
CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
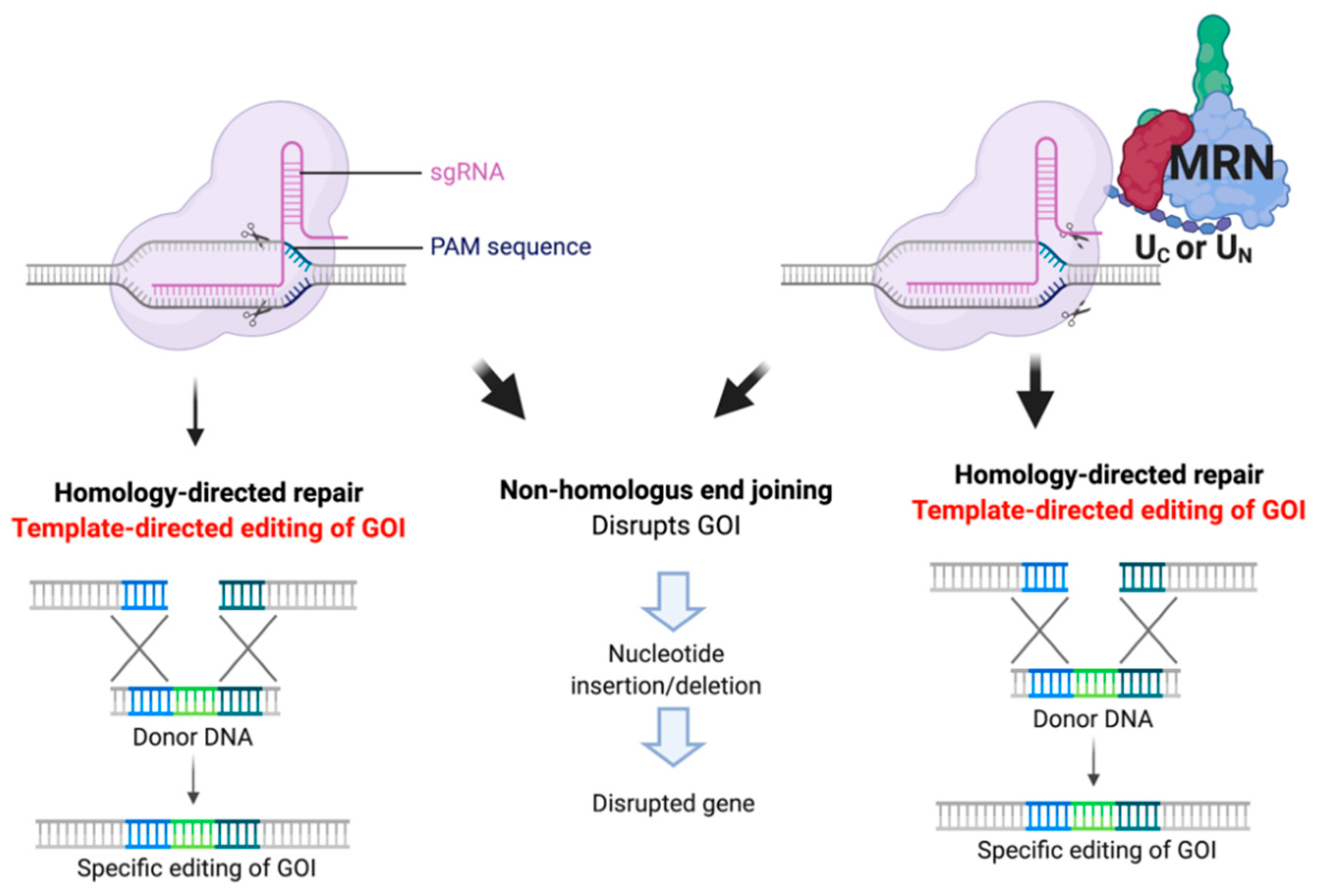
Abstract
:1. Introduction
2. Results
2.1. Characterization of TAF1 ts Cell Lines
2.2. Co-Editing of the TAF1 ts Cells
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Calculation of Cell Doubling Time
4.3. Plasmids and Transfection
4.4. SgRNA Design and Analysis of Editing
4.5. RNA Extraction and cDNA Preparation and Analysis
4.6. Protein Gels and Immunoblotting
4.7. Proteasome Activity Assay
4.8. Statistical Analysis
4.9. Flow Cytometry
4.10. Figures
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kuhn, R. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet. 2019, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.; Adler, J.; Broennimann, K.; Myers, N.; Shaul, Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules 2019, 9, 584. [Google Scholar] [CrossRef] [Green Version]
- Li, X.L.; Li, G.H.; Fu, J.; Fu, Y.W.; Zhang, L.; Chen, W.; Arakaki, C.; Zhang, J.P.; Wen, W.; Zhao, M.; et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018, 46, 10195–10215. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Chiang, C.M. The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Antonova, S.V.; Boeren, J.; Timmers, H.T.M.; Snel, B. Epigenetics and transcription regulation during eukaryotic diversification: The saga of TFIID. Genes Dev. 2019, 33, 888–902. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, T.; Sekiguchi, T.; Noguchi, E.; Sunamoto, H.; Ohba, T.; Nishimoto, T. The CCG1/TAFII250 gene is mutated in thermosensitive G1 mutants of the BHK21 cell line derived from golden hamster. Gene 1994, 141, 267–270. [Google Scholar]
- Wang, P.J.; Page, D.C. Functional substitution for TAF(II)250 by a retroposed homolog that is expressed in human spermatogenesis. Hum. Mol. Genet. 2002, 11, 2341–2346. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356. [Google Scholar] [CrossRef]
- Landry, J.J.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stutz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 Genes Genomes Genet. 2013, 3, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Arendt, C.S.; Hochstrasser, M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef] [Green Version]
