
Rho GTPases in Retinal Vascular Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
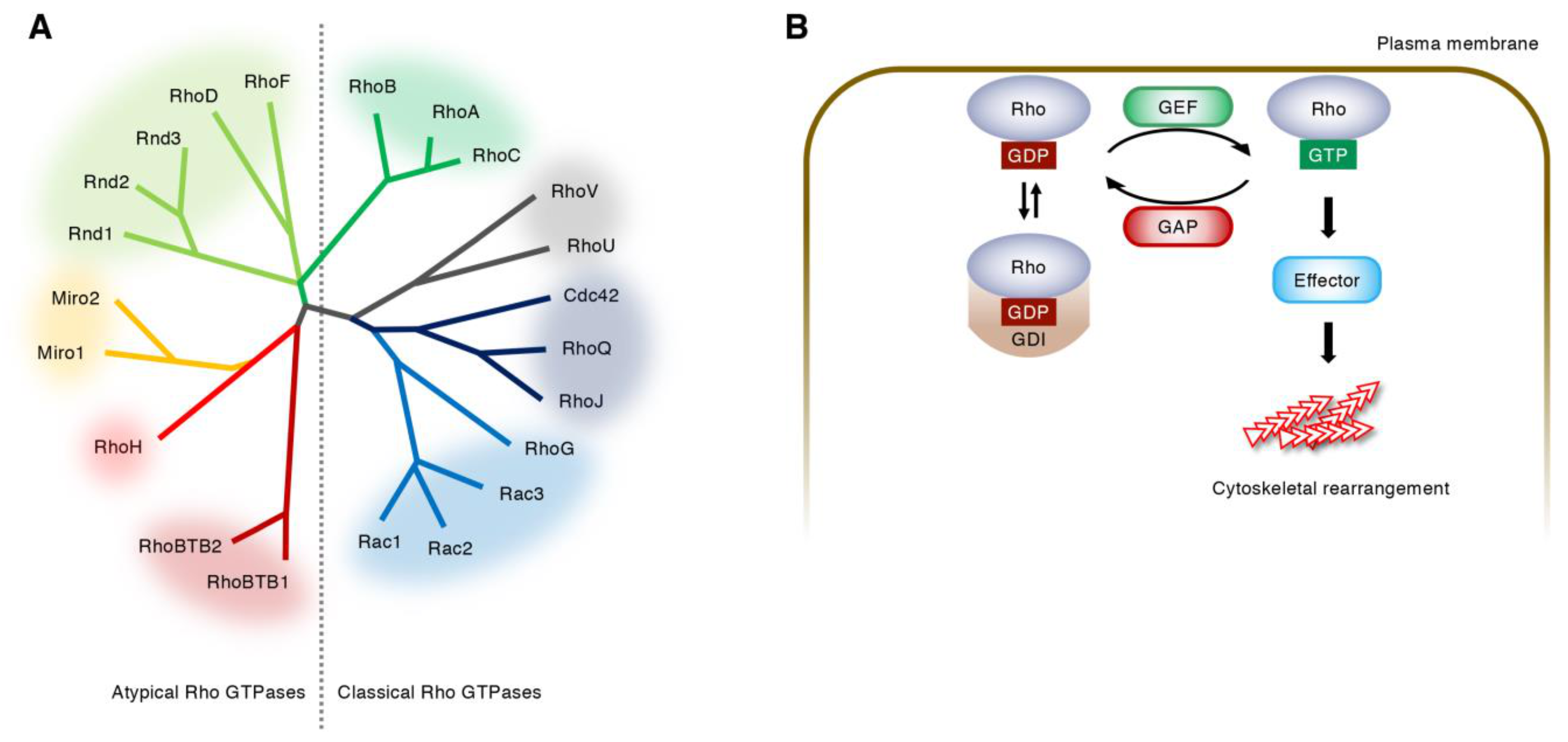
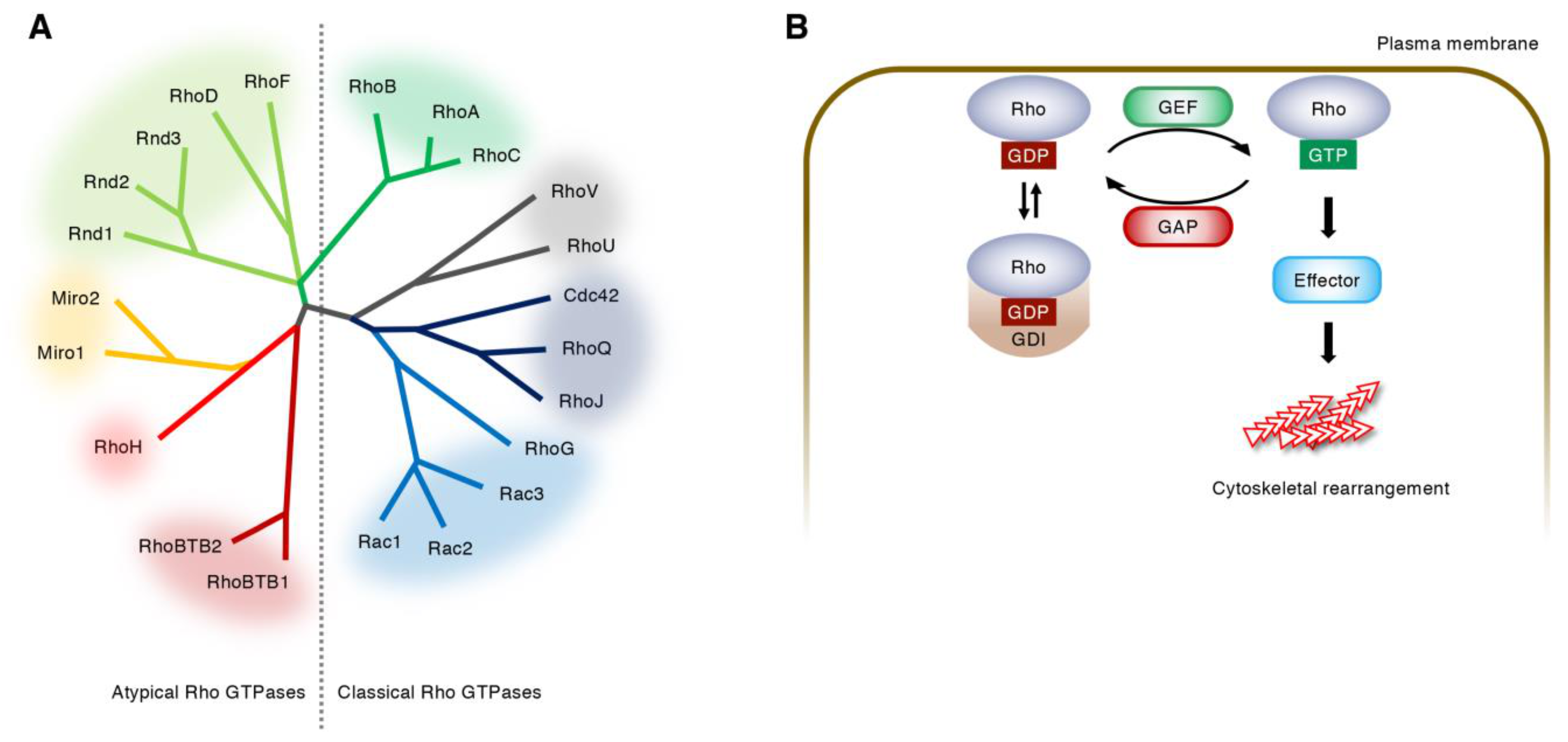
2. Regulation of Rho GTPase Activity
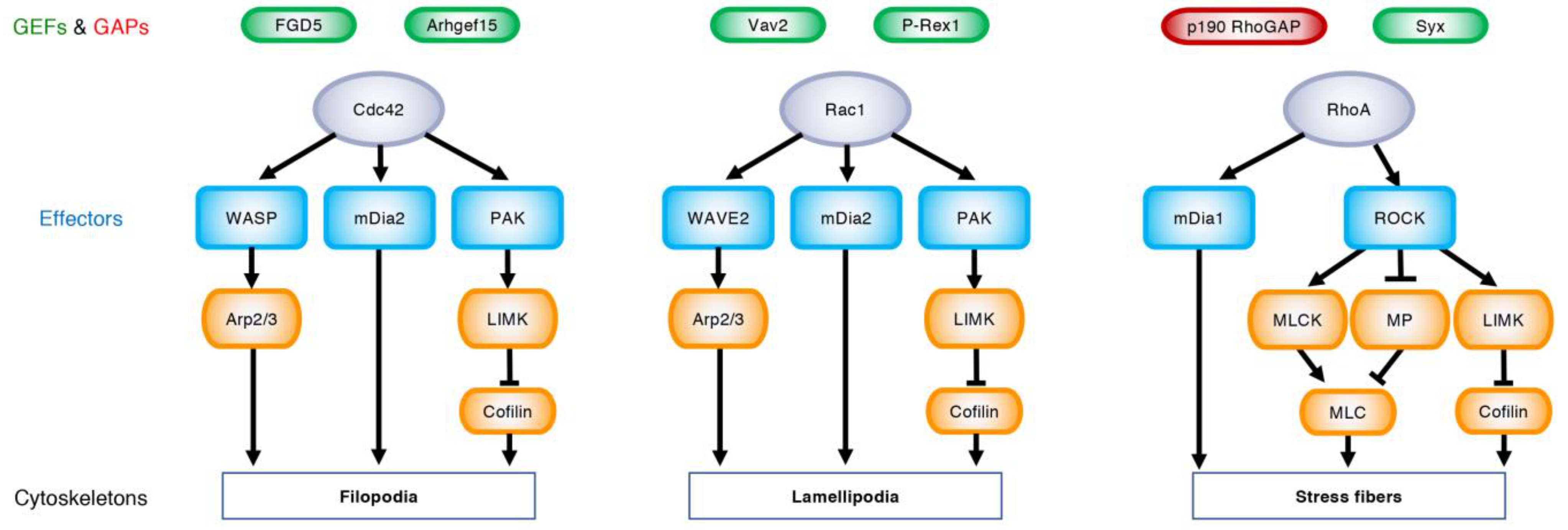
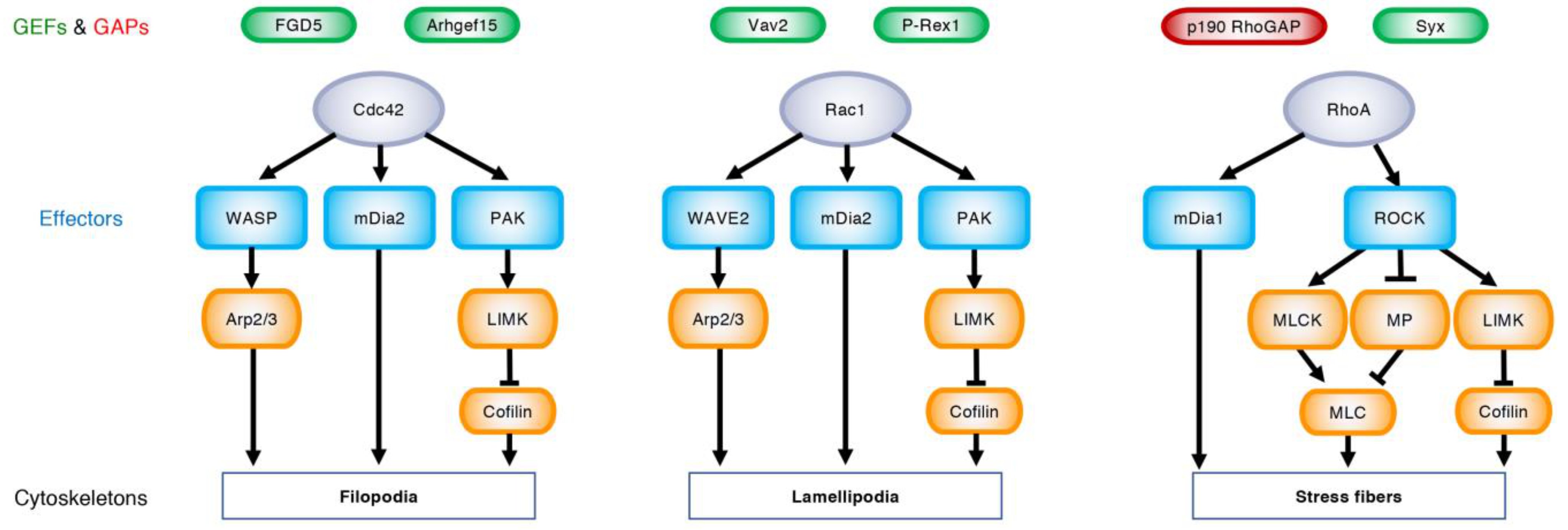
3. Regulation of Angiogenesis by Rho GTPases
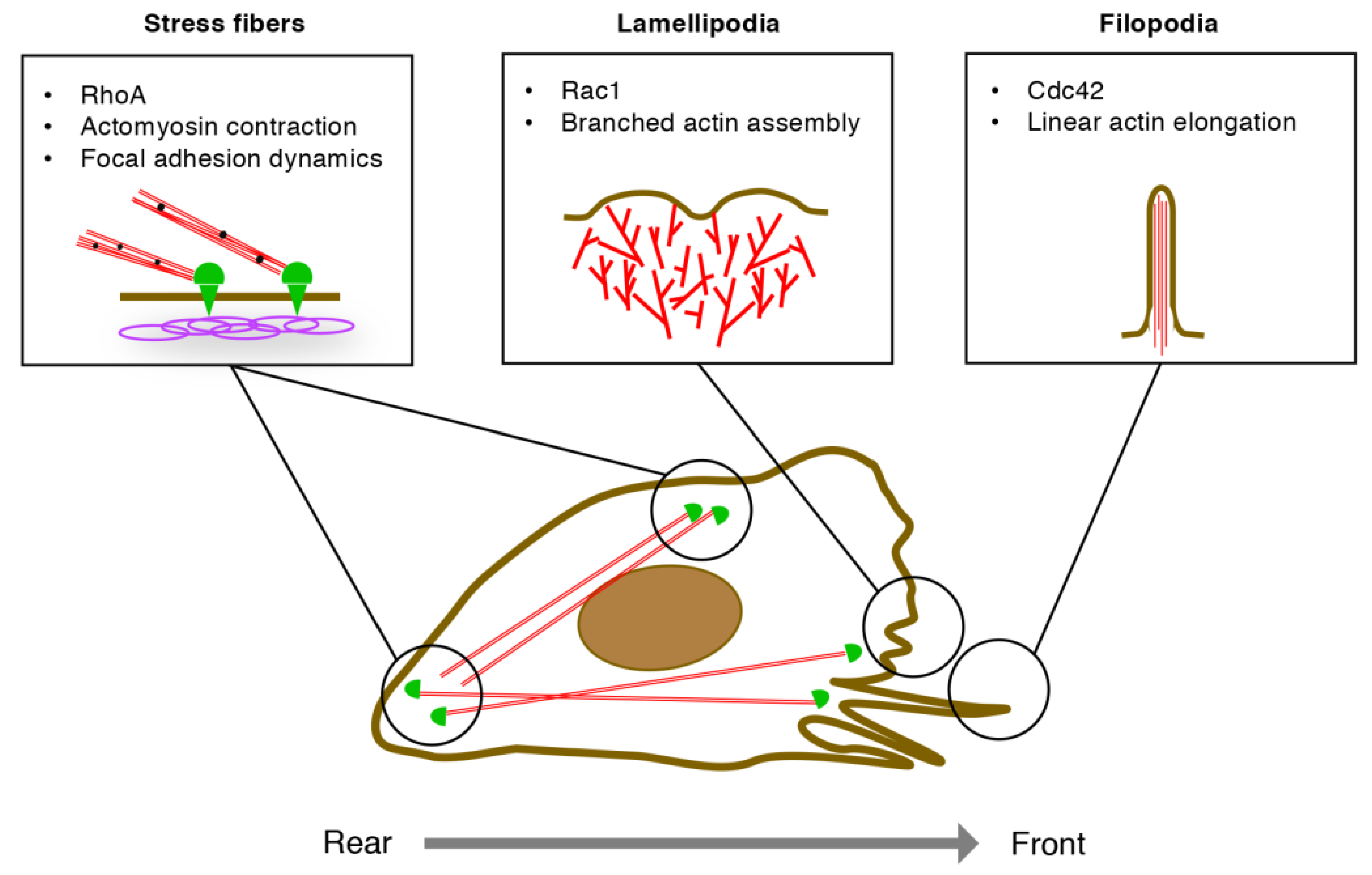
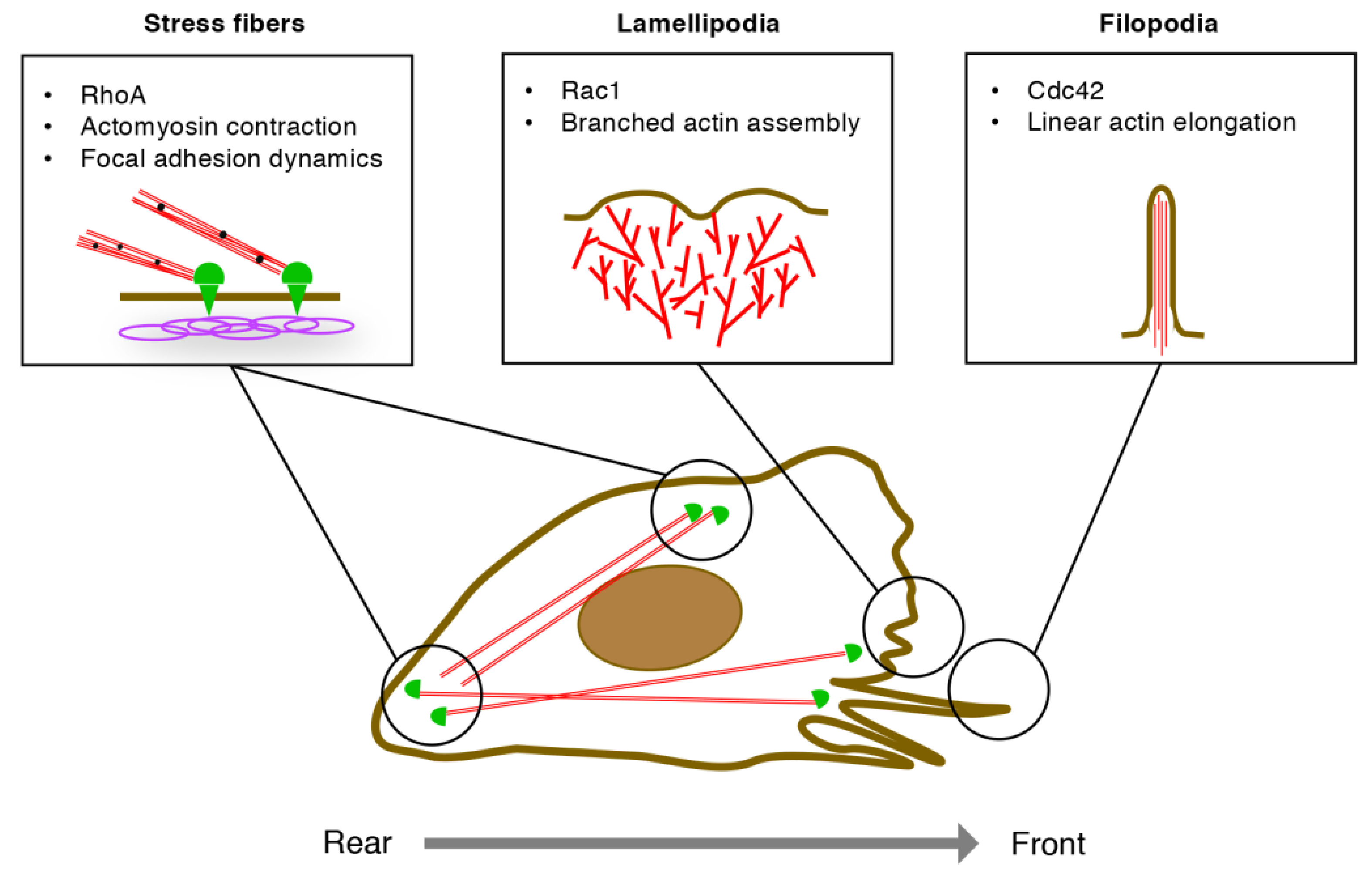
3.1. EC Migration
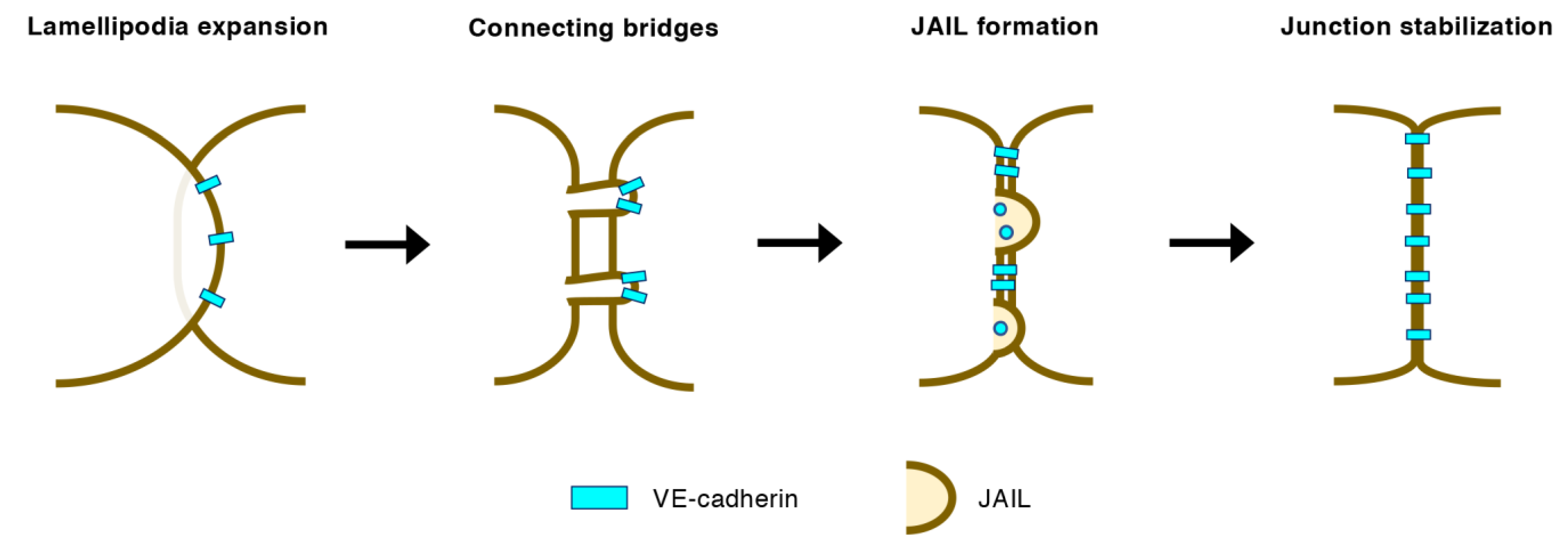
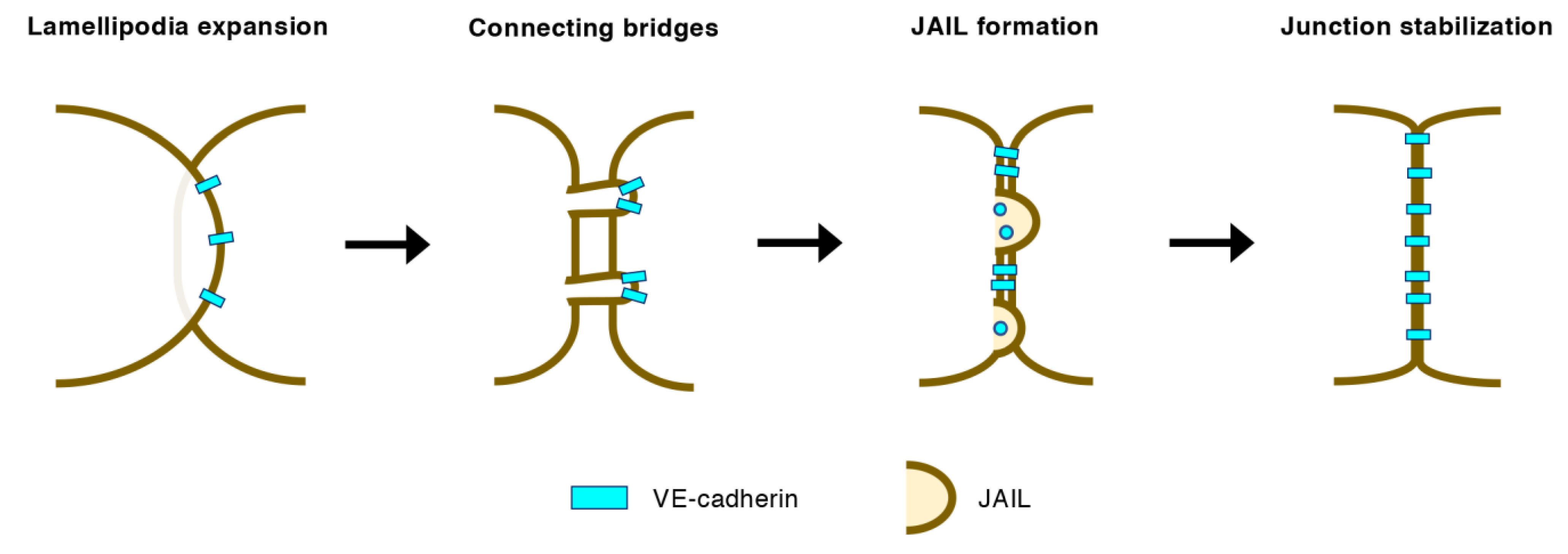
3.2. EC–EC Junctions in Angiogenic Vessels
3.3. Retinal Vascular Development
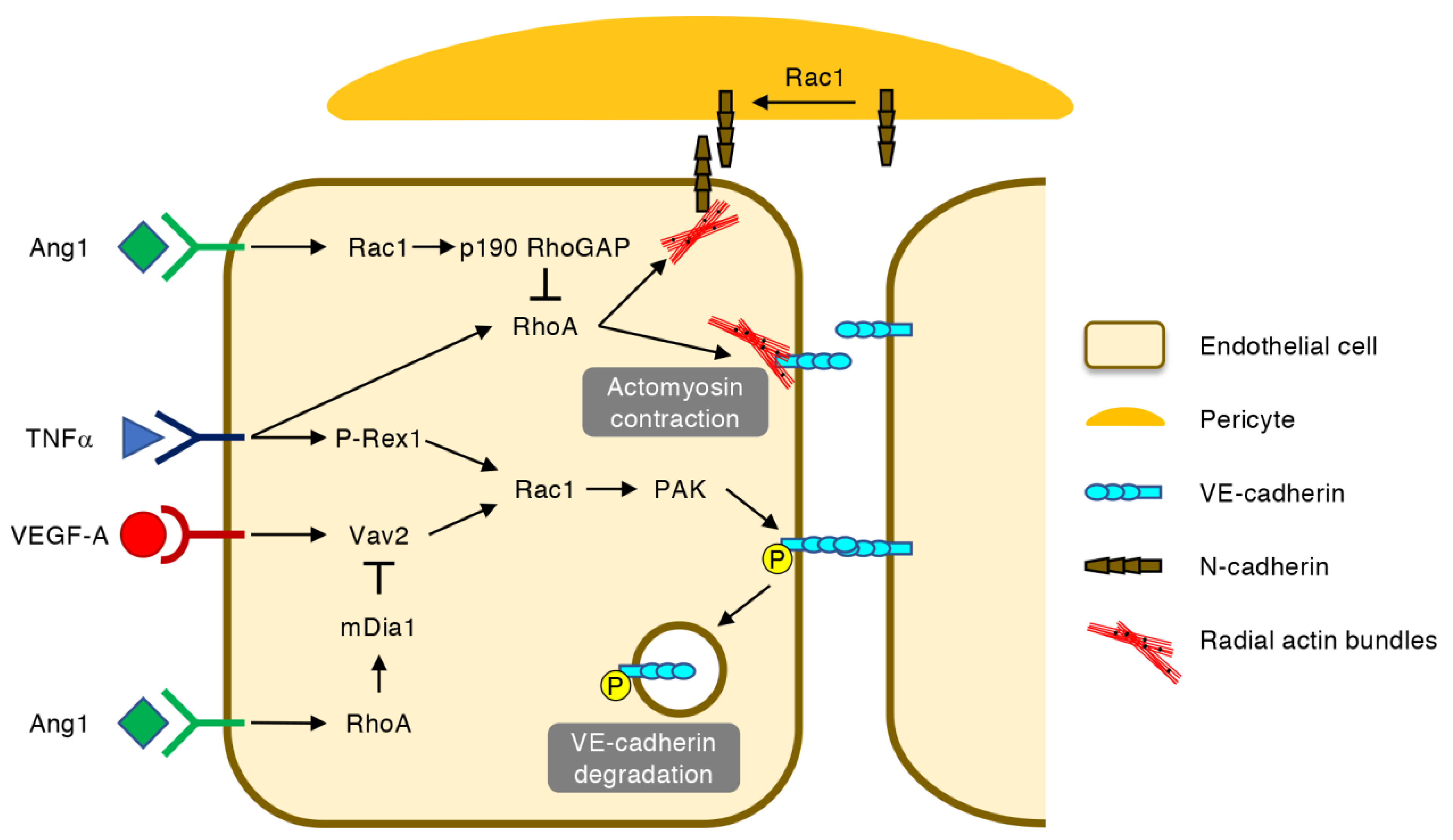
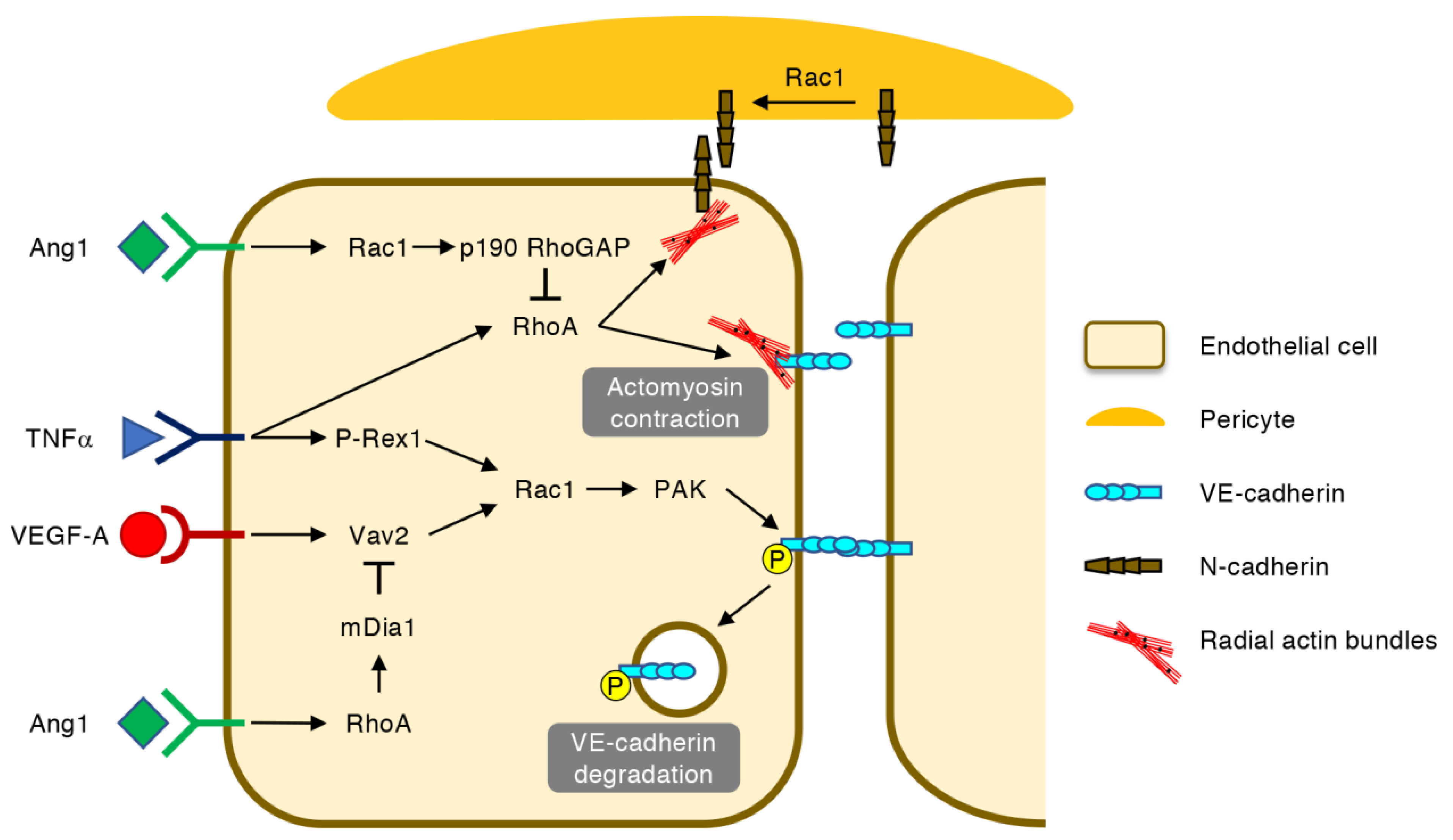
4. Regulation of Vascular Permeability by Rho GTPases
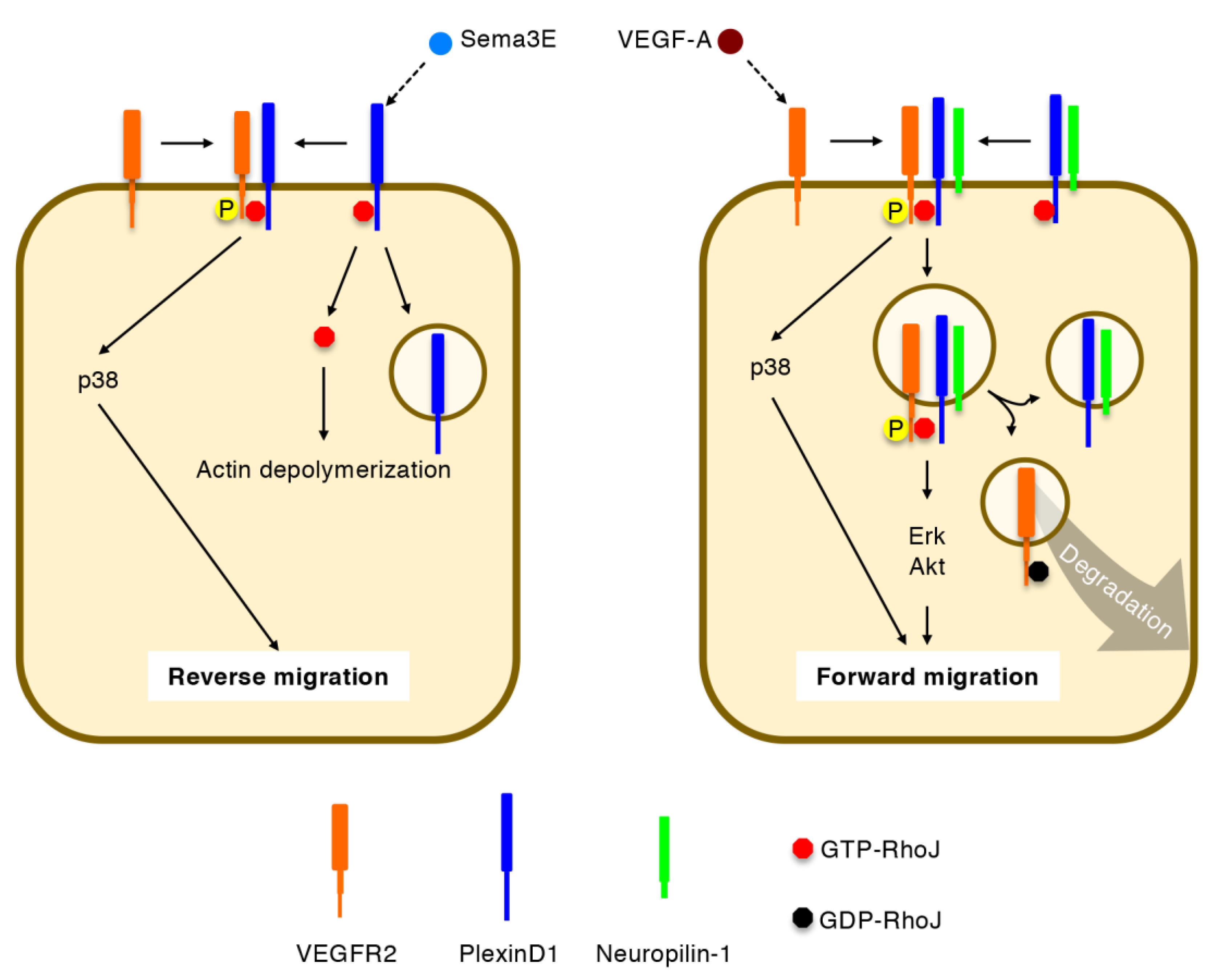
5. Regulation of Directional EC Migration by RhoJ
5.1. Identification of RhoJ as an EC-Enriched Rho GTPase
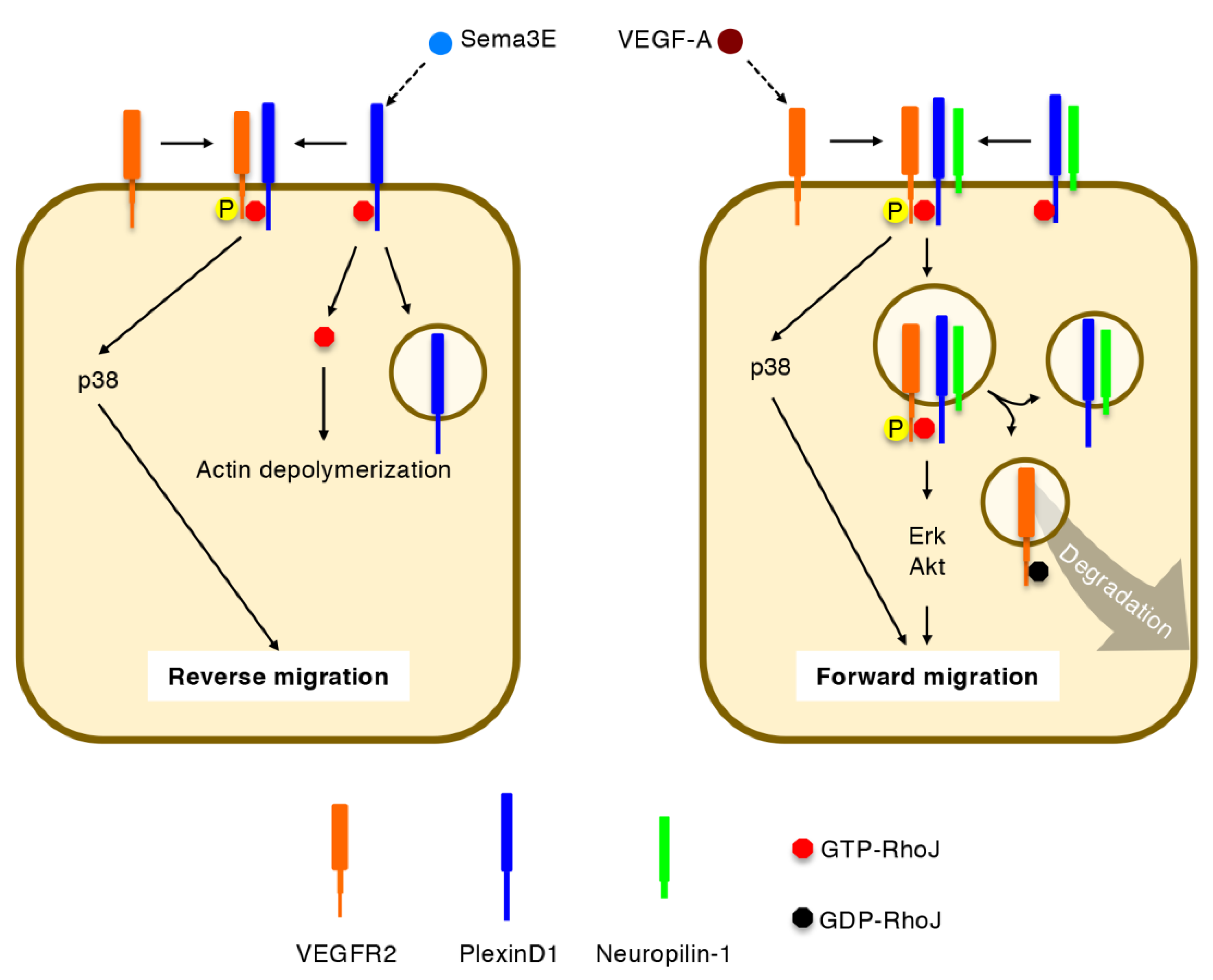
5.2. Integration of VEGF-A and Sema3E Signals by RhoJ
5.3. RhoJ in Retinal Angiogenesis
6. Rho GTPases as Therapeutic Targets of Retinal Vascular Diseases
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang | Angiopoietin |
| AJ | Adherens junction |
| Arp2/3 | Actin-related protein-2/3 |
| BRB | Blood-retina barrier |
| CRIB | Cdc42/Rac interactive binding |
| EC | Endothelial cell |
| Erk | Extracellular signal-regulated kinase |
| GAP | GTPase activating protein |
| GDI | Guanine dissociation inhibitor |
| GEF | Guanine nucleotide exchange factor |
| mDia | Mammalian diaphanous |
| N | Neural |
| MLC | Myosin light chain |
| PAK | p21-activated kinase |
| ROCK | Rho-associated coiled-coil-containing protein kinase |
| Sema3E | Semaphorin 3E |
| TNF | Tumor necrosis factor |
| VE | Vascular endothelial |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| WASP | Wiskott-Aldrich syndrome protein |
| WAVE | WASP-family verprolin homologous protein |
References
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. 2016, 11, 251–281. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2020. [Google Scholar] [CrossRef]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef]
