, R.E.; Milberg, W.P.; et al. BDNF genotype is associated with hippocampal volume in mild traumatic brain injury. Genes Brain Behav. 2018, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, E.; Hashimoto, K.; Iyo, M. Ethnic difference of the BDNF 196G/A (val66met) polymorphism frequencies: The possibility to explain ethnic mental traits. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 126B, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Petryshen, T.L.; Sabeti, P.C.; Aldinger, K.A.; Fry, B.; Fan, J.B.; Schaffner, S.F.; Waggoner, S.G.; Tahl, A.R.; Sklar, P. Population genetic study of the brain-derived neurotrophic factor (BDNF) gene. Mol. Psychiatry 2010, 15, 810–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dretsch, M.N.; Williams, K.; Emmerich, T.; Crynen, G.; Ait-Ghezala, G.; Chaytow, H.; Mathura, V.; Crawford, F.C.; Iverson, G.L. Brain-derived neurotropic factor polymorphisms, traumatic stress, mild traumatic brain injury, and combat exposure contribute to postdeployment traumatic stress. Brain Behav. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson-Dupuis, C.; Chamard, É.; Falardeau, V.; Frasnelli, J.; Beaulieu, C.; Poirier, J.; Carrier, J.; Lassonde, M.; Théoret, H.; Bacon, B.A.; et al. Impact of BDNF Val66Met polymorphism on olfactory functions of female concussed athletes. Brain Inj. 2015, 29, 963–970. [Google Scholar] [CrossRef]
- Hariri, A.R.; Goldberg, T.E.; Mattay, V.S.; Kolachana, B.S.; Callicott, J.H.; Egan, M.F.; Weinberger, D.R. Brain-Derived Neurotrophic Factor val 66 met Polymorphism Affects Human Memory-Related Hippocampal Activity and Predicts Memory Performance. J. Neurosci. 2003, 23, 6690–6694. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, V.; Veeramuthu, V.; Ahmad-Annuar, A.; Ramli, N.; Waran, V.; Chinna, K.; Bondi, M.W.; Delano-Wood, L.; Ganesan, D. Missense mutation of Brain Derived Neurotrophic Factor (BDNF) alters neurocognitive performance in patients with mild traumatic brain injury: A longitudinal study. PLoS ONE 2016, 11, e0158838. [Google Scholar]
- Merritt, V.C.; Clark, A.L.; Evangelista, N.D.; Sorg, S.F.; Schiehser, D.M.; Delano-Wood, L. Dissociation of BDNF Val66Met polymorphism on neurocognitive functioning in military veterans with and without a history of remote mild traumatic brain injury. Clin. Neuropsychol. 2020, 34, 1226–1247. [Google Scholar] [CrossRef]
- Mendez, M.F. What is the Relationship of Traumatic Brain Injury to Dementia? J. Alzheimers Dis. 2017, 57, 667–681. [Google Scholar] [CrossRef]
- Nordström, A.; Nordström, P. Traumatic brain injury and the risk of dementia diagnosis: A nationwide cohort study. PLoS Med. 2018, 15, e1002496. [Google Scholar] [CrossRef]
- Barnes, D.E.; Byers, A.L.; Gardner, R.C.; Seal, K.H.; Boscardin, W.J.; Yaffe, K. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol. 2018, 75, 1055–1061. [Google Scholar] [CrossRef]
- Hunter, L.E.; Freudenberg-Hua, Y.; Davies, P.; Kim, M.; Fleysher, R.; Stewart, W.F.; Lipton, R.B.; Lipton, M.L. BDNF Val66Met Positive Players Demonstrate Diffusion Tensor Imaging Consistent with Impaired Myelination Associated With High Levels of Soccer Heading: Indication of a Potential Gene-Environment Interaction Mechanism. Front. Neurol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Failla, M.D.; Conley, Y.P.; Wagner, A.K. Brain-Derived Neurotrophic Factor (BDNF) in Traumatic Brain Injury-Related Mortality: Interrelationships between Genetics and Acute Systemic and Central Nervous System BDNF Profiles. Neurorehabilit. Neural Repair 2016, 30, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Van Praag, D.L.G.; Cnossen, M.C.; Polinder, S.; Wilson, L.; Maas, A.I.R. Post-Traumatic Stress Disorder after Civilian Traumatic Brain Injury: A Systematic Review and Meta-Analysis of Prevalence Rates. J. Neurotrauma 2019, 36, 3220–3232. [Google Scholar] [CrossRef]
