
Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles



{kind=link}
Abstract
1. Introduction
2. Mitochondria-ER Contacts (MERCs): A Signalling Platform for Mitochondrial Ca2+ Homeostasis
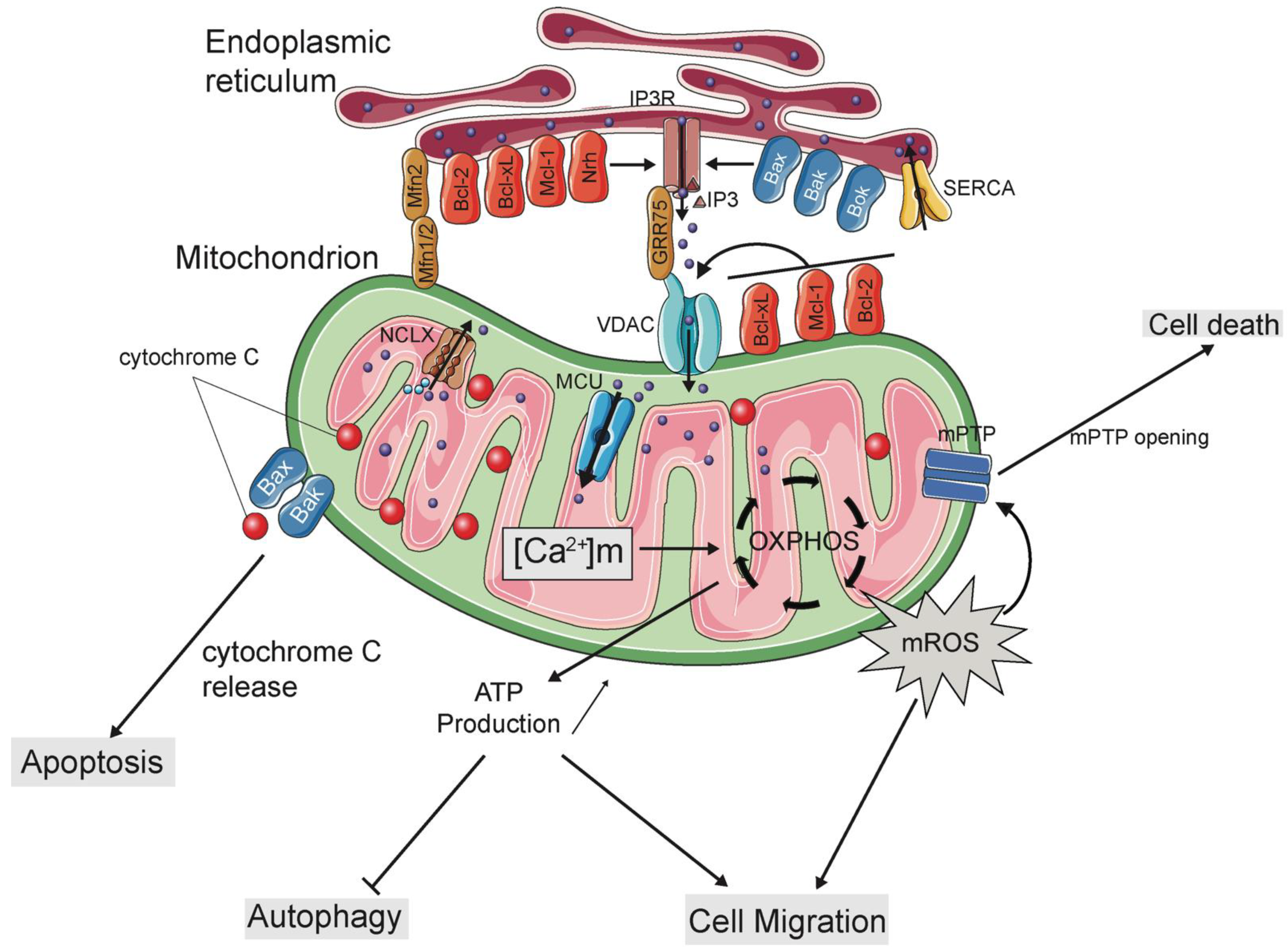
3. Regulation of Mitochondrial Ca2+ Uptake Machinery by Bcl-2 Family Proteins
4. Remote Control of Mitochondrial Ca2+ Signalling by ER-Based Bcl-2 Proteins
5. Bridging the Gap between Mitochondria and ER during Cell Death and Survival
6. Role of Bcl-2 Family Proteins in Ca2+-Dependent Cell Migration
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Letai, A. Apoptosis and Cancer. Annu. Rev. Cancer Biol. 2017, 1, 275–294. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.E.; Reid, G.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A Serine Protease, HtrA2, Is Released from the Mitochondria and Interacts with XIAP, Inducing Cell Death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Croce, C.M. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 5214–5218. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 2017, 25, 13–20. [Google Scholar] [CrossRef]
- Green, D.R.; Fitzgerald, P. Just So Stories about the Evolution of Apoptosis. Curr. Biol. 2016, 26, R620–R627. [Google Scholar] [CrossRef]
- Hengartner, M.O.; Ellis, R.; Horvitz, R. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nat. Cell Biol. 1992, 356, 494–499. [Google Scholar] [CrossRef]
- Hengartner, M.O.; Horvitz, H.C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 1994, 76, 665–676. [Google Scholar] [CrossRef]
- Veis, D.J.; Sorenson, C.M.; Shutter, J.R.; Korsmeyer, S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993, 75, 229–240. [Google Scholar] [CrossRef]
- Knudson, C.M.; Tung, K.S.; Tourtellotte, W.G.; Brown, G.A.; Korsmeyer, S.J. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 1995, 270, 96–99. [Google Scholar] [CrossRef]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genome Res. 2000, 14, 23–27. [Google Scholar]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Alavian, K.N.; Lazrove, E.; Mehta, N.; Jones, A.; Zhang, P.; Licznerski, P.; Graham, M.; Uo, T.; Guo, J.; et al. A Bcl-Xl-Drp1 Complex Regulates Synaptic Vesicle Membrane Dynamics During Endocytosis. Nat Cell Biol 2013, 15, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.-S.; Toombs, J.; Kuo, Y.-C.; Piazza, J.T.; Tuladhar, R.; Barrett, Q.; Fan, C.-W.; Zhang, X.; Walensky, L.D.; et al. Extra-mitochondrial prosurvival BCL-2 proteins regulate gene transcription by inhibiting the SUFU tumour suppressor. Nat. Cell Biol. 2017, 19, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Bonneau, B.; Ferri, K.F.; Prudent, J.; Thibaut, J.; Gillet, G. The Apoptotic Regulator Nrz Controls Cytoskeletal Dynamics via the Regulation of Ca2+ Trafficking in the Zebrafish Blastula. Dev. Cell 2011, 20, 663–676. [Google Scholar] [CrossRef]
- Prudent, J.; Popgeorgiev, N.; Bonneau, B.; Thibaut, J.; Gadet, R.; Lopez, J.; Gonzalo, P.; Rimokh, R.; Manon, S.; Houart, C.; et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat. Commun. 2013, 4, 2330. [Google Scholar] [CrossRef]
- Krajewski, S.; Tanaka, S.; Takayama, S.; Schibler, M.J.; Fenton, W.; Reed, J.C. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993, 53, 4701–4714. [Google Scholar]