- Van der Meel, R.; Symons, M.H.; Kudernatsch, R.; Kok, R.J.; Schiffelers, R.M.; Storm, G.; Gallagher, W.M.; Byrne, A.T. The VEGF/Rho GTPase signalling pathway: A promising target for anti-angiogenic/anti-invasion therapy. Drug Discov. Today 2011, 16, 219–228. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Leszczynska, K.; Kaur, S.; Wilson, E.; Bicknell, R.; Heath, V.L. The role of RhoJ in endothelial cell biology and angiogenesis. Biochem. Soc. Trans. 2011, 39, 1606–1611. [Google Scholar] [CrossRef]
- Fukushima, Y.; Okada, M.; Kataoka, H.; Hirashima, M.; Yoshida, Y.; Mann, F.; Gomi, F.; Nishida, K.; Nishikawa, S.; Uemura, A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Investig. 2011, 121, 1974–1985. [Google Scholar] [CrossRef]
- Fukushima, Y.; Nishiyama, K.; Kataoka, H.; Fruttiger, M.; Fukuhara, S.; Nishida, K.; Mochizuki, N.; Kurihara, H.; Nishikawa, S.I.; Uemura, A. RhoJ integrates attractive and repulsive cues in directional migration of endothelial cells. EMBO J. 2020, 39, e102930. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ’invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.M.; Rademacher, J.; Bagshaw, R.D.; Wortmann, C.; Barth, C.; van Unen, J.; Alp, K.M.; Giudice, G.; Eccles, R.L.; Heinrich, L.E.; et al. Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol. 2020, 22, 498–511. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Rotty, J.D.; Wu, C.; Haynes, E.M.; Suarez, C.; Winkelman, J.D.; Johnson, H.E.; Haugh, J.M.; Kovar, D.R.; Bear, J.E. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 2015, 32, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Suarez, C.; Carroll, R.T.; Burke, T.A.; Christensen, J.R.; Bestul, A.J.; Sees, J.A.; James, M.L.; Sirotkin, V.; Kovar, D.R. Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev. Cell 2015, 32, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.; Tremblay, M.L.; Yamada, K.M. Phosphatases in cell–matrix adhesion and migration. Nat. Rev. Mol. Cell Biol. 2003, 4, 700–711. [Google Scholar] [CrossRef]
