
Improved CRISPR/Cas9 Tools for the Rapid Metabolic Engineering of Clostridium acetobutylicum

Abstract
:1. Introduction
2. Results and Discussion
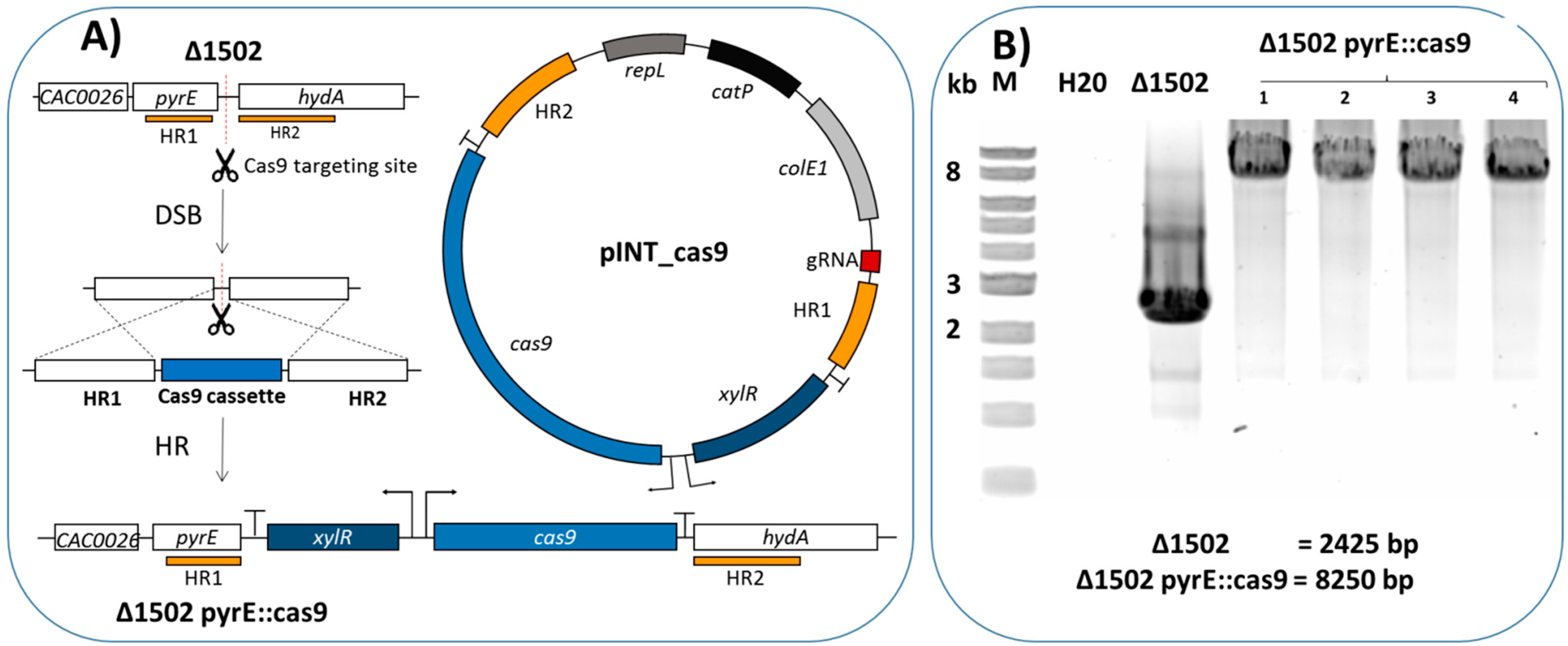
2.1. Integration of Cas9 into the Genome
2.2. Determination of the Efficiency of the CRISPR/Cas9 System
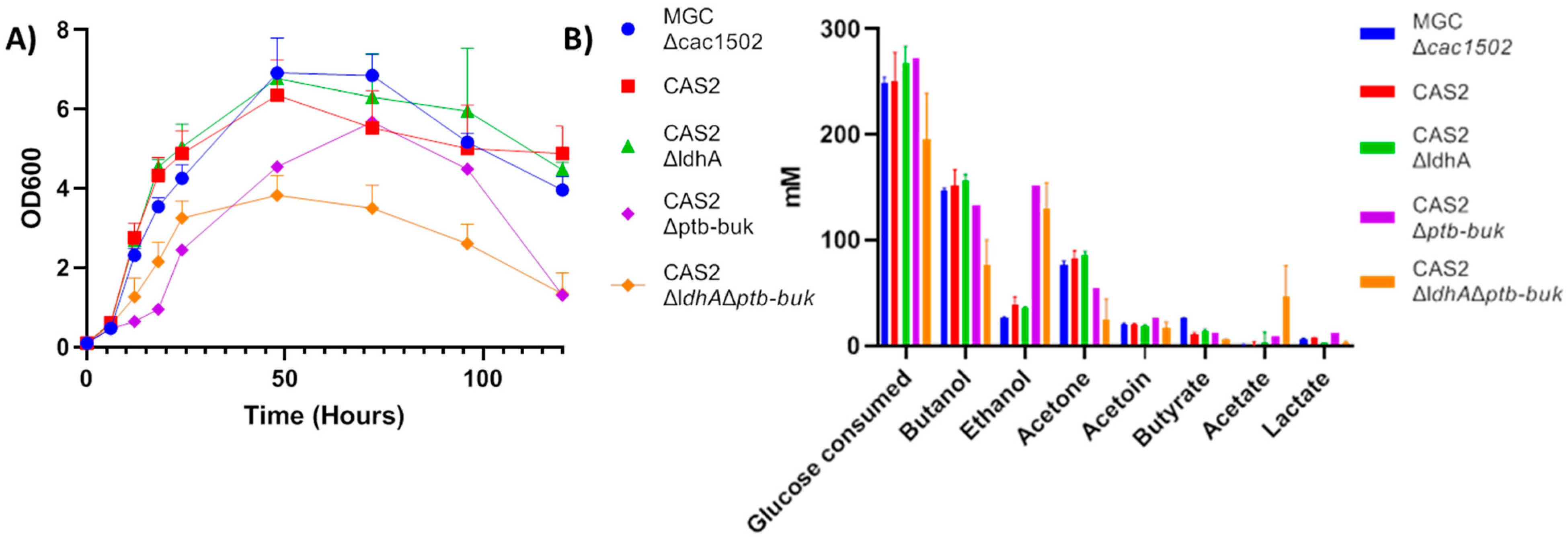
2.3. Deletion of ldhA and the ptb-buk Operon
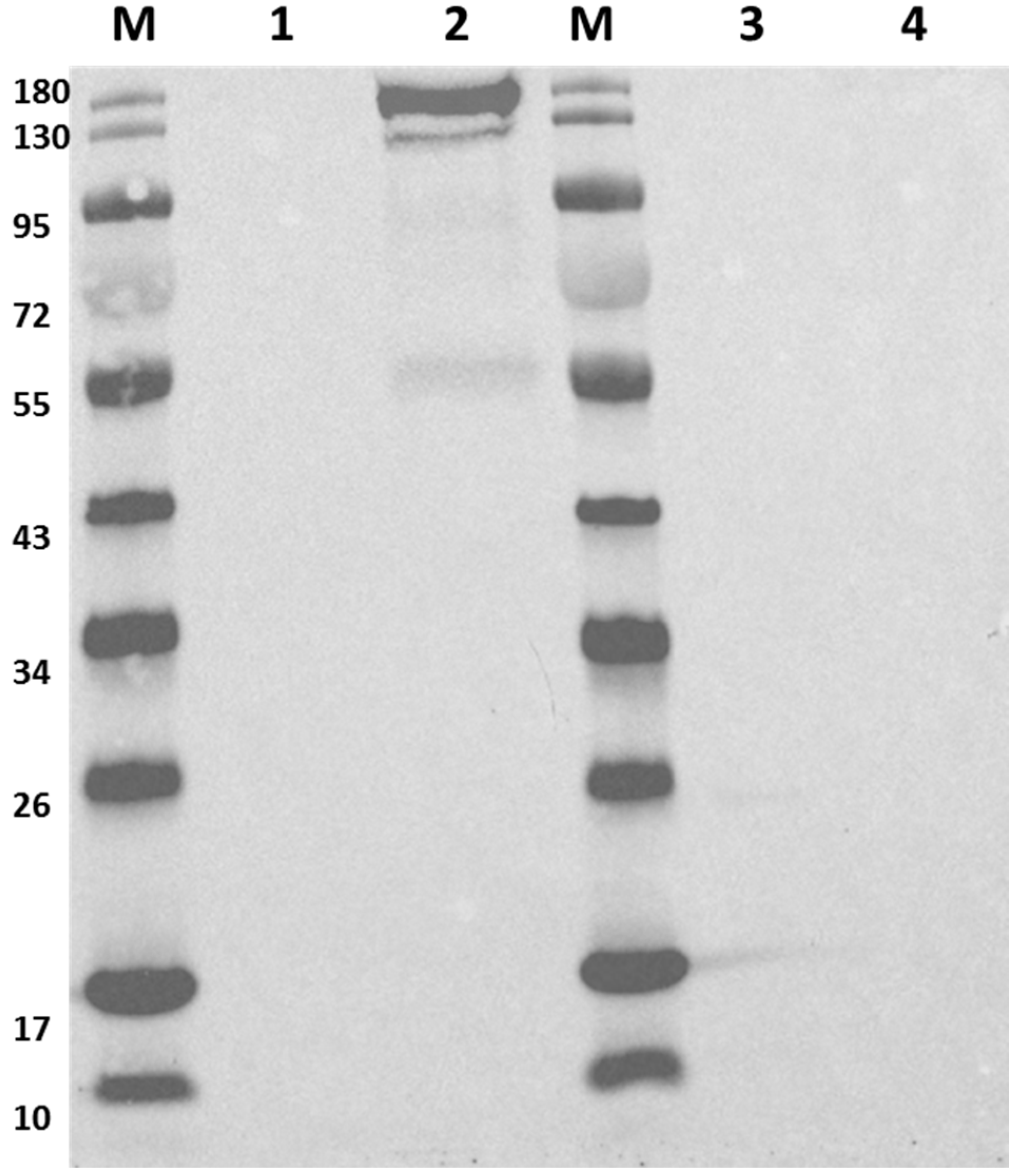
2.4. Removal of Cas9
3. Conclusions
4. Materials and Methods
4.1. Growth Conditions
4.2. DNA Isolation and Manipulation
4.3. Analytical Methods
4.4. Western Blot
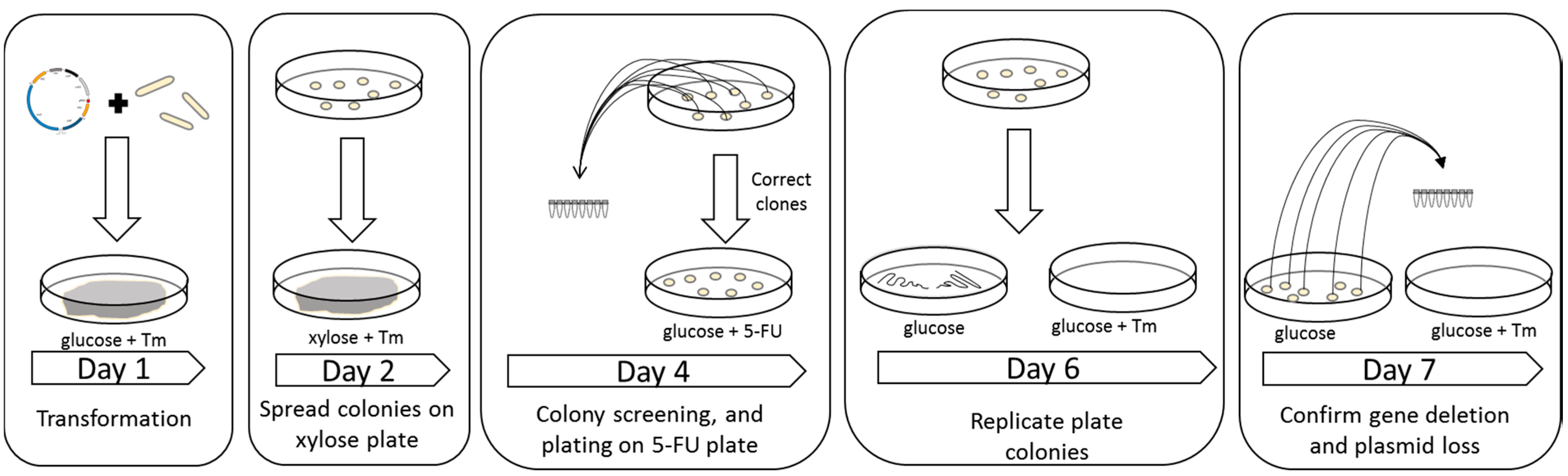
4.5. Construction of Strains
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shao, L.; Hu, S.; Yang, Y.; Gu, Y.; Chen, J.; Yang, Y.; Jiang, W.; Yang, S. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 2007, 17. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The ClosTron: A universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 2007, 70, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, C.; Huang, C.-N.; Nguyen, N.-P.-T.; Thiel, A.; Wilding-Steel, T.; Soula, J.; Yoo, M.; Ehrenreich, A.; Meynial-Salles, I.; Liebl, W.; et al. An efficient method for markerless mutant generation by allelic exchange in Clostridium acetobutylicum and Clostridium saccharobutylicum using suicide vectors. Biotechnol. Biofuels 2019, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Al-Hinai, M.A.; Fast, A.G.; Papoutsakis, E.T. Novel system for efficient isolation of clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl. Environ. Microbiol. 2012, 78, 8112–8121. [Google Scholar] [CrossRef] [Green Version]