- Yang, T.; Kozopas, K.M.; Craig, R.W. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 1995, 128, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Schlipf, S.; Sanz, J.; Neubert, K.; Stein, R.; Borner, C. Characterization of the signal that directs Bcl-xL, but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 2003, 160, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Lucendo, E.; Sancho, M.; Lolicato, F.; Javanainen, M.; Kulig, W.; Leiva, D.; Duart, G.; Andreu-Fernández, V.; Mingarro, I.; Orzáez, M. Mcl-1 and Bok transmembrane domains: Unexpected players in the modulation of apoptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 27980–27988. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.-P.; Aromolaran, A.S.; Bultynck, G.; Zhong, F.; Li, X.; McColl, K.; Matsuyama, S.; Herlitze, S.; Roderick, H.L.; Bootman, M.D.; et al. Targeting Bcl-2-IP3 Receptor Interaction to Reverse Bcl-2’s Inhibition of Apoptotic Calcium Signals. Mol. Cell 2008, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The Endoplasmic Reticulum Gateway to Apoptosis by Bcl-X(L) Modulation of the Insp3r. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Li, X.X.; Gottleib, E.; Hill, R.B.; Thompson, C.B.; Colombini, M. Bcl-Xl Promotes the Open Configuration of the Voltage-Dependent Anion Channel and Metabolite Passage through the Outer Mitochondrial Membrane. J. Biol. Chem. 2001, 276, 19414–19419. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, B.; Prudent, J.; Popgeorgiev, N.; Gillet, G. Non-apoptotic roles of Bcl-2 family: The calcium connection. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1833, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Bernardi, P.; von Stockum, S. The Permeability Transition Pore as a Ca(2+) Release Channel: New Answers to an Old Question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef] [PubMed]
- Vallese, F.; Barazzuol, L.; Maso, L.; Brini, M.; Calì, T. ER-Mitochondria Calcium Transfer, Organelle Contacts and Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2019, 1131, 719–746. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Prudent, J.; McBride, H.M. The mitochondria–endoplasmic reticulum contact sites: A signalling platform for cell death. Curr. Opin. Cell Biol. 2017, 47, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- De La Fuente, S.; Fernandez-Sanz, C.; Vail, C.; Agra, E.J.; Holmstrom, K.; Sun, J.; Mishra, J.; Williams, D.; Finkel, T.; Murphy, E.; et al. Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. J. Biol. Chem. 2016, 291, 23343–23362. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 Domain of Anti-apoptotic Bcl-XL, but Not That of the Related Bcl-2, Limits the Voltage-dependent Anion Channel 1 (VDAC1)-mediated Transfer of Pro-apoptotic Ca2+ Signals to Mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef]
- Lalier, L.; Mignard, V.; Joalland, M.-P.; Lanoé, D.; Cartron, P.-F.; Manon, S.; Vallette, F.M. TOM20-mediated transfer of Bcl2 from ER to MAM and mitochondria upon induction of apoptosis. Cell Death Dis. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Williams, A.; Hayashi, T.; Wolozny, D.; Yin, B.; Su, T.C.; Betenbaugh, M.J.; Su, T.P. The Non-Apoptotic Action of Bcl-Xl: Regulating Ca(2+) Signaling and Bioenergetics at the Er-Mitochondrion Interface. J. Bioenerg. Biomembr. 2016, 48, 211–225. [Google Scholar] [CrossRef]