- Wu, C.; Agrawal, S.; Vasanji, A.; Drazba, J.; Sarkaria, S.; Xie, J.; Welch, C.M.; Liu, M.; Anand-Apte, B.; Horowitz, A. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J. Biol. Chem. 2011, 286, 23511–23520. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef]
- Arima, S.; Nishiyama, K.; Ko, T.; Arima, Y.; Hakozaki, Y.; Sugihara, K.; Koseki, H.; Uchijima, Y.; Kurihara, Y.; Kurihara, H. Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development 2011, 138, 4763–4776. [Google Scholar] [CrossRef] [Green Version]
- Hoelzle, M.K.; Svitkina, T. The cytoskeletal mechanisms of cell–cell junction formation in endothelial cells. Mol. Biol. Cell 2012, 23, 310–323. [Google Scholar] [CrossRef]
- Abu Taha, A.; Taha, M.; Seebach, J.; Schnittler, H.J. ARP2/3-mediated junction-associated lamellipodia control VE-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol. Biol. Cell 2014, 25, 245–256. [Google Scholar] [CrossRef]
- Cao, J.; Ehling, M.; Marz, S.; Seebach, J.; Tarbashevich, K.; Sixta, T.; Pitulescu, M.E.; Werner, A.C.; Flach, B.; Montanez, E.; et al. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat. Commun. 2017, 8, 2210. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, J.R.; Fortunato, I.C.; Fonseca, C.G.; Pezzarossa, A.; Barbacena, P.; Dominguez-Cejudo, M.A.; Vasconcelos, F.F.; Santos, N.C.; Carvalho, F.A.; Franco, C.A. Non-canonical Wnt signaling regulates junctional mechanocoupling during angiogenic collective cell migration. eLife 2019, 8, e45853. [Google Scholar] [CrossRef]
- Abraham, S.; Yeo, M.; Montero-Balaguer, M.; Paterson, H.; Dejana, E.; Marshall, C.J.; Mavria, G. VE-Cadherin-mediated cell–cell interaction suppresses sprouting via signaling to MLC2 phosphorylation. Curr. Biol. 2009, 19, 668–674. [Google Scholar] [CrossRef]
- Pitulescu, M.E.; Schmidt, I.; Benedito, R.; Adams, R.H. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 2010, 5, 1518–1534. [Google Scholar] [CrossRef]
- Zahra, F.T.; Sajib, M.S.; Ichiyama, Y.; Akwii, R.G.; Tullar, P.E.; Cobos, C.; Minchew, S.A.; Doci, C.L.; Zheng, Y.; Kubota, Y.; et al. Endothelial RhoA GTPase is essential for in vitro endothelial functions but dispensable for physiological in vivo angiogenesis. Sci. Rep. 2019, 9, 11666. [Google Scholar] [CrossRef] [Green Version]
