1. Introduction
Sjögren’s disease (SD) is a chronic autoimmune disease that targets the lacrimal and salivary glands. Early pathogenesis is characterized by the infiltration of lymphocytes in these tissues leading to foci of inflammation and glandular destruction. In later stages of the disease, the autoimmune response leads to reduced production of tears and saliva resulting in progressive dry eyes and mouth and associated complications including poor oral health and vision-threatening ocular surface damage. Extra-glandular manifestations may occur targeting nearly any organ, and individuals with SD often experience significant pain and fatigue. Individuals with SD are also at risk for lymphoma, pregnancy complications such as neonatal lupus, and overall decreased quality of life. Treatments may provide some improvement in symptoms, but no treatments have demonstrated consistent reversal of clinical manifestations.
Type I interferons (IFNs) are cytokines that have broad effects on the immune system including stimulation and modulation of both innate and adaptive immune cells to drive inflammatory responses. While the IFN system is believed to have developed as a key anti-viral mechanism, the role of IFNs in multiple autoimmune diseases, including SD, has been demonstrated [
1,
2,
3,
4]. IFNs drive both innate and adaptive immune cell activities including T and B cell proliferation, macrophage activation and NK cell function [
5]. In SD, IFN-stimulated genes are upregulated in the minor salivary glands, ocular epithelial cells, peripheral mononuclear cells, monocytes and B cells [
6,
7,
8,
9,
10,