- Carpio, M.A.; Means, R.E.; Brill, A.L.; Sainz, A.; Ehrlich, B.E.; Katz, S.G. BOK controls apoptosis by Ca2+ transfer through ER-mitochondrial contact sites. Cell Rep. 2021, 34, 108827. [Google Scholar] [CrossRef]
- Saxena, N.; Katiyar, S.P.; Liu, Y.; Grover, A.; Gao, R.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Molecular Interactions of Bcl-2 and Bcl-Xl with Mortalin: Identification and Functional Characterization. Biosci. Rep. 2013, 33, e00073. [Google Scholar] [CrossRef]
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, e1800013. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nat. Cell Biol. 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.A.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.; Sheiko, T.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 1477–1485. [Google Scholar] [CrossRef]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. Vdac1 Selectively Transfers Apoptotic Ca2+ Signals to Mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef]
- Shimizu, H.; Schredelseker, J.; Huang, J.; Lu, K.; Naghdi, S.; Lu, F.; Franklin, S.; Fiji, H.D.; Wang, K.; Zhu, H.; et al. Mitochondrial Ca2+ uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity. eLife 2015, 4, e04801. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Schumacker, P.T.; Thompson, C.B. Bcl-Xl Prevents Cell Death Following Growth Factor Withdrawal by Facilitating Mitochondrial Atp/Adp Exchange. Mol. Cell 1999, 3, 159–167. [Google Scholar] [CrossRef]
- Roy, S.S.; Madesh, M.; Davies, E.; Antonsson, B.; Danial, N.; Hajnóczky, G. Bad Targets the Permeability Transition Pore Independent of Bax or Bak to Switch between Ca2+-Dependent Cell Survival and Death. Mol. Cell 2009, 33, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nat. Cell Biol. 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Eno, C.O.; Eckenrode, E.F.; Olberding, K.E.; Zhao, G.; White, C.; Li, C. Distinct Roles of Mitochondria- and Er-Localized Bcl-Xl in Apoptosis Resistance and Ca2+ Homeostasis. Mol. Biol. Cell 2012, 23, 2605–2618. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An Interaction between Bcl-Xl and the Voltage-Dependent Anion Channel (Vdac) Promotes Mitochondrial Ca2+ Uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the Antiapoptotic Activity of Bcl-Xl Protein Upon Interaction with Vdac1 Protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef]
- Jiao, J.; Huang, X.; Feit-Leithman, R.A.; Neve, R.L.; Snider, W.; Dartt, D.A.; Chen, D.F. Bcl-2 enhances Ca2+ signaling to support the intrinsic regenerative capacity of CNS axons. EMBO J. 2005, 24, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Basset, O.; Boittin, F.-X.; Cognard, C.; Constantin, B.; Ruegg, U.T. Bcl-2 overexpression prevents calcium overload and subsequent apoptosis in dystrophic myotubes. Biochem. J. 2006, 395, 267–276. [Google Scholar] [CrossRef]