- Bombardier, C.H.; Fann, J.R.; Temkin, N.R.; Esselman, P.C.; Barber, J.; Dikmen, S.S. Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA 2010, 303, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.J.; Chen, K.Y.; Kuo, L.N.; Wang, W.C.; Hsu, Y.W.; Wong, H.S.C.; Lin, C.M.; Liao, K.H.; Zhang, Y.F.; Chiang, Y.H.; et al. The association between BDNF Val66Met polymorphism and emotional symptoms after mild traumatic brain injury. BMC Med. Genet. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Clasen, P.C.; Wells, T.T.; Knopik, V.S.; Mcgeary, J.E.; Beevers, C.G. 5-HTTLPR and BDNF Val66Met polymorphisms moderate effects of stress on rumination. Genes Brain Behav. 2011, 10, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Lanctt, K.L.; Rapoport, M.J.; Chan, F.; Rajaram, R.D.; Strauss, J.; Sicard, T.; McCullagh, S.; Feinstein, A.; Kiss, A.; Kennedy, J.L.; et al. Genetic predictors of response to treatment with citalopram in depression secondary to traumatic brain injury. Brain Inj. 2010, 24, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, S.; Minafra, L.; Bravatà, V.; Boccagni, C.; Sant’Angelo, A.; Castiglione, A.; Andriolo, M.; Lucca, L.F.; De Tanti, A.; Pistarini, C.; et al. Brain-derived neurotrophic factor (Val66Met) polymorphism does not influence recovery from a post-traumatic vegetative state: A blinded retrospective multi-centric study. J. Neurotrauma 2012, 29, 2050–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbey, A.K.; Colom, R.; Paul, E.; Forbes, C.; Krueger, F.; Goldman, D.; Grafman, J. Preservation of general intelligence following traumatic brain injury: Contributions of the Met66 brain-derived neurotrophic factor. PLoS ONE 2014, 9, e88733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.; Nascimento, R.I.M.D.; Filho, E.M.R.; Bencke, J.; Regner, A. Plasma brain-derived neurotrophic factor levels after severe traumatic brain injury. Brain Inj. 2016, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Antonelli, A.; Riccardi, R.; Genovese, O.; Pezzotti, P.; Di Rocco, C.; Tortorolo, L.; Piedimonte, G. Nerve growth factor expression correlates with severity and outcome of traumatic brain injury in children. Eur. J. Paediatr. Neurol. 2008, 12, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, T.; Cine, N.; Bamac, B.; Kurtas, O.; Ozbek, A.; Bicer, U.; Sunnetci, D.; Savli, H. Microarray-based gene expression analysis of an animal model for closed head injury. Injury 2012, 43, 1264–1270. [Google Scholar] [CrossRef]
- Wang, Y.; Hameed, M.Q.; Rakhade, S.N.; Iglesias, A.H.; Muller, P.A.; Mou, D.L.; Rotenberg, A. Hippocampal immediate early gene transcription in the rat fluid percussion traumatic brain injury model. Neuroreport 2014, 25, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.R.; Sell, S.L.; Micci, M.A.; Crookshanks, J.M.; Parsley, M.; Uchida, T.; Prough, D.S.; DeWitt, D.S.; Hellmich, H.L. Traumatic brain injury-induced dysregulation of the circadian clock. PLoS ONE 2012, 7, e46204. [Google Scholar] [CrossRef] [PubMed]
- Matzilevich, D.A.; Rall, J.M.; Moore, A.N.; Grill, R.J.; Dash, P.K. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J. Neurosci. Res. 2002, 67, 646–663. [Google Scholar] [CrossRef]
- Yang, K.; Perez-Polo, J.R.; Mu, X.S.; Yan, H.Q.; Xue, J.J.; Iwamoto, Y.; Liu, S.J.; Dixon, C.E.; Hayes, R.L. Increased expression of brain-derived neurotrophic factor but not neurotrophin-3 mRNA in rat brain after cortical impact injury. J. Neurosci. Res. 1996, 44, 157–164. [Google Scholar] [CrossRef]
- Griesbach, G.S.; Sutton, R.L.; Hovda, D.A.; Ying, Z.; Gomez-Pinilla, F. Controlled contusion injury alters molecular systems associated with cognitive performance. J. Neurosci. Res. 2009, 87, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Sakakima, H.; Dhammu, T.S.; Shunmugavel, A.; Im, Y.B.; Gilg, A.G.; Singh, A.K.; Singh, I. S-nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J. Neuroinflamm. 2011, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Dobrachinski, F.; Gerbatin, R.R.; Sartori, G.; Golombieski, R.M.; Antoniazzi, A.; Nogueira, C.W.; Royes, L.F.; Fighera, M.R.; Porciuncula, L.O.; Cunha, R.A.; et al. Guanosine Attenuates Behavioral Deficits After Traumatic Brain Injury by Modulation of Adenosinergic Receptors. Mol. Neurobiol. 2019, 56, 3145–3158. [Google Scholar] [CrossRef]