11]. Both type I and type II IFNs have been implicated in the pathogenesis of SD. Gene expression studies of target tissue from humans with SD have demonstrated heterogeneity in the expression of the IFN signature. Labial minor salivary gland biopsy specimens from individuals with SD demonstrated an IFN signature in 58% of individuals, with further analyses identifying three IFN-related patterns nearly evenly distributed: patients with type I IFN dominant signature, those with type II IFN dominant signature, and those with a combined type I and type II IFN signature [
12,
13,
14]. Whether these subsets represent multiple different pathways driving the early immune attack on salivary and lacrimal glands in the context of SD or, rather, different temporal stages of the inflammatory response is not known.
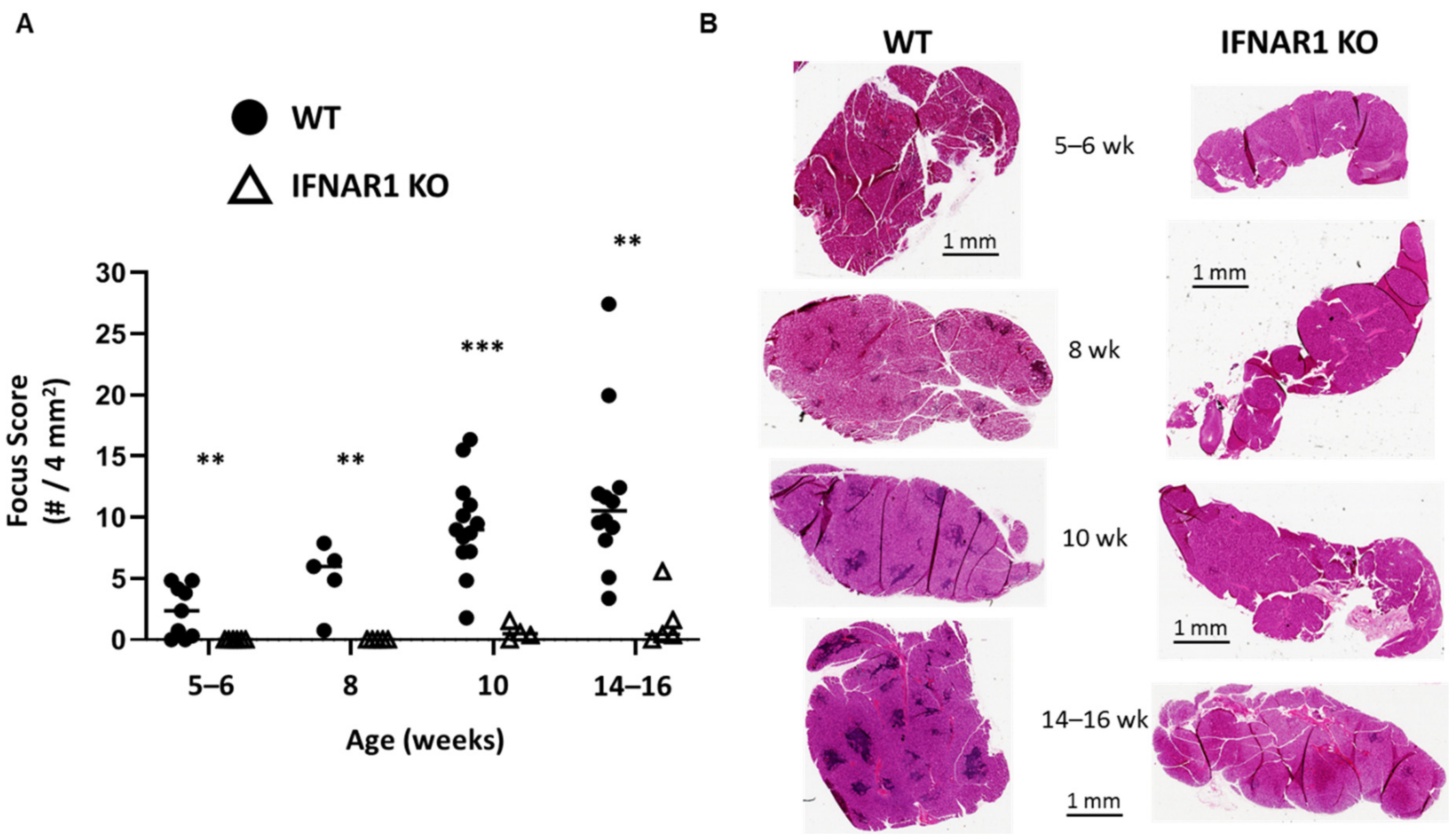
Nonobese diabetic (NOD) mice develop spontaneous SD-like autoimmunity including similar immunohistopathology of lacrimal and salivary glands as observed in humans [
15,
16]. However, NOD mice develop lacrimal and salivary gland disease independently in a sex-dependent manner with males spontaneously developing lacrimal gland inflammation and females spontaneously developing salivary gland inflammation [
17,
18]. Prior studies have demonstrated a pathogenic role for IFN signaling in the development of SD-like manifestations in NOD mice with clear distinctions in IFN requirements based on sex and gland affected. Lacrimal gland disease (spontaneous in males) required type I IFN signaling [
19], while salivary gland disease (spontaneous in females) required type II IFN signaling [
20]. Thus, spontaneous, sex-based, organ-specific, SD-like autoimmunity in NOD mice models two of the IFN-dependencies observed in humans with lacrimal gland disease representing the type I IFN-dominant disease and salivary gland disease representing the type II IFN-dominant disease [
19,
20]. This provides a unique tool to define immunopathogenic mechanisms of disease development that may then be translated to human SD.