- Croux, C.; Nguyen, N.-P.-T.; Lee, J.; Raynaud, C.; Saint-Prix, F.; Gonzalez-Pajuelo, M.; Meynial-Salles, I.; Soucaille, P. Construction of a restriction-less, marker-less mutant useful for functional genomic and metabolic engineering of the biofuel producer Clostridium acetobutylicum. Biotechnol. Biofuels 2016, 9, 23. [Google Scholar] [CrossRef]
- Heap, J.T.; Ehsaan, M.; Cooksley, C.M.; Ng, Y.-K.; Cartman, S.T.; Winzer, K.; Minton, N.P. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res. 2012, 40, e59. [Google Scholar] [CrossRef]
- Ehsaan, M.; Kuit, W.; Zhang, Y.; Cartman, S.T.; Heap, J.T.; Winzer, K.; Minton, N.P. Mutant generation by allelic exchange and genome resequencing of the biobutanol organism Clostridium acetobutylicum ATCC 824. Biotechnol. Biofuels 2016, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Z.-T.; Seo, S.-O.; Lynn, P.; Lu, T.; Jin, Y.-S.; Blaschek, H.P. Bacterial Genome Editing with CRISPR-Cas9: Deletion, Integration, Single Nucleotide Modification, and Desirable “Clean” Mutant Selection inClostridium beijerinckiias an Example. ACS Synth. Biol. 2016, 5, 721–732. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Minton, N.P.; Zhang, Y.; Wen, Z.; Liu, J.; Yang, H.; Zeng, Z.; Ren, X.; Yang, J.; et al. CRISPR-based genome editing and expression control systems inClostridium acetobutylicumandClostridium beijerinckii. Biotechnol. J. 2016, 11, 961–972. [Google Scholar] [CrossRef]
- Wasels, F.; Jean-Marie, J.; Collas, F.; López-Contreras, A.M.; Ferreira, N.L. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J. Microbiol. Methods 2017, 140, 5–11. [Google Scholar] [CrossRef]