- Krishna, G.; Ying, Z.; Gomez-Pinilla, F. Blueberry Supplementation Mitigates Altered Brain Plasticity and Behavior after Traumatic Brain Injury in Rats. Mol. Nutr. Food Res. 2019, 63, e1801055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, S.Z.; Motamedi, S.; Royo, N.C.; LeBold, D.; Watson, D.J. Identification of potentially neuroprotective genes upregulated by neurotrophin treatment of CA3 neurons in the injured brain. J. Neurotrauma 2011, 28, 415–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp. Neurol. 2006, 197, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, E.; Agrawal, R.; Ying, Z.; Gomez-Pinilla, F. TBI and sex: Crucial role of progesterone protecting the brain in an omega-3 deficient condition. Exp. Neurol. 2014, 253, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J. Neurotrauma 2011, 28, 2113–2122. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Luo, W.; Sun, X.; Hao, S.; Zhang, Y.; Xu, F.; Wang, Z.; Liu, B. Hydrogen-rich saline protects against oxidative damage and cognitive deficits after mild traumatic brain injury. Brain Res. Bull. 2012, 88, 560–565. [Google Scholar] [CrossRef]
- Alder, J.; Fujioka, W.; Giarratana, A.; Wissocki, J.; Thakkar, K.; Vuong, P.; Patel, B.; Chakraborty, T.; Elsabeh, R.; Parikh, A.; et al. Genetic and pharmacological intervention of the p75NTR pathway alters morphological and behavioural recovery following traumatic brain injury in mice. Brain Inj. 2016, 30, 48–65. [Google Scholar] [CrossRef] [Green Version]
- Griesbach, G.S.; Hovda, D.A.; Molteni, R.; Wu, A.; Gomez-Pinilla, F. Voluntary exercise following traumatic brain injury: Brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience 2004, 125, 129–139. [Google Scholar] [CrossRef]
- Griesbach, G.S.; Gomez-Pinilla, F.; Hovda, D.A. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J. Neurotrauma 2007, 24, 1161–1171. [Google Scholar] [CrossRef]
- Aiguo, W.; Zhe, Y.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Ying, Z.; Schubert, D.; Gomez-Pinilla, F. Brain and spinal cord interaction: A dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil. Neural Repair 2011, 25, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chytrova, G.; Ying, Z.; Gomez-Pinilla, F. Exercise normalizes levels of MAG and Nogo—A growth inhibitors after brain trauma. Eur. J. Neurosci. 2008, 27, 1–11. [Google Scholar] [CrossRef]
- Griesbach, G.S.; Tio, D.L.; Vincelli, J.; McArthur, D.L.; Taylor, A.N. Differential effects of voluntary and forced exercise on stress responses after traumatic brain injury. J. Neurotrauma 2012, 29, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience 2013, 248, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Hao, S.; Zhu, Z.; Zhang, H.; Wu, W.; Xu, F.; Liu, B. Procyanidins protects against oxidative damage and cognitive deficits after traumatic brain injury. Brain Inj. 2015, 29, 86–92. [Google Scholar] [CrossRef]
- Da Silva Fiorin, F.; de Oliveira Ferreira, A.P.; Ribeiro, L.R.; Silva, L.F.; de Castro, M.R.; da Silva, L.R.; da Silveira, M.E., Jr.; Zemolin, A.P.; Dobrachinski, F.; Marchesan de Oliveira, S.; et al. The Impact of Previous Physical Training on Redox Signaling after Traumatic Brain Injury in Rats: A Behavioral and Neurochemical Approach. J. Neurotrauma 2016, 33, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Hovda, D.A.; Gomez-Pinilla, F. Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. 2009, 1288, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Li, A.; Zhang, Y.; Dong, X.; Shan, T.; Wu, Y.; Jia, J.; Hu, Y. The effect of different intensities of treadmill exercise on cognitive function deficit following a severe controlled cortical impact in rats. Int. J. Mol. Sci. 2013, 14, 21598–21612. [Google Scholar] [CrossRef]
- Chou, W.; Liu, Y.F.; Lin, C.H.; Lin, M.T.; Chen, C.C.; Liu, W.P.; Chang, C.P.; Chio, C.C. Exercise Rehabilitation Attenuates Cognitive Deficits in Rats with Traumatic Brain Injury by Stimulating the Cerebral HSP20/BDNF/TrkB Signalling Axis. Mol. Neurobiol. 2018, 55, 8602–8611. [Google Scholar] [CrossRef]
- Kobori, N.; Clifton, G.L.; Dash, P. Altered expression of novel genes in the cerebral cortex following experimental brain injury. Brain Res. Mol. Brain Res. 2002, 104, 148–158. [Google Scholar] [CrossRef]