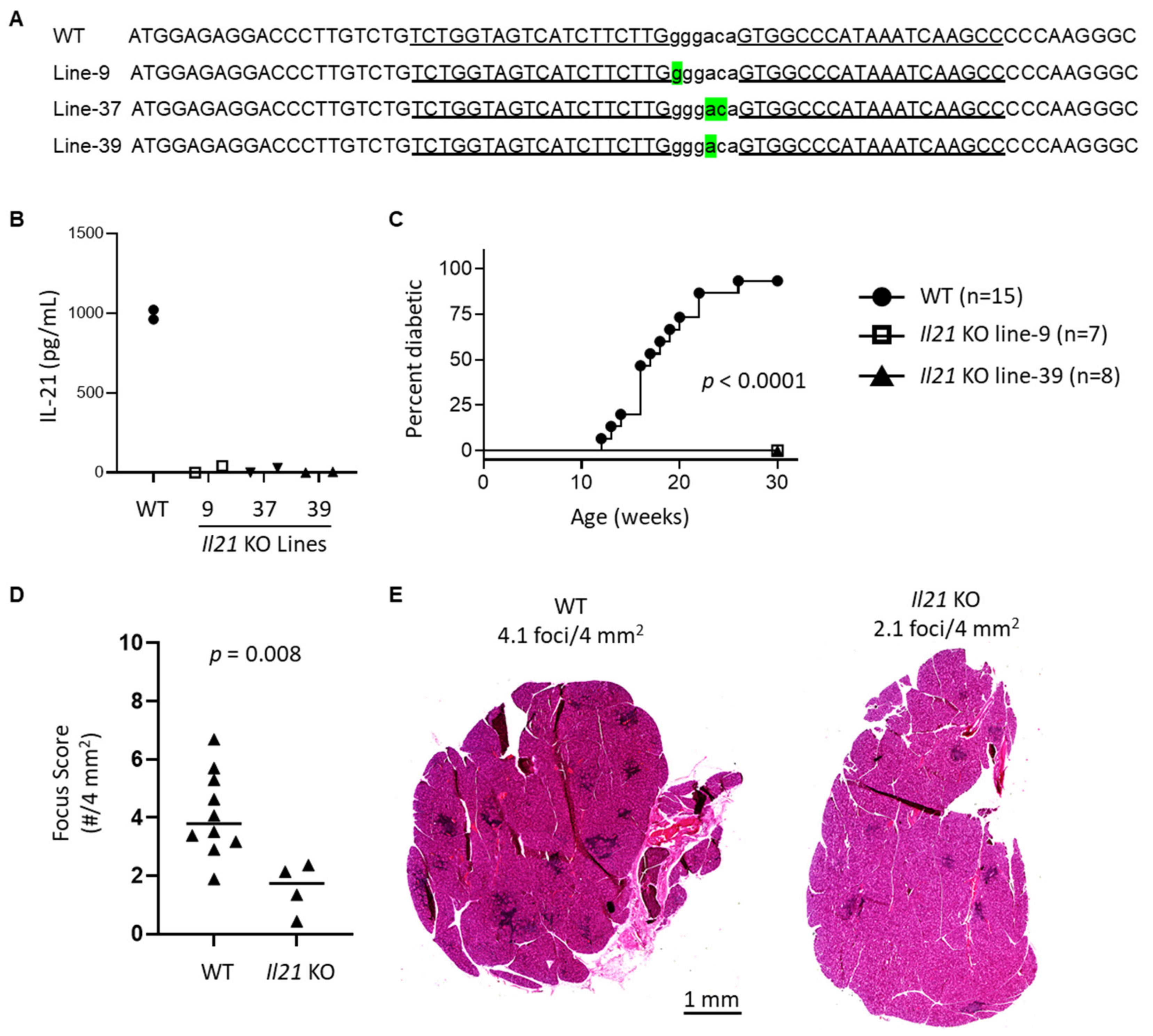
In this study, we have used type I IFN signaling-deficient NOD mice to study the role of type I IFN signaling in lacrimal gland autoimmunity. Through lacrimal gland tissue RNA sequencing studies, we identified type I IFN-dependent immune pathways associated with lacrimal gland inflammation largely dominated by cytokines and chemokines. The top cytokine upregulated in a type I IFN-dependent manner was interleukin (IL)-21. New Il21 knockout (KO) NOD mice were created and demonstrated significant protection in the development of lacrimal gland inflammation. Additional gene expression and flow cytometry studies implicate multiple chemokines including ligands for CXCR3 and enrichment of CXCR3-expressing T cells within inflamed lacrimal glands. Ultimately, defining the role of type I IFN signaling in the NOD mouse model of SD will provide insight into the early type I IFN-dependent pathogenic mechanisms in the development of lacrimal gland autoimmunity.
3. Discussion
Type I IFNs have repeatedly been implicated in the pathogenesis of systemic autoimmune diseases such as SD [
1,
2,
3,
4]. However, the downstream consequences of such IFN signaling have not been clearly defined for each disease. We had previously identified a requisite role for type I IFN in lacrimal gland inflammation in NOD mice [
19], and a pathogenic role for type I IFN in lacrimal and salivary gland inflammation was reported in another mouse model of SD [
26]. Here, we identified potentially disease-relevant genes upregulated in lacrimal glands of NOD mice in a type I IFN-dependent manner to further define the downstream consequences of type I IFN signaling in the context of spontaneous lacrimal gland inflammation in the NOD mouse model of SD.