- Nohata, N.; Uchida, Y.; Stratman, A.N.; Adams, R.H.; Zheng, Y.; Weinstein, B.M.; Mukouyama, Y.S.; Gutkind, J.S. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J.; et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef] [Green Version]
- Barry, D.M.; Xu, K.; Meadows, S.M.; Zheng, Y.; Norden, P.R.; Davis, G.E.; Cleaver, O. Cdc42 is required for cytoskeletal support of endothelial cell adhesion during blood vessel formation in mice. Development 2015, 142, 3058–3070. [Google Scholar] [CrossRef] [Green Version]
- Lavina, B.; Castro, M.; Niaudet, C.; Cruys, B.; Alvarez-Aznar, A.; Carmeliet, P.; Bentley, K.; Brakebusch, C.; Betsholtz, C.; Gaengel, K. Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development 2018, 145, dev161182. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naikawadi, R.P.; Cheng, N.; Vogel, S.M.; Qian, F.; Wu, D.; Malik, A.B.; Ye, R.D. A critical role for phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1 in endothelial junction disruption and vascular hyperpermeability. Circ. Res. 2012, 111, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef] [Green Version]
- Alimperti, S.; Mirabella, T.; Bajaj, V.; Polacheck, W.; Pirone, D.M.; Duffield, J.; Eyckmans, J.; Assoian, R.K.; Chen, C.S. Three-dimensional biomimetic vascular model reveals a RhoA, Rac1, and N-cadherin balance in mural cell-endothelial cell-regulated barrier function. Proc. Natl. Acad. Sci. USA 2017, 114, 8758–8763. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, C.; Ridley, A.J. Endothelial cell–cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Ngok, S.P.; Geyer, R.; Liu, M.; Kourtidis, A.; Agrawal, S.; Wu, C.; Seerapu, H.R.; Lewis-Tuffin, L.J.; Moodie, K.L.; Huveldt, D.; et al. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J. Cell Biol. 2012, 199, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Mammoto, T.; Parikh, S.M.; Mammoto, A.; Gallagher, D.; Chan, B.; Mostoslavsky, G.; Ingber, D.E.; Sukhatme, V.P. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J. Biol. Chem. 2007, 282, 23910–23918. [Google Scholar] [CrossRef] [Green Version]