- Huang, H.; Shah, K.H.A.; Bradbury, N.; Li, C.; White, C.M. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, Y.; Chua, B.H.; Ho, Y.-S.; Kuo, T.H. Regulation of Sodium–Calcium Exchange and Mitochondrial Energetics by Bcl-2 in the Heart of Transgenic Mice. J. Mol. Cell. Cardiol. 2001, 33, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Seo, M.-D.; Enomoto, M.; Ishiyama, N.; Stathopulos, P.B.; Ikura, M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1853, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.A.; Mignery, G.; Südhof, T.C. Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J. Biol. Chem. 1994, 269, 28613–28619. [Google Scholar] [CrossRef]
- Taylor, C.W.; Tovey, S.C. IP3 Receptors: Toward Understanding Their Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [PubMed]
- Nougarède, A.; Popgeorgiev, N.; Kassem, L.; Omarjee, S.; Borel, S.; Mikaelian, I.; Lopez, J.; Gadet, R.; Marcillat, O.; Treilleux, I.; et al. Breast Cancer Targeting through Inhibition of the Endoplasmic Reticulum-Based Apoptosis Regulator Nrh/BCL2L10. Cancer Res. 2018, 78, 1404–1417. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.-P.; Barr, P.; Yee, V.C.; Distelhorst, C.W. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 971–978. [Google Scholar] [CrossRef][Green Version]
- Yang, J.; Vais, H.; Gu, W.; Foskett, J.K. Biphasic Regulation of Insp3 Receptor Gating by Dual Ca2+ Release Channel Bh3-Like Domains Mediates Bcl-Xl Control of Cell Viability. Proc. Natl. Acad. Sci. USA 2016, 113, E1953–E1962. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis Protection by Mcl-1 and Bcl-2 Modulation of Inositol 1,4,5-Trisphosphate Receptor-dependent Ca2+ Signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef]
- Minagawa, N.; Kruglov, E.A.; Dranoff, J.A.; Robert, M.E.; Gores, G.J.; Nathanson, M.H. The Anti-apoptotic Protein Mcl-1 Inhibits Mitochondrial Ca2+ Signals. J. Biol. Chem. 2005, 280, 33637–33644. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, B.; Ando, H.; Kawaai, K.; Hirose, M.; Takahashi-Iwanaga, H.; Mikoshiba, K. IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. eLife 2016, 5, e19896. [Google Scholar] [CrossRef]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Decrock, E.; Molgó, J.; Sorrentino, V.; Missiaen, L.; Leybaert, L.; De Smedt, H.; Kasri, N.N.; Parys, J.B.; Bultynck, G. Bcl-2 binds to and inhibits ryanodine receptors. J. Cell Sci. 2014, 127, 2782–2792. [Google Scholar] [CrossRef]
- Vervliet, T.; Lemmens, I.; Vandermarliere, E.; Decrock, E.; Ivanova, H.; Monaco, G.; Sorrentino, V.; Kasri, N.N.; Missiaen, L.; Martens, L.; et al. Ryanodine receptors are targeted by anti-apoptotic Bcl-XL involving its BH4 domain and Lys87 from its BH3 domain. Sci. Rep. 2015, 5, 9641. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta BBA Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- CsordásG, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.-S.; Schwall, C.T.; Pazarentzos, E.; Datler, C.; Alder, N.N.; Grimm, S. Mitochondrial Ca2+ influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death Differ. 2014, 21, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Clerix, E.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G. Modulation of Ca2+ Signaling by Anti-apoptotic B-Cell Lymphoma 2 Proteins at the Endoplasmic Reticulum–Mitochondrial Interface. Front. Oncol. 2017, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, I.; Kerkhofs, M.; Veettil, S.P.; Dehaen, W.; Bultynck, G. Cancer cell death strategies by targeting Bcl-2’s BH4 domain. Biochim. Biophys. Acta BBA Bioenerg. 