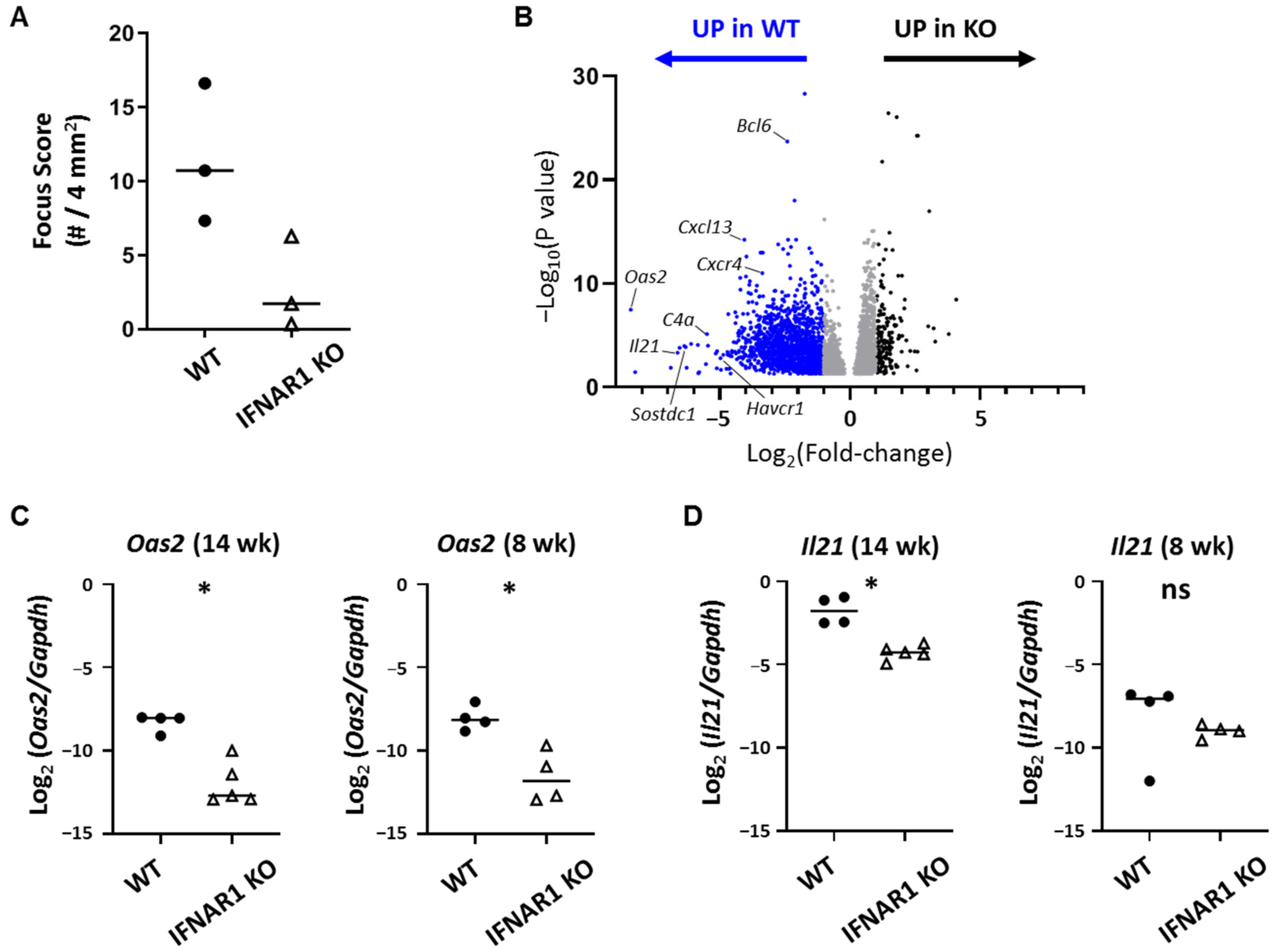
The most highly DE gene overexpressed in WT lacrimal glands in a type I IFN-dependent manner was
Oas2, which is a member of the oligoadenylate synthase (OAS) family of enzymes that work in conjunction with RNase L to degrade viral RNA in infected cells [
27]. This process results in cell death as a means to prevent viral survival and spread. While the association of OAS family members with autoimmunity is not new [
27,
28,
29,
30,
31], the specific mechanisms by which OAS family members may contribute to autoimmunity is not well-described. In an individual with defects in clearing apoptotic cells, an anti-viral response via OAS family members could conceivably provide increased antigen or ligand for innate immune receptors such as Toll-like receptor 7 (TLR7) or other pattern recognition receptors. Alternatively, perhaps some dysregulation of the OAS family of proteins may lead to aberrant responses targeting self-RNA in the absence of an infectious trigger. Regardless, the associations of OAS members with autoimmunity encompasses multiple autoimmune diseases. OAS members were upregulated in muscle biopsies from children with juvenile dermatomyositis [
28].