- Vignal, E.; De Toledo, M.; Comunale, F.; Ladopoulou, A.; Gauthier-Rouviere, C.; Blangy, A.; Fort, P. Characterization of TCL, a new GTPase of the rho family related to TC10 andCcdc42. J. Biol Chem 2000, 275, 36457–36464. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Kato, M.; Miki, H.; Takenawa, T.; Endo, T. Small GTPase Tc10 and its homologue RhoT induce N-WASP-mediated long process formation and neurite outgrowth. J. Cell Sci. 2003, 116, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Nishizuka, M.; Arimoto, E.; Tsuchiya, T.; Nishihara, T.; Imagawa, M. Crucial role of TCL/TC10beta L, a subfamily of Rho GTPase, in adipocyte differentiation. J. Biol. Chem. 2003, 278, 15279–15284. [Google Scholar] [CrossRef] [Green Version]
- De Toledo, M.; Senic-Matuglia, F.; Salamero, J.; Uze, G.; Comunale, F.; Fort, P.; Blangy, A. The GTP/GDP cycling of rho GTPase TCL is an essential regulator of the early endocytic pathway. Mol. Biol. Cell 2003, 14, 4846–4856. [Google Scholar] [CrossRef]
- Herbert, J.M.; Stekel, D.; Sanderson, S.; Heath, V.L.; Bicknell, R. A novel method of differential gene expression analysis using multiple cDNA libraries applied to the identification of tumour endothelial genes. BMC Genom. 2008, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Sacharidou, A.; Stratman, A.N.; Le Bras, A.; Zwiers, P.J.; Spokes, K.; Bhasin, M.; Shih, S.C.; Nagy, J.A.; Molema, G.; et al. RhoJ is an endothelial cell-restricted Rho GTPase that mediates vascular morphogenesis and is regulated by the transcription factor ERG. Blood 2011, 118, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Leszczynska, K.; Abraham, S.; Scarcia, M.; Hiltbrunner, S.; Marshall, C.J.; Mavria, G.; Bicknell, R.; Heath, V.L. RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arter. Thromb. Vasc. Biol. 2011, 31, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.; Hetheridge, C.; Mellor, H. The Formin FMNL3 Controls Early Apical Specification in Endothelial Cells by Regulating the Polarized Trafficking of Podocalyxin. Curr. Biol. 2015, 25, 2325–2331. [Google Scholar] [CrossRef] [Green Version]