- Zhao, Z.; Sabirzhanov, B.; Wu, J.; Faden, A.I.; Stoica, B.A. Voluntary Exercise Preconditioning Activates Multiple Antiapoptotic Mechanisms and Improves Neurological Recovery after Experimental Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Yang, B.; Li, D.; Ma, S.; Tian, Y.; Qu, R.; Zhang, W.; Zhang, Y.; Hu, K.; Guan, F.; et al. Wharton’s Jelly Transplantation Improves Neurologic Function in a Rat Model of Traumatic Brain Injury. Cell. Mol. Neurobiol. 2015, 35, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crupi, R.; Paterniti, I.; Campolo, M.; Di Paola, R.; Cuzzocrea, S.; Esposito, E. Exogenous T3 administration provides neuroprotection in a murine model of traumatic brain injury. Pharmacol. Res. 2013, 70, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shao, A.; Zhao, M.; Chen, S.; Yu, J.; Zhou, J.; Liang, F.; Shi, L.; Dixon, B.J.; Wang, Z.; et al. Melatonin attenuates neuronal apoptosis through up-regulation of K(+) -Cl(-) cotransporter KCC2 expression following traumatic brain injury in rats. J. Pineal Res. 2016, 61, 241–250. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.S.; Wu, C.H.; Chen, S.F.; Yang, F.Y. Transcranial ultrasound stimulation promotes brain-derived neurotrophic factor and reduces apoptosis in a mouse model of traumatic brain injury. Brain Stimul. 2017, 10, 1032–1041. [Google Scholar] [CrossRef]
- Ji, X.; Peng, D.; Zhang, Y.; Zhang, J.; Wang, Y.; Gao, Y.; Lu, N.; Tang, P. Astaxanthin improves cognitive performance in mice following mild traumatic brain injury. Brain Res. 2017, 1659, 88–95. [Google Scholar] [CrossRef]
- Griesbach, G.S.; Gomez-Pinilla, F.; Hovda, D.A. The upregulation of plasticity-related proteins following TBI is disrupted with acute voluntary exercise. Brain Res. 2004, 1016, 154–162. [Google Scholar] [CrossRef]
- Xiong, L.L.; Hu, Y.; Zhang, P.; Zhang, Z.; Li, L.H.; Gao, G.D.; Zhou, X.F.; Wang, T.H. Neural Stem Cell Transplantation Promotes Functional Recovery from Traumatic Brain Injury via Brain Derived Neurotrophic Factor-Mediated Neuroplasticity. Mol. Neurobiol. 2018, 55, 2696–2711. [Google Scholar] [CrossRef]
- Meng, Y.; Chopp, M.; Zhang, Y.; Liu, Z.; An, A.; Mahmood, A.; Xiong, Y. Subacute intranasal administration of tissue plasminogen activator promotes neuroplasticity and improves functional recovery following traumatic brain injury in rats. PLoS ONE 2014, 9, e106238. [Google Scholar] [CrossRef]
- Feng, Y.; Ju, Y.; Cui, J.; Wang, L. Bone marrow stromal cells promote neuromotor functional recovery, via upregulation of neurotrophic factors and synapse proteins following traumatic brain injury in rats. Mol. Med. Rep. 2017, 16, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lee, J.H.; Chen, D.; Gu, X.; Caslin, A.; Li, J.; Yu, S.P.; Wei, L. DL-3-n-butylphthalide induced neuroprotection, regenerative repair, functional recovery and psychological benefits following traumatic brain injury in mice. Neurochem. Int. 2017, 111, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Gupta, R.; Kaiser, H.; Sen, N. Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 after Traumatic Brain Injury. J. Neurosci. 2017, 37, 5900–5911. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Williams, A.M.; Bhatti, U.F.; Biesterveld, B.E.; Liu, B.; Nikolian, V.C.; Dennahy, I.S.; Lee, J.; Li, Y.; Alam, H.B. Valproic Acid and Neural Apoptosis, Inflammation, and Degeneration 30 Days after Traumatic Brain Injury, Hemorrhagic Shock, and Polytrauma in a Swine Model. J. Am. Coll. Surg. 2019, 228, 265–275. [Google Scholar] [CrossRef]
- Ma, D.; Wang, N.; Fan, X.; Zhang, L.; Luo, Y.; Huang, R.; Zhang, L.; Li, Y.; Zhao, G.; Li, L. Protective Effects of Cornel Iridoid Glycoside in Rats After Traumatic Brain Injury. Neurochem. Res. 2018, 43, 959–971. [Google Scholar] [CrossRef]
- Yin, R.; Zhao, S.; Qiu, C. Brain-derived neurotrophic factor fused with a collagen-binding domain inhibits neuroinflammation and promotes neurological recovery of traumatic brain injury mice via TrkB signalling. J. Pharm. Pharmacol. 2020, 72, 539–550. [Google Scholar] [CrossRef]
- Della-Pace, I.D.; Souza, T.L.; Grauncke, A.C.B.; Rambo, L.M.; Ribeiro, L.R.; Cipolatto, R.P.; Severo, L.; Papalia, W.L.; Santos, A.R.S.; Facundo, V.A.; et al. Modulation of Na(+)/K(+)- ATPase activity by triterpene 3beta, 6beta, 16beta-trihidroxilup-20 (29)-ene (TTHL) limits the long-term secondary degeneration after traumatic brain injury in mice. Eur. J. Pharmacol. 2019, 854, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Cekic, M.; Johnson, S.J.; Bhatt, V.H.; Stein, D.G. Progesterone treatment alters neurotrophin/proneurotrophin balance and receptor expression in rats with traumatic brain injury. Restor. Neurol. Neurosci. 2012, 30, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto-Combs, K.; McNeal, D.W.; Morecraft, R.J.; Combs, C.K. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 2007, 24, 1719–1742. [Google Scholar] [CrossRef]