OAS2 was among a limited set of genes upregulated in T cells of individuals with systemic lupus erythematosus (SLE), and this limited set of genes was able to reliably differentiate SLE from healthy controls and was associated with disease activity [
32]. In SD, among genes with differential methylation patterns in labial minor salivary gland biopsy specimens,
OAS2 was found to have the strongest association with disease [
33]. Genetic variant of another OAS family member,
OAS1, was associated with SD and the variant resulted in isoforms purported to dysregulate type I IFN signaling [
34]. The NOD mouse model of spontaneous lacrimal gland autoimmunity may help to elucidate the role of the OAS family in autoimmunity to determine whether their upregulation represents a pathogenic role versus a consequence of a dysregulated system that drives the aberrant immune response.
Among the most highly DE genes and the highest DE cytokine gene was
Il21, which encodes IL-21, a pleiotropic cytokine with varied roles in innate and adaptive immunity and implicated in autoimmunity [
22,
35]. Specifically in SD, IL-21 was elevated in both serum [
36,
37] and labial minor salivary gland biopsy specimens of individuals with SD compared to controls [
37] and in saliva of children with SD compared to controls [
38]. A role for IL-21 in ocular inflammation in SD was demonstrated by elevation of IL-21 in tears and increased expression of
IL21 in conjunctival epithelial cells of individuals with SD compared to controls [
39]. Moreover, the elevated tear IL-21 correlated with measures of increased ocular surface inflammation or tear dysfunction suggesting a pathogenic role.
Il21 expression was also elevated in cornea and conjunctiva in the CD25 KO mouse model of SD [
40]. In NOD mice,
Il21 was detected in salivary glands and suppression with shRNA led to some improvement in salivary gland inflammation and function [
41]. In lacrimal glands of WT NOD mice, IL-21-producing CD4
+ T cells have been identified through single cell studies of gland-infiltrating T cells [
42]. Phenotypic analyses suggested these IL-21-producing CD4
+ T cells expressed some T follicular helper (Tfh) cell markers such as ICOS and PD1 but they did not express
Cxcr5 or
Bcl6. Notably, they also expressed
Sostdc1, which we found upregulated nearly 80-fold in WT lacrimal glands in a type I IFN-dependent manner. Whether the
Il21 upregulation in our WT NOD mouse lacrimal glands represents these Tfh-like cells, true Tfh cells, or another IL-21-producing population remains to be determined. Of note, though,
Cxcr5,
Bcl6,
Icos and
Pdcd1 were upregulated in our WT lacrimal glands (5–23-fold) (
Supplemental Table S1), and pathogenic contributions of Tfh cells in SD have been implicated in humans and animal models [
43]. B cell hyperactivity is a hallmark in many SD patients, and the role of Tfh cells in driving B cell immune responses makes Tfh–B cell interactions a likely relevant immune axis in SD pathogenesis. However, the role of B cells in early SD is less clear given that B cell-deficient NOD mice are not completely protected from development of exocrine gland inflammation [
44]. Early gland infiltrates are dominated by T cells, but B cells increase within the glands over time [
42,
45] suggesting the role of B cells may be greater later in disease. This may be especially relevant for the subset of SD patients who develop ectopic germinal centers (GC) within salivary glands providing a niche for chronic autoreactive B cell stimulation that increases risk for development of lymphoma. Recent transcriptomic analyses of salivary gland biopsy specimens from SD patients identified Tfh gene signature and increased
IL21 in specimens with ectopic GC and increased IL-21-producing Tfh in those with mucosa-associated lymphoid tissue lymphomas [
46]. Our findings that
Il21 KO NOD mice are protected from WT-levels of lacrimal gland inflammation suggest a potential earlier role for IL-21 in development of lacrimal gland inflammation, but we did not detect significant upregulation of
Il21 in lacrimal glands at the earlier time point suggesting that IL-21 was not the only downstream signal mediating early disease development in an IFNAR1-dependent manner. This is not surprising given the many genes upregulated in lacrimal glands in an IFNAR1-dependent manner. Given the multiple cellular sources of IL-21, additional studies are needed to more directly assess the contributions of Tfh and other immune cell subsets in the production of IL-21 within lacrimal glands of NOD mice.