- Sundararaman, A.; Fukushima, Y.; Norman, J.C.; Uemura, A.; Mellor, H. RhoJ Regulates alpha5beta1 Integrin Trafficking to Control Fibronectin Remodeling during Angiogenesis. Curr. Biol. 2020, 30, 2146–2155.e2145. [Google Scholar] [CrossRef]
- Kusuhara, S.; Fukushima, Y.; Fukuhara, S.; Jakt, L.M.; Okada, M.; Shimizu, Y.; Hata, M.; Nishida, K.; Negi, A.; Hirashima, M.; et al. Arhgef15 promotes retinal angiogenesis by mediating VEGF-induced Cdc42 activation and potentiating RhoJ inactivation in endothelial cells. PLoS ONE 2012, 7, e45858. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Bruning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Wilson, E.; Leszczynska, K.; Poulter, N.S.; Edelmann, F.; Salisbury, V.A.; Noy, P.J.; Bacon, A.; Rappoport, J.Z.; Heath, J.K.; Bicknell, R.; et al. RhoJ interacts with the GIT-PIX complex and regulates focal adhesion disassembly. J. Cell Sci. 2014, 127, 3039–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takase, H.; Matsumoto, K.; Yamadera, R.; Kubota, Y.; Otsu, A.; Suzuki, R.; Ishitobi, H.; Mochizuki, H.; Kojima, T.; Takano, S.; et al. Genome-wide identification of endothelial cell-enriched genes in the mouse embryo. Blood 2012, 120, 914–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Yang, H.; Fukushima, Y.; Saw, P.E.; Lee, J.; Park, J.S.; Park, I.; Jung, J.; Kataoka, H.; Lee, D.; et al. Vascular RhoJ is an effective and selective target for tumor angiogenesis and vascular disruption. Cancer Cell 2014, 25, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Li, H.; Xia, L.; Xu, P.; Ding, Y.; Huo, D.; Hu, Y. Anti-RhoJ antibody functionalized Au@I nanoparticles as CT-guided tumor vessel-targeting radiosensitizers in patient-derived tumor xenograft model. Biomaterials 2017, 141, 1–12. [Google Scholar] [CrossRef]