- Corne, R.; Leconte, C.; Ouradou, M.; Fassina, V.; Zhu, Y.; Deou, E.; Besson, V.; Plotkine, M.; Marchand-Leroux, C.; Mongeau, R. Spontaneous resurgence of conditioned fear weeks after successful extinction in brain injured mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 276–286. [Google Scholar] [CrossRef]
- Griesbach, G.S.; Tio, D.L.; Nair, S.; Hovda, D.A. Recovery of stress response coincides with responsiveness to voluntary exercise after traumatic brain injury. J. Neurotrauma 2014, 31, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, R.R.; Boggs, A.; Leider, D.; Kraemer, P.; Brown, R.; Scheff, S.W.; Seroogy, K.B. Effects of Exercise Following Lateral Fluid Percussion Brain Injury in Rats. Restor. Neurol. Neurosci. 1998, 12, 41–47. [Google Scholar] [PubMed]
- Shin, M.S.; Park, H.K.; Kim, T.W.; Ji, E.S.; Lee, J.M.; Choi, H.S.; Kim, M.Y.; Kim, Y.P. Neuroprotective Effects of Bone Marrow Stromal Cell Transplantation in Combination with Treadmill Exercise Following Traumatic Brain Injury. Int. Neurourol. J. 2016, 20 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, N.J.; Van Landingham, J.W.; Figueiroa, S.; Seth, R.; Corniola, R.S.; Levenson, C.W. Chronic caloric restriction reduces tissue damage and improves spatial memory in a rat model of traumatic brain injury. J. Neurosci. Res. 2010, 88, 2933–2939. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, A.; Aksan, B.; Heuvel, F.O.; Forstner, P.; Sinske, D.; Rehman, R.; Palmer, A.; Ludolph, A.; Huber-Lang, M.; Bockers, T.; et al. Neuroprotective effect of acute ethanol intoxication in TBI is associated to the hierarchical modulation of early transcriptional responses. Exp. Neurol. 2018, 302, 34–45. [Google Scholar] [CrossRef]
- Ignowski, E.; Winter, A.N.; Duval, N.; Fleming, H.; Wallace, T.; Manning, E.; Koza, L.; Huber, K.; Serkova, N.J.; Linseman, D.A. The cysteine-rich whey protein supplement, Immunocal(R), preserves brain glutathione and improves cognitive, motor, and histopathological indices of traumatic brain injury in a mouse model of controlled cortical impact. Free Radic. Biol. Med. 2018, 124, 328–341. [Google Scholar] [CrossRef]
- Ren, Y.Z.; Zhang, B.Z.; Zhao, X.J.; Zhang, Z.Y. Resolvin D1 ameliorates cognitive impairment following traumatic brain injury via protecting astrocytic mitochondria. J. Neurochem. 2020, 154, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Portbury, S.D.; Hare, D.J.; Finkelstein, D.I.; Adlard, P.A. Trehalose improves traumatic brain injury-induced cognitive impairment. PLoS ONE 2017, 12, e0183683. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Vergnes, L.; Reue, K.; Gomez-Pinilla, F. Coupling energy homeostasis with a mechanism to support plasticity in brain trauma. Biochim. Biophys. Acta 2014, 1842, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, A.; Lu, D.; Chopp, M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J. Neurotrauma 2004, 21, 33–39. [Google Scholar] [CrossRef]
- Mahmood, A.; Lu, D.; Qu, C.; Goussev, A.; Chopp, M. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J. Neurosurg. 2006, 104, 272–277. [Google Scholar] [CrossRef]
- Deng, Q.J.; Xu, X.F.; Ren, J. Effects of SDF-1/CXCR4 on the Repair of Traumatic Brain Injury in Rats by Mediating Bone Marrow Derived Mesenchymal Stem Cells. Cell. Mol. Neurobiol. 2018, 38, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.H.; Kim, S.H. Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: Secretion of neurotrophic factors and inhibition of apoptosis. J. Neurotrauma 2010, 27, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Xue, X.; Sun, J.; Wu, Q.; Wang, H.; Guo, Y.; Sun, B. The Promising Effects of Transplanted Umbilical Cord Mesenchymal Stem Cells on the Treatment in Traumatic Brain Injury. J. Craniofac. Surg. 2018, 29, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, D.; Yang, X.; Huang, P.; Ye, H.; Hui, Y.; Wang, X.; Sun, W.; Wu, H.; Zhang, S.; et al. Study on Umbilical Cord-Matrix Stem Cells Transplantation for Treatment of Acute Traumatic Brain Injury in Rats. Turk. Neurosurg. 2019, 29, 750–758. [Google Scholar] [PubMed]