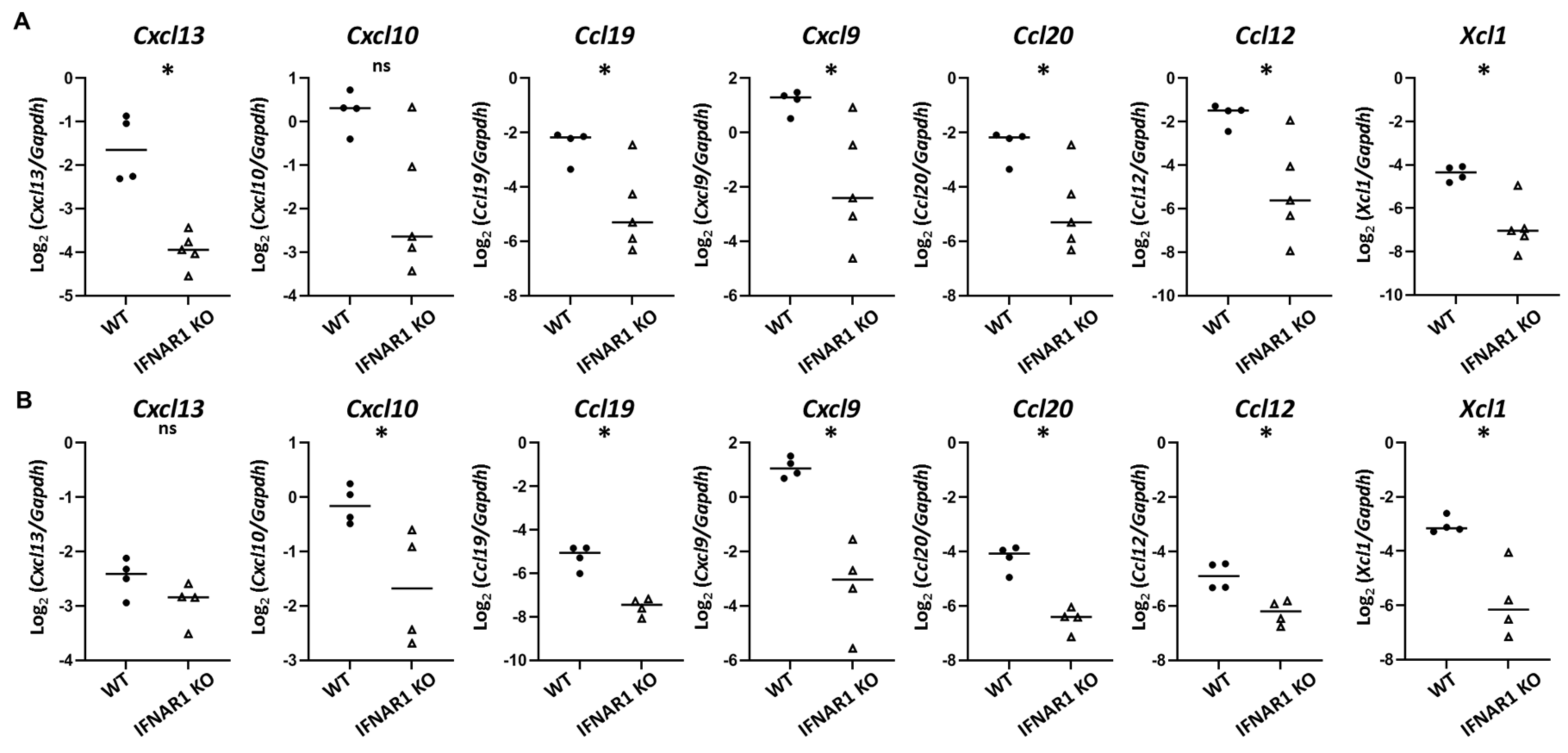
Immune cell trafficking is directed largely by chemokines. Among chemokines upregulated in WT NOD mouse lacrimal glands in a type I IFN-dependent manner, the most highly DE was
Cxcl13. CXCL13 plays a role in directing Tfh cells into B cell follicles in the process of GC formation, which is consistent with the likely role for Tfh in lacrimal gland inflammation in NOD mice. CXCL13 has been implicated in SD in numerous human and animal studies [
47]. However, the role of CXCL13 may be more significant in later propagation of the inflammatory process as
Cxcl13 was upregulated at the later, but not earlier, time point by qPCR in our studies. Additional chemokines upregulated in a type I IFN-dependent manner included multiple chemokines that were upregulated even at the earlier eight-week time point. These include
Cxcl9 and
Ccl19, which we had previously identified as upregulated in lacrimal glands of older NOD mice in a type I IFN-dependent manner [
19], and other cytokines including
Cxcl10,
Ccl20,
Ccl12, and
Xcl1. XCL1 (lymphotactin) is produced by T cells (especially activated CD8
+ T cells and Th1 CD4
+ T cells), NK cells, and NKT cells to recruit XCR1-expressing cells with recent evidence suggesting a role in promoting cross-presenting dendritic cells to stimulate cytotoxic CD8
+ T cells [
48,
49,
50].