- Ho, H.; Aruri, J.; Kapadia, R.; Mehr, H.; White, M.A.; Ganesan, A.K. RhoJ regulates melanoma chemoresistance by suppressing pathways that sense DNA damage. Cancer Res. 2012, 72, 5516–5528. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R.; Jahid, S.; Harris, M.; Marzese, D.M.; Espitia, F.; Vasudeva, P.; Chen, C.F.; de Feraudy, S.; Wu, J.; Gillen, D.L.; et al. The RhoJ-BAD signaling network: An Achilles’ heel for BRAF mutant melanomas. PLoS Genet. 2017, 13, e1006913. [Google Scholar] [CrossRef]
- Kim, C.; Yang, H.; Park, I.; Chon, H.J.; Kim, J.H.; Kwon, W.S.; Lee, W.S.; Kim, T.S.; Rha, S.Y. Rho GTPase RhoJ is Associated with Gastric Cancer Progression and Metastasis. J. Cancer 2016, 7, 1550–1556. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jiang, X.; Yang, Y.; Chen, H.; Zhang, C.; Xu, H.; Qi, B.; Yao, C.; Xia, H. Rhoj Is a Novel Target for Progression and Invasion of Glioblastoma by Impairing Cytoskeleton Dynamics. Neurotherapeutics 2020, 17, 2028–2040. [Google Scholar] [CrossRef]
- Chen, B.; Yuan, Y.; Sun, L.; Chen, J.; Yang, M.; Yin, Y.; Xu, Y. MKL1 Mediates TGF-beta Induced RhoJ Transcription to Promote Breast Cancer Cell Migration and Invasion. Front. Cell Dev. Biol 2020, 8, 832. [Google Scholar] [CrossRef]
- Jampol, L.M.; Glassman, A.R.; Sun, J. Evaluation and Care of Patients with Diabetic Retinopathy. N. Engl. J. Med. 2020, 382, 1629–1637. [Google Scholar] [CrossRef]
- Scott, I.U.; Campochiaro, P.A.; Newman, N.J.; Biousse, V. Retinal vascular occlusions. Lancet 2020, 396, 1927–1940. [Google Scholar] [CrossRef]
- Chan-Ling, T.; Gole, G.A.; Quinn, G.E.; Adamson, S.J.; Darlow, B.A. Pathophysiology, screening and treatment of ROP: A multi-disciplinary perspective. Prog. Retin Eye Res. 2018, 62, 77–119. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [Green Version]
- Tanna, A.P.; Johnson, M. Rho Kinase Inhibitors as a Novel Treatment for Glaucoma and Ocular Hypertension. Ophthalmology 2018, 125, 1741–1756. [Google Scholar] [CrossRef] [Green Version]
- Al-Humimat, G.; Marashdeh, I.; Daradkeh, D.; Kooner, K. Investigational Rho Kinase Inhibitors for the Treatment of Glaucoma. J. Exp. Pharmacol. 2021, 13, 197–212. [Google Scholar] [CrossRef]