- Khalin, I.; Alyautdin, R.; Wong, T.W.; Gnanou, J.; Kocherga, G.; Kreuter, J. Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv. 2016, 23, 3520–3528. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Hung, T.H.; Chen, C.C.; Ke, C.H.; Lee, C.Y.; Wang, P.Y.; Chen, S.F. Post-injury treatment with 7,8-dihydroxyflavone, a TrkB receptor agonist, protects against experimental traumatic brain injury via PI3K/Akt signaling. PLoS ONE 2014, 9, e113397. [Google Scholar] [CrossRef]
- Krishna, G.; Agrawal, R.; Zhuang, Y.; Ying, Z.; Paydar, A.; Harris, N.G.; Royes, L.F.F.; Gomez-Pinilla, F. 7,8-Dihydroxyflavone facilitates the action exercise to restore plasticity and functionality: Implications for early brain trauma recovery. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1204–1213. [Google Scholar] [CrossRef]
- Delbary-Gossart, S.; Lee, S.; Baroni, M.; Lamarche, I.; Arnone, M.; Canolle, B.; Lin, A.; Sacramento, J.; Salegio, E.A.; Castel, M.N.; et al. A novel inhibitor of p75-neurotrophin receptor improves functional outcomes in two models of traumatic brain injury. Brain 2016, 139 Pt 6, 1762–1782. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Teng, S.; Reddi, S.; Zheng, C.; Adler, D.; Thakker-Varia, S.; Alder, J. BDNF Val66Met Genetic Polymorphism Results in Poor Recovery Following Repeated Mild Traumatic Brain Injury in a Mouse Model and Treatment With AAV-BDNF Improves Outcomes. Front. Neurol. 2019, 10, 1175. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Chen, J. Conditional knockout of brain-derived neurotrophic factor in the hippocampus increases death of adult-born immature neurons following traumatic brain injury. J. Neurotrauma 2009, 26, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Yu, Z.; Zhao, S.; Liao, Z.; Xing, C.; Jiang, Y.; Yang, Y.G.; Whalen, M.J.; Lo, E.H.; Sun, X.; et al. Thrombospondin-1 Gene Deficiency Worsens the Neurological Outcomes of Traumatic Brain Injury in Mice. Int. J. Med. Sci. 2017, 14, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, B., 3rd; Elkin, B.S.; Dolle, J.P.; Yarmush, M.L. In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 2011, 13, 91–126. [Google Scholar] [CrossRef]
- Thelin, E.P.; Hall, C.E.; Tyzack, G.E.; Frostell, A.; Giorgi-Coll, S.; Alam, A.; Carpenter, K.L.H.; Mitchell, J.; Tajsic, T.; Hutchinson, P.J.; et al. Delineating Astrocytic Cytokine Responses in a Human Stem Cell Model of Neural Trauma. J. Neurotrauma 2020, 37, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, S.A.; Phillips, J.K.; Costa, J.T.; Cho, F.S.; Oungoulian, S.R.; Finan, J.D. Stretch Injury of Human Induced Pluripotent Stem Cell Derived Neurons in a 96 Well Format. Sci. Rep. 2016, 6, 34097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zander, N.E.; Piehler, T.; Hogberg, H.; Pamies, D. Explosive Blast Loading on Human 3D Aggregate Minibrains. Cell. Mol. Neurobiol. 2017, 37, 1331–1334. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.D.; Berlind, J.E.; Fricklas, G.; Maria, N.S.; Jacobs, R.; Yu, V.; Ichida, J.K. A model of traumatic brain injury using human iPSC-derived cortical brain organoids. BioRxiv 2020. [Google Scholar] [CrossRef]
- Xu, X.; Garcia, J.; Ewalt, R.; Nason, S.; Pozzo-Miller, L. The BDNF val-66-met Polymorphism Affects Neuronal Morphology and Synaptic Transmission in Cultured Hippocampal Neurons from Rett Syndrome Mice. Front. Cell. Neurosci. 2017, 11, 203. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; You, Y.; Joseph, C.; Mirzaei, M.; Klistorner, A.; Graham, S.L.; Gupta, V. BDNF Polymorphism: A Review of Its Diagnostic and Clinical Relevance in Neurodegenerative Disorders. Aging Dis. 2018, 9, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Gabrys, R.L.; Dixon, K.; Holahan, M.R.; Anisman, H. Self-Reported Mild Traumatic Brain Injuries in Relation to Rumination and Depressive Symptoms: Moderating Role of Sex Differences and a Brain-Derived Neurotrophic Factor Gene Polymorphism. Clin. J. Sport Med. 2019, 29, 494–499. [Google Scholar] [CrossRef]