Xcl1 expression in NOD mouse lacrimal glands was previously reported [
51]. CCL12 (monocyte chemotactic protein 5, MCP-5) is produced by innate immune cells and binds CCR2 to recruit innate and adaptive immune cells. CCL12 has been implicated in animal models of SD.
Ccl12 was upregulated in salivary glands in NZB/W mice when salivary gland inflammation was accelerated by TLR3 agonist-treatment [
52]. In lacrimal glands,
Ccl12 was detected, albeit at low levels, in the MRL model [
53]. CCL20 (macrophage inflammatory protein 3α, MIP-3α) is produced by several innate and adaptive immune cells and binds to CCR6, which is largely expressed on lymphocytes. CCL20 produced by keratinocytes recruits Th17 cells contributing to inflammation in psoriasis [
54] but may contribute to an immunosuppressive environment through recruitment of immunomodulatory immune cells in the tumor microenvironment [
55]. Whether CCL20 plays an inflammatory or compensatory anti-inflammatory role in NOD lacrimal glands is not known. CCL19 (macrophage inflammatory protein 3, MIP-3) binds CCR7 and plays a key role in localization of lymphocytes to secondary lymphoid organs or to ectopic lymphoid structures such as GC within exocrine glands in SD, a role it may play along with CXCL13 [
47]. We and others have recently discussed the role of CCL19 in SD [
19,
47].
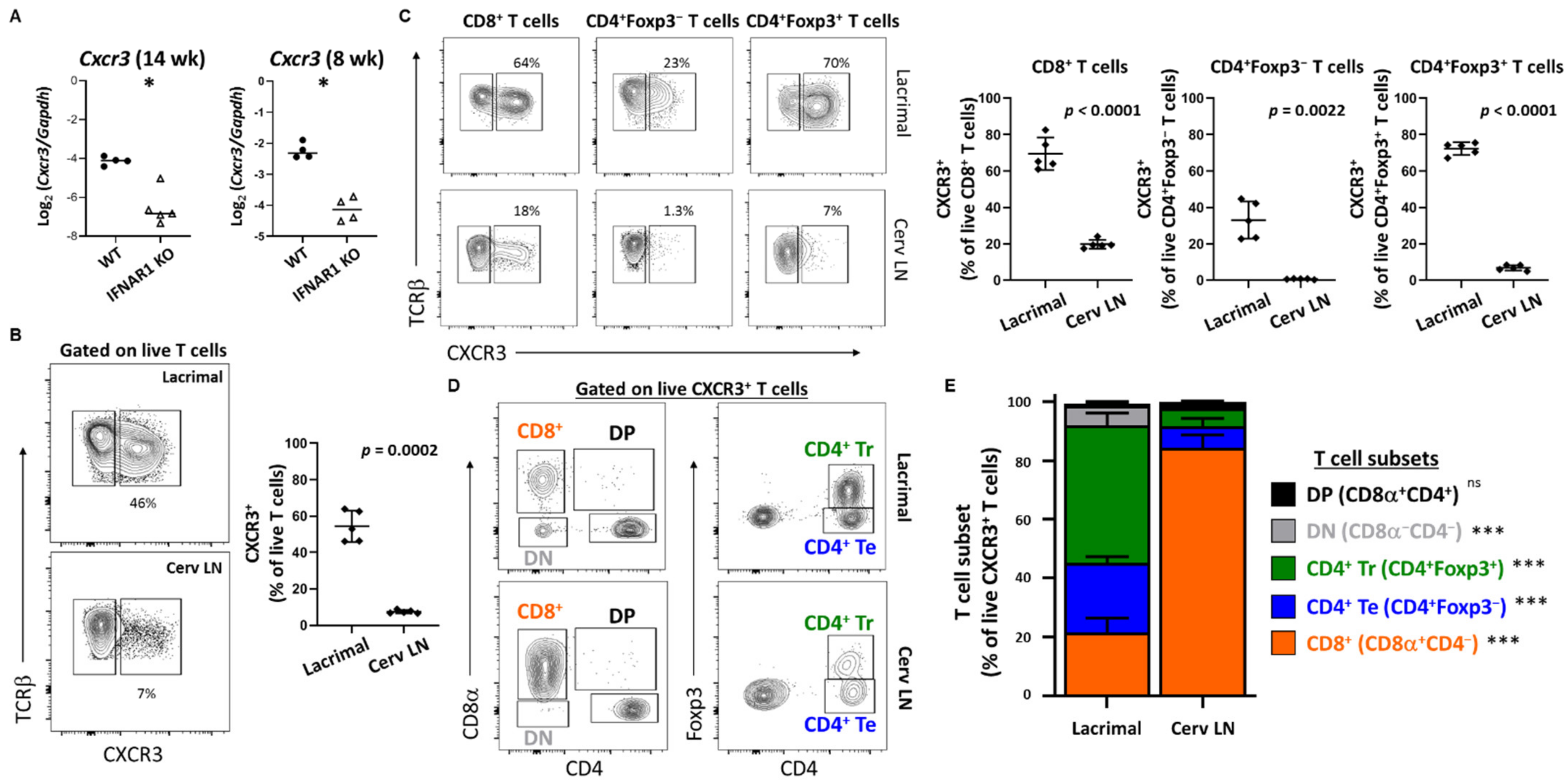
CXCL9 (monokine induced by gamma-IFN, MIG) and CXCL10 (IFN-γ induced protein 10, IP-10) each bind CXCR3 and have been implicated in SD in multiple human and animal studies [
47]. Here we have extended our previous findings of upregulation of
Cxcl9 in lacrimal glands of older mice in a type I IFN-dependent manner [
19] to demonstrate upregulation of
Cxcl9 and
Cxcl10 in a type I IFN-dependent manner at an early stage of lacrimal gland inflammation. Moreover, we detected CXCR3-expressing T cell populations enriched within lacrimal glands of WT NOD mice compared to the lacrimal gland-draining cervical lymph nodes. While all gland-infiltrating T cell subsets have larger CXCR3