2021, 1868, 118983. [Google Scholar] [CrossRef]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.-P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2–IP3 receptor interaction. Blood 2011, 117, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgo, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Greenberg, E.F.; McColl, K.S.; Zhong, F.; Wildey, G.; Dowlati, A.; Distelhorst, C.W. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015, 6, e2034. [Google Scholar] [CrossRef]
- Lavik, A.R.; Zhong, F.; Chang, M.-J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388–27402. [Google Scholar] [CrossRef]
- Kerkhofs, M.; La Rovere, R.; Welkenhuysen, K.; Janssens, A.; Vandenberghe, P.; Madesh, M.; Parys, J.B.; Bultynck, G. BIRD-2, a BH4-domain-targeting peptide of Bcl-2, provokes Bax/Bak-independent cell death in B-cell cancers through mitochondrial Ca2+-dependent mPTP opening. Cell Calcium 2021, 94, 102333. [Google Scholar] [CrossRef]
- Schulman, J.J.; Wright, F.A.; Kaufmann, T.; Wojcikiewicz, R.J.H. The Bcl-2 Protein Family Member Bok Binds to the Coupling Domain of Inositol 1,4,5-Trisphosphate Receptors and Protects Them from Proteolytic Cleavage. J. Biol. Chem. 2013, 288, 25340–25349. [Google Scholar] [CrossRef]
- Schulman, J.J.; Wright, F.A.; Han, X.; Zluhan, E.J.; Szczesniak, L.M.; Wojcikiewicz, R.J.H. The Stability and Expression Level of Bok Are Governed by Binding to Inositol 1,4,5-Trisphosphate Receptors. J. Biol. Chem. 2016, 291, 11820–11828. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.J.; Szczesniak, L.M.; Bunker, E.N.; Nelson, H.A.; Roe, M.W.; Wagner, L.E., 2nd; Yule, D.I.; Wojcikiewicz, R.J.H. Bok Regulates Mitochondrial Fusion and Morphology. Cell Death Differ. 2019, 26, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Nutt, L.K.; Pataer, A.; Pahler, J.; Fang, B.; Roth, J.; McConkey, D.J.; Swisher, S.G. Bax and Bak Promote Apoptosis by Modulating Endoplasmic Reticular and Mitochondrial Ca2+ Stores. J. Biol. Chem. 2002, 277, 9219–9225. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK Regulation of Endoplasmic Reticulum Ca2+: A Control Point for Apoptosis. Sci. 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.-X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.-C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 105–110. [Google Scholar] [CrossRef]
- D’Orsi, B.; Kilbride, S.M.; Chen, G.; Alvarez, S.P.; Bonner, H.P.; Pfeiffer, S.; Plesnila, N.; Engel, T.; Henshall, D.C.; Düssmann, H.; et al. Bax Regulates Neuronal Ca2+ Homeostasis. J. Neurosci. 2015, 35, 1706–1722. [Google Scholar] [CrossRef]
- Wang, X.E.; Olberding, K.; White, C.; Li, C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2010, 18, 38–47. [Google Scholar] [CrossRef]
- Kanekura, K.; Ma, X.; Murphy, J.T.; Zhu, L.J.; Diwan, A.; Urano, F. IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci. Signal. 2015, 8, ra62. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; García-Sáez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef]