- Wasels, F.; Chartier, G.; Hocq, R.; Ferreira, N.L. A CRISPR/Anti-CRISPR Genome Editing Approach Underlines the Synergy of Butanol Dehydrogenases in Clostridium acetobutylicum DSM 792. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Cañadas, I.C.; Groothuis, D.; Zygouropoulou, M.; Rodrigues, R.; Minton, N.P. RiboCas: A Universal CRISPR-Based Editing Tool for Clostridium. ACS Synth. Biol. 2019, 8, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Li, Y.; Shi, Z.; Hemme, C.L.; Li, Y.; Zhu, Y.; Van Nostrand, J.D.; He, Z.; Zhou, J. Efficient Genome Editing in Clostridium cellulolyticum via CRISPR-Cas9 Nickase. Appl. Environ. Microbiol. 2015, 81, 4423–4431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Seys, F.M.; Minton, N.P.; Yang, J.; Jiang, Y.; Jiang, W.; Yang, S. CRISPR-Cas9 D10A nickase-assisted base editing in the solvent producer Clostridium beijerinckii. Biotechnol. Bioeng. 2019, 116, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; de Gennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, E.M.D.O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35. [Google Scholar] [CrossRef]
- Zhang, J.; Hong, W.; Zong, W.; Wang, P.; Wang, Y. Markerless genome editing in Clostridium beijerinckii using the CRISPR-Cpf1 system. J. Biotechnol. 2018, 284, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Zhang, J.; Cui, G.; Wang, L.; Wang, Y. Multiplexed CRISPR-Cpf1-Mediated Genome Editing in Clostridium difficile toward the Understanding of Pathogenesis of C. difficile Infection. ACS Synth. Biol. 2018, 7, 1588–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hong, W.; Guo, L.; Wang, Y.; Wang, Y. Enhancing plasmid transformation efficiency and enabling CRISPR-Cas9/Cpf1—based genome editing in Clostridium tyrobutyricum. Biotechnol. Bioeng. 2020, 117, 2911–2917. [Google Scholar] [CrossRef]
- Zhang, J.; Zong, W.; Hong, W.; Zhang, Z.-T.; Wang, Y. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metab. Eng. 2018, 47, 49–59. [Google Scholar] [CrossRef]