+ populations compared to the same subsets within the organ-draining cervical lymph nodes, we found significantly different distributions in T cell subsets within the CXCR3
+ populations with CD8
+ T cells dominating the CXCR3
+ T cells in the lymph nodes but CD4
+Foxp3
+ regulatory T cells dominating the CXCR3
+ T cell population within lacrimal glands. CXCR3
+ regulatory T cells were previously found enriched within pancreatic islets in NOD mice and expressed
Tbx21 (encodes T-BET), and these T-BET
+CXCR3
+ regulatory T cells were shown to limit the development and progression of T1D [
56]. Specifically, while the proportion of pancreatic regulatory T cells was relatively unchanged in the absence of these CXCR3
+ regulatory T cells, their absence resulted in accelerated progression of T1D demonstrating their importance in modulating autoimmunity. Of note, T cells infiltrating lacrimal glands of NOD mice produce IFN-γ and TNF suggesting a strong type 1 immune response [
57]. T-BET drives expression of CXCR3 in regulatory T cells to optimize modulation of type 1 inflammation [
58,
59]. Together, these data suggest that the CXCR3
+ regulatory T cells enriched within lacrimal glands may limit the immunopathology during the lacrimal gland autoimmune response in NOD mice. In accordance with this, adoptive transfer of cervical lymph node cells depleted of CD4
+Foxp3
+ regulatory T cells resulted in extensive inflammation with diffuse rather than focal infiltrates in NOD-SCID recipient mice (our unpublished observations). However, in both T1D and SD-like manifestations in NOD mice, autoimmunity develops spontaneously suggesting that while CXCR3
+ regulatory T cells may limit immunopathology in affected organs, they are incapable of preventing disease development in the first place. Whether this regulatory T cell dysfunction is driven by type I IFN is under investigation.
In summary, type I IFN-signaling promoted upregulation of