- Edut, S.; Rubovitch, V.; Rehavi, M.; Schreiber, S.; Pick, C.G. A study on the mechanism by which MDMA protects against dopaminergic dysfunction after minimal traumatic brain injury (mTBI) in mice. J. Mol. Neurosci. 2014, 54, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, C.; Yang, X.; Wang, J.; Zhao, M.L.; Sun, H.; Zhang, S.; Tu, Y. Acupuncture Improved Neurological Recovery after Traumatic Brain Injury by Activating BDNF/TrkB Pathway. Evid. Based Complement. Alternat. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shen, M.; Sun, K.; Wang, Y.; Wang, X.; Jin, X.; Xu, J.; Ding, L.; Sun, X. Aminoguanidine reverses cognitive deficits and activation of cAMP/CREB/BDNF pathway in mouse hippocampus after traumatic brain injury (TBI). Brain Inj. 2018, 32, 1858–1865. [Google Scholar] [CrossRef]
- Umschweif, G.; Liraz-Zaltsman, S.; Shabashov, D.; Alexandrovich, A.; Trembovler, V.; Horowitz, M.; Shohami, E. Angiotensin receptor type 2 activation induces neuroprotection and neurogenesis after traumatic brain injury. Neurotherapeutics 2014, 11, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Umschweif, G.; Shabashov, D.; Alexandrovich, A.G.; Trembovler, V.; Horowitz, M.; Shohami, E. Neuroprotection after traumatic brain injury in heat-acclimated mice involves induced neurogenesis and activation of angiotensin receptor type 2 signaling. J. Cereb. Blood Flow Metab. 2014, 34, 1381–1390. [Google Scholar] [CrossRef]
- Lee, I.N.; Lin, M.H.; Chung, C.Y.; Lee, M.H.; Weng, H.H.; Yang, J.T. Chronic cigarette smoke exposure enhances brain-derived neurotrophic factor expression in rats with traumatic brain injury. Metab Brain Dis. 2012, 27, 197–204. [Google Scholar] [CrossRef]
- Gu, Y.L.; Zhang, L.W.; Ma, N.; Ye, L.L.; Wang de, X.; Gao, X. Cognitive improvement of mice induced by exercise prior to traumatic brain injury is associated with cytochrome c oxidase. Neurosci. Lett. 2014, 570, 86–91. [Google Scholar] [CrossRef]
- Yoon, K.J.; Lee, Y.T.; Chae, S.W.; Park, C.R.; Kim, D.Y. Effects of anodal transcranial direct current stimulation (tDCS) on behavioral and spatial memory during the early stage of traumatic brain injury in the rats. J. Neurol. Sci. 2016, 362, 314–320. [Google Scholar] [CrossRef]
- Liraz-Zaltsman, S.; Slusher, B.; Atrakchi-Baranes, D.; Rosenblatt, K.; Friedman Levi, Y.; Kesner, E.; Silva, A.J.; Biegon, A.; Shohami, E. Enhancement of Brain d-Serine Mediates Recovery of Cognitive Function after Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1667–1680. [Google Scholar] [CrossRef]
- Gatson, J.W.; Liu, M.M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Simpkins, J.W.; Minei, J.P. Estrone is neuroprotective in rats after traumatic brain injury. J. Neurotrauma 2012, 29, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Grundy, P.L.; Patel, N.; Harbuz, M.S.; Lightman, S.L.; Sharples, P.M. Glucocorticoids modulate BDNF mRNA expression in the rat hippocampus after traumatic brain injury. Neuroreport 2000, 11, 3381–3384. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Kong, X.; Acosta, S.; Sava, V.; Borlongan, C.; Sanchez-Ramos, J. Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J. Neurosci. Res. 2016, 94, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algamal, M.; Saltiel, N.; Pearson, A.J.; Ager, B.; Burca, I.; Mouzon, B.; Diamond, D.M.; Mullan, M.; Ojo, J.O.; Crawford, F. Impact of Repetitive Mild Traumatic Brain Injury on Behavioral and Hippocampal Deficits in a Mouse Model of Chronic Stress. J. Neurotrauma 2019, 36, 2590–2607. [Google Scholar] [CrossRef] [PubMed]
- Ko, I.G.; Kim, S.E.; Hwang, L.; Jin, J.J.; Kim, C.J.; Kim, B.K.; Kim, H. Late starting treadmill exercise improves spatial leaning ability through suppressing CREP/BDNF/TrkB signaling pathway following traumatic brain injury in rats. J. Exerc. Rehabil. 2018, 14, 327–334. [Google Scholar] [CrossRef]
- Mahmood, A.; Goussev, A.; Kazmi, H.; Qu, C.; Lu, D.; Chopp, M. Long-term benefits after treatment of traumatic brain injury with simvastatin in rats. Neurosurgery 2009, 65, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Xuan, W.; Agrawal, T.; Huang, L.; Gupta, G.K.; Hamblin, M.R. Low-level laser therapy for traumatic brain injury in mice increases brain derived neurotrophic factor (BDNF) and synaptogenesis. J. Biophotonics 2015, 8, 502–511. [Google Scholar] [CrossRef]
- Shah, S.A.; Prough, D.S.; Garcia, J.M.; DeWitt, D.S.; Hellmich, H.L. Molecular correlates of age-specific responses to traumatic brain injury in mice. Exp. Gerontol. 2006, 41, 1201–1205. [Google Scholar] [CrossRef]