- Maikova, A.; Kreis, V.; Boutserin, A.; Severinov, K.; Soutourina, O. Using an Endogenous CRISPR-Cas System for Genome Editing in the Human PathogenClostridium difficile. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E.; Lanahan, A.A.; Zheng, T.; Toruno, C.; Lynd, L.R.; Cameron, J.C.; Olson, D.G.; Eckert, C.A. Development of both type I–B and type II CRISPR/Cas genome editing systems in the cellulolytic bacterium Clostridium thermocellum. Metab. Eng. Commun. 2020, 10, e00116. [Google Scholar] [CrossRef]
- Nariya, H.; Miyata, S.; Kuwahara, T.; Okabe, A. Development and Characterization of a Xylose-Inducible Gene Expression System for Clostridium perfringens. Appl. Environ. Microbiol. 2011, 77, 8439–8441. [Google Scholar] [CrossRef] [Green Version]
- Diallo, M.; Hocq, R.; Collas, F.; Chartier, G.; Wasels, F.; Wijaya, H.S.; Werten, M.W.; Wolbert, E.J.; Kengen, S.W.; Van Der Oost, J.; et al. Adaptation and application of a two-plasmid inducible CRISPR-Cas9 system in Clostridium beijerinckii. Methods 2020, 172, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Croux, C.; Meynial-Salles, I.; Soucaille, P. Metabolic flexibility of a butyrate pathway mutant of Clostridium acetobutylicum. Metab. Eng. 2017, 40, 138–147. [Google Scholar] [CrossRef]
- Green, E.M.; Boynton, Z.L.; Harris, L.M.; Rudolph, F.B.; Papoutsakis, E.T.; Bennett, G.N. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology 1996, 142, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, D.; Radomski, N.; Lütke-Eversloh, T. New insights into the butyric acid metabolism of Clostridium acetobutylicum. Appl. Microbiol. Biotechnol. 2012, 96, 1325–1339. [Google Scholar] [CrossRef]