- Große, L.A.; Wurm, C.; Brüser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, K.-I.; Miyata, N.; Mukai, S.; Furuki, S.; Okumoto, K.; Cheng, E.H.; Fujiki, Y. The VDAC2–BAK axis regulates peroxisomal membrane permeability. J. Cell Biol. 2017, 216, 709–722. [Google Scholar] [CrossRef]
- Csordás, G.; Madesh, M.; Antonsson, B.; Hajnóczky, G. tcBid promotes Ca2+ signal propagation to the mitochondria: Control of Ca2+ permeation through the outer mitochondrial membrane. EMBO J. 2002, 21, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Klee, M.; Pallauf, K.; Alcalá, S.; Fleischer, A.; Pimentel-Muiños, F.X. Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J. 2009, 28, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Mathai, J.P.; Germain, M.; Shore, G.C. BH3-only BIK Regulates BAX,BAK-dependent Release of Ca2+ from Endoplasmic Reticulum Stores and Mitochondrial Apoptosis during Stress-induced Cell Death. J. Biol. Chem. 2005, 280, 23829–23836. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Leyva-Baca, I.; Wathelet, M.G.; Lacey, N.; Chand, H.S.; Choi, A.M.K.; Tesfaigzi, Y. Bik reduces hyperplastic cells by increasing Bak and activating DAPk1 to juxtapose ER and mitochondria. Nat. Commun. 2017, 8, 803. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Prudent, J.; Popgeorgiev, N.; Gadet, R.; Deygas, M.; Rimokh, R.; Gillet, G. Mitochondrial Ca2+ uptake controls actin cytoskeleton dynamics during cell migration. Sci. Rep. 2016, 6, 36570. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF -1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Ishikawa, K.; Koshikawa, N.; Takenaga, K.; Nakada, K.; Hayashi, J.-I. Reversible regulation of metastasis by ROS-generating mtDNA mutations. Mitochondrion 2008, 8, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Bessou, M.J.; Lopez, R.; Gadet, M.; Deygas, N.; Popgeorgiev, D.; Poncet, A.; Nougarede, P.; Billard, I.; Mikaelian, P.; Gonzalo, R.; et al. The Apoptosis Inhibitor Bcl-Xl Controls Breast Cancer Cell Migration through Mitochondria-Dependent Reactive Oxygen Species Production. Oncogene 2020, 39, 3056–3074. [Google Scholar] [CrossRef] [PubMed]
- Fouqué, A.; Lepvrier, E.; Debure, L.; Gouriou, Y.; Malleter, M.; Delcroix, V.; Ovize, M.; Ducret, T.; Li, C.; Hammadi, M.; et al. The apoptotic members CD95, BclxL, and Bcl-2 cooperate to promote cell migration by inducing Ca(2+) flux from the endoplasmic reticulum to mitochondria. Cell Death Differ. 2016, 23, 1702–1716. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, J.L.; Gillet, G.; Prudent, J.; Popgeorgiev, N. Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. Int. J. Mol. Sci. 2021, 22, 3730. https://doi.org/10.3390/ijms22073730
Morris JL, Gillet G, Prudent J, Popgeorgiev N. Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. International Journal of Molecular Sciences. 2021; 22(7):3730. https://doi.org/10.3390/ijms22073730
Chicago/Turabian StyleMorris, Jordan L., Germain Gillet, Julien Prudent, and Nikolay Popgeorgiev. 2021. "Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles" International Journal of Molecular Sciences 22, no. 7: 3730. https://doi.org/10.3390/ijms22073730
APA StyleMorris, J. L., Gillet, G., Prudent, J., & Popgeorgiev, N. (2021). Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. International Journal of Molecular Sciences, 22(7), 3730. https://doi.org/10.3390/ijms22073730