- Hida, Y.; Nakamura, S.; Nishinaka, A.; Inoue, Y.; Shimazawa, M.; Hara, H. Effects of ripasudil, a ROCK inhibitor, on retinal edema and nonperfusion area in a retinal vein occlusion murine model. J. Pharmacol. Sci. 2018, 137, 129–136. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Nakao, S.; Arita, R.; Kaizu, Y.; Arima, M.; Zhou, Y.; Kita, T.; Yoshida, S.; Kimura, K.; Isobe, T.; et al. Vascular Normalization by ROCK Inhibitor: Therapeutic Potential of Ripasudil (K-115) Eye Drop in Retinal Angiogenesis and Hypoxia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2264–2276. [Google Scholar] [CrossRef] [Green Version]
- Ahmadieh, H.; Nourinia, R.; Hafezi-Moghadam, A.; Sabbaghi, H.; Nakao, S.; Zandi, S.; Yaseri, M.; Tofighi, Z.; Akbarian, S. Intravitreal injection of a Rho-kinase inhibitor (fasudil) combined with bevacizumab versus bevacizumab monotherapy for diabetic macular oedema: A pilot randomised clinical trial. Br. J. Ophthalmol. 2019, 103, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Song, Y.S.; Ishibazawa, A.; Omae, T.; Ro-Mase, T.; Ishiko, S.; Yoshida, A. Effect of ripasudil on diabetic macular edema. Sci. Rep. 2019, 9, 3703. [Google Scholar] [CrossRef] [Green Version]
- O’Bryan, J.P. Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol. Res. 2019, 139, 503–511. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uemura, A.; Fukushima, Y. Rho GTPases in Retinal Vascular Diseases. Int. J. Mol. Sci. 2021, 22, 3684. https://doi.org/10.3390/ijms22073684
Uemura A, Fukushima Y. Rho GTPases in Retinal Vascular Diseases. International Journal of Molecular Sciences. 2021; 22(7):3684. https://doi.org/10.3390/ijms22073684
Chicago/Turabian StyleUemura, Akiyoshi, and Yoko Fukushima. 2021. "Rho GTPases in Retinal Vascular Diseases" International Journal of Molecular Sciences 22, no. 7: 3684. https://doi.org/10.3390/ijms22073684
APA StyleUemura, A., & Fukushima, Y. (2021). Rho GTPases in Retinal Vascular Diseases. International Journal of Molecular Sciences, 22(7), 3684. https://doi.org/10.3390/ijms22073684