- Nguyen, N.-P.-T.; Raynaud, C.; Meynial-Salles, I.; Soucaille, P. Reviving the Weizmann process for commercial n-butanol production. Nat. Commun. 2018, 9, 3682. [Google Scholar] [CrossRef]
- Dusséaux, S.; Croux, C.; Soucaille, P.; Meynial-Salles, I. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for the high-yield production of a biofuel composed of an isopropanol/butanol/ethanol mixture. Metab. Eng. 2013, 18, 1–8. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | pCons2-1 | pGRNAminiPthl Δupp_HA1000 | pGRNAminiPthlΔupp | pGRNAJ23119ΔuppHA1000 | pGRNAJ23119Δupp |
|---|---|---|---|---|---|
| CFU/μg of DNA | 5.13 (±0.51) × 103 | 5.45 (±0.67) × 103 | 7.81 (±0.84) × 103 | 7.53 (±0.58) × 103 | 7.66 (±0.72) × 103 |
| Promoter | J23119 | MiniPthl | |||
|---|---|---|---|---|---|
| Size of homology arms | 1000 bp | 1000 bp | 500 bp | 250 bp | 100 bp |
| Editing efficiency | 100% | 100% | 96% | 91% | 0% |
| Gene | Integration of Cas9 Cassette | Deletion of upp | Deletion of ldhA | Deletion of ptb-buk Operon | Removal of Cas9 Cassette |
|---|---|---|---|---|---|
| Efficiency | 100% | 100% | 100% | 93% | 100% |
| size ofdeletion/insertion | 5872 bp | 630 bp | 942 bp | 2001 bp | 5872 bp |
| Plasmids | Relevant Characteristics | Source or Reference |
|---|---|---|
| pCons2-1 | Cmr, repL | [5] |
| pCons::upp | Cmr, repL, upp | [5] |
| pINT_cas9 | Cmr, repL, pyrE::pxyl_cas9 | This study |
| pGRNAminiPthl Δupp_HA1000 | Cmr, repL, miniPthlgRNAupp, Δupp | This study |
| pGRNAminiPthlΔupp | Cmr, repL, miniPthlgRNAupp, | This study |
| pGRNAJ23119ΔuppHA1000 | Cmr, repL, J23119PthlgRNAupp | This study |
| pGRNAJ23119Δupp | Cmr, repL, J23119PthlgRNAupp, Δupp | This study |
| pGRNAminiPthl Δupp_HA500 | Cmr, repL, miniPthlgRNAupp, Δupp | This study |