- Esenaliev, R.O.; Petrov, I.Y.; Petrov, Y.; Guptarak, J.; Boone, D.R.; Mocciaro, E.; Weisz, H.; Parsley, M.A.; Sell, S.L.; Hellmich, H.; et al. Nano-Pulsed Laser Therapy Is Neuroprotective in a Rat Model of Blast-Induced Neurotrauma. J. Neurotrauma 2018, 35, 1510–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.; Mahesh, R.; Jindal, A.; Devadoss, T. Neuropharmacological and neurochemical evaluation of N-n-propyl-3-ethoxyquinoxaline-2-carboxamide (6n): A novel serotonergic 5-HT3 receptor antagonist for co-morbid antidepressant- and anxiolytic-like potential using traumatic brain injury model in rats. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 93–100. [Google Scholar] [CrossRef]
- Sonmez, A.; Sayin, O.; Gurgen, S.G.; Calisir, M. Neuroprotective effects of MK-801 against traumatic brain injury in immature rats. Neurosci. Lett. 2015, 597, 137–142. [Google Scholar] [CrossRef]
- Shang, J.L.; Cheng, Q.; Yang, W.F.; Zhang, M.; Cui, Y.; Wang, Y.F. Possible roles of COX-1 in learning and memory impairment induced by traumatic brain injury in mice. Br. J. Med. Biol. Res. 2014, 47, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Ghadiri, T.; Vakilzadeh, G.; Hajali, V.; Khodagholi, F. Progesterone modulates post-traumatic epileptogenesis through regulation of BDNF-TrkB signaling and cell survival-related pathways in the rat hippocampus. Neurosci. Lett. 2019, 709, 134384. [Google Scholar] [CrossRef]
- Oyesiku, N.M.; Evans, C.O.; Houston, S.; Darrell, R.S.; Smith, J.S.; Fulop, Z.L.; Dixon, C.E.; Stein, D.G. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999, 833, 161–172. [Google Scholar] [CrossRef]
- Golz, C.; Kirchhoff, F.P.; Westerhorstmann, J.; Schmidt, M.; Hirnet, T.; Rune, G.M.; Bender, R.A.; Schafer, M.K.E. Sex hormones modulate pathogenic processes in experimental traumatic brain injury. J. Neurochem. 2019, 150, 173–187. [Google Scholar] [CrossRef]
- Cutler, S.M.; Vanlandingham, J.W.; Stein, D.G. Tapered progesterone withdrawal promotes long-term recovery following brain trauma. Exp. Neurol. 2006, 200, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Mychasiuk, R.; Hehar, H.; Candy, S.; Ma, I.; Esser, M.J. The direction of the acceleration and rotational forces associated with mild traumatic brain injury in rodents effect behavioural and molecular outcomes. J. Neurosci. Methods 2016, 257, 168–178. [Google Scholar] [CrossRef]
- Xing, P.; Ma, K.; Li, L.; Wang, D.; Hu, G.; Long, W. The protection effect and mechanism of hyperbaric oxygen therapy in rat brain with traumatic injury. Acta Cir. Bras. 2018, 33, 341–353. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Campolo, M.; Bruschetta, G.; Crupi, R.; Cordaro, M.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Traumatic Brain Injury Leads to Development of Parkinson’s Disease Related Pathology in Mice. Front. Neurosci. 2016, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, A.; Lu, D.; Qu, C.; Goussev, A.; Zhang, Z.G.; Lu, C.; Chopp, M. Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J. Neurosurg. 2007, 107, 392–397. [Google Scholar] [CrossRef]
- Davidson, J.; Cusimano, M.D.; Bendena, W.G. Post-traumatic brain injury: Genetic susceptibility to outcome. Neuroscientist 2015, 21, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Finan, J.D.; Udani, S.V.; Patel, V.; Bailes, J.E. The Influence of the Val66Met Polymorphism of Brain-Derived Neurotrophic Factor on Neurological Function after Traumatic Brain Injury. J. Alzheimers Dis. 2018, 65, 1055–1064. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustafsson, D.; Klang, A.; Thams, S.; Rostami, E. The Role of BDNF in Experimental and Clinical Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 3582. https://doi.org/10.3390/ijms22073582
Gustafsson D, Klang A, Thams S, Rostami E. The Role of BDNF in Experimental and Clinical Traumatic Brain Injury. International Journal of Molecular Sciences. 2021; 22(7):3582. https://doi.org/10.3390/ijms22073582
Chicago/Turabian StyleGustafsson, David, Andrea Klang, Sebastian Thams, and Elham Rostami. 2021. "The Role of BDNF in Experimental and Clinical Traumatic Brain Injury" International Journal of Molecular Sciences 22, no. 7: 3582. https://doi.org/10.3390/ijms22073582
APA StyleGustafsson, D., Klang, A., Thams, S., & Rostami, E. (2021). The Role of BDNF in Experimental and Clinical Traumatic Brain Injury. International Journal of Molecular Sciences, 22(7), 3582. https://doi.org/10.3390/ijms22073582