- Heinemeyer, W.; Fischer, M.; Krimmer, T.; Stachon, U.; Wolf, D.H. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 1997, 272, 25200–25209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehnal, D.; Rose, A.S.; Koča, J.; Burley, S.K.; Velankar, S. Mol*: Towards a common library and tools for web molecular graphics. In Workshop on Molecular Graphics and Visual Analysis of Molecular Data; The Eurographics Association: Geneva, Switzerland, 2018; Volume 10, pp. 29–33. [Google Scholar] [CrossRef]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Benitez, E.K.; Lomova Kaufman, A.; Cervantes, L.; Clark, D.N.; Ayoub, P.G.; Senadheera, S.; Osborne, K.; Sanchez, J.M.; Crisostomo, R.V.; Wang, X.; et al. Global and Local Manipulation of DNA Repair Mechanisms to Alter Site-Specific Gene Editing Outcomes in Hematopoietic Stem Cells. Front. Genome Ed. 2020, 2. [Google Scholar] [CrossRef]
- Agudelo, D.; Duringer, A.; Bozoyan, L.; Huard, C.C.; Carter, S.; Loehr, J.; Synodinou, D.; Drouin, M.; Salsman, J.; Dellaire, G.; et al. Marker-free coselection for CRISPR-driven genome editing in human cells. Nat. Methods 2017, 14, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Akrap, N.; Cerboni, S.; Porritt, M.J.; Wimberger, S.; Lundin, A.; Moller, C.; Firth, M.; Gordon, E.; Lazovic, B.; et al. Universal toxin-based selection for precise genome engineering in human cells. Nat. Commun. 2021, 12, 497. [Google Scholar] [CrossRef]
- Yan, N.; Sun, Y.; Fang, Y.; Deng, J.; Mu, L.; Xu, K.; Mymryk, J.S.; Zhang, Z. A Universal Surrogate Reporter for Efficient Enrichment of CRISPR/Cas9-Mediated Homology-Directed Repair in Mammalian Cells. Mol. Ther. Nucleic Acids 2019, 19, 775–789. [Google Scholar] [CrossRef]
- Liao, S.; Tammaro, M.; Yan, H. Enriching CRISPR-Cas9 targeted cells by co-targeting the HPRT gene. Nucleic Acids Res. 2015, 43, e134. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell. Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; Goldberg, A.L. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzym. 2005, 398, 364–378. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuven, N.; Adler, J.; Myers, N.; Shaul, Y. CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing. Int. J. Mol. Sci. 2021, 22, 3741. https://doi.org/10.3390/ijms22073741
Reuven N, Adler J, Myers N, Shaul Y. CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing. International Journal of Molecular Sciences. 2021; 22(7):3741. https://doi.org/10.3390/ijms22073741
Chicago/Turabian StyleReuven, Nina, Julia Adler, Nadav Myers, and Yosef Shaul. 2021. "CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing" International Journal of Molecular Sciences 22, no. 7: 3741. https://doi.org/10.3390/ijms22073741
APA StyleReuven, N., Adler, J., Myers, N., & Shaul, Y. (2021). CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing. International Journal of Molecular Sciences, 22(7), 3741. https://doi.org/10.3390/ijms22073741