| pGRNAminiPthl Δupp_HA250 | Cmr, repL, miniPthlgRNAupp, Δupp | This study |
| pGRNAminiPthl Δupp_HA100 | Cmr, repL, miniPthlgRNAupp, Δupp | This study |
| pGRNAΔldhA | Cmr, repL, upp, J23119gRNAldhA, ΔldhA | This study |
| pGRNAΔptb-buk | Cmr, repL, upp, miniPthlgRNAbuk, Δbuk-ptb | This study |
| pGRNAΔcas9 | Cmr, repL, upp, J23119gRNAcas9, Δcas9 | This study |
| Strain | Relevant Characteristics | Source or Reference |
|---|---|---|
| Bacterial strains | ||
| E. coli TOP10 | Invitrogen | |
| C. acetobutylicum | ||
| MGCΔcac1502 | ΔCA_C1502 | [5] |
| CAS1 | ΔCA_C1502, pyrE::pXyl_cas9 | This study |
| CAS2 | ΔCA_C1502, pyrE::pXyl_cas9, ΔCA_C2879 | This study |
| CAS2ΔldhA | ΔCA_C1502, pyrE::pxyl_cas9, ΔCA_C2879, ΔCA_C0267 | This study |
| CAS2Δptb-buk | ΔCA_C1502, pyrE::pxyl_cas9, ΔCA_C2879, ΔCA_C3075, ΔCA_C3076 | This study |
| CAS2ΔldhAΔptb-buk | ΔCA_C1502, pyrE::pxyl_cas9, ΔCA_C2879, ΔCA_C0267, ΔCA_C3075, ΔCA_C3076 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilding-Steele, T.; Ramette, Q.; Jacottin, P.; Soucaille, P. Improved CRISPR/Cas9 Tools for the Rapid Metabolic Engineering of Clostridium acetobutylicum. Int. J. Mol. Sci. 2021, 22, 3704. https://doi.org/10.3390/ijms22073704
Wilding-Steele T, Ramette Q, Jacottin P, Soucaille P. Improved CRISPR/Cas9 Tools for the Rapid Metabolic Engineering of Clostridium acetobutylicum. International Journal of Molecular Sciences. 2021; 22(7):3704. https://doi.org/10.3390/ijms22073704
Chicago/Turabian StyleWilding-Steele, Tom, Quentin Ramette, Paul Jacottin, and Philippe Soucaille. 2021. "Improved CRISPR/Cas9 Tools for the Rapid Metabolic Engineering of Clostridium acetobutylicum" International Journal of Molecular Sciences 22, no. 7: 3704. https://doi.org/10.3390/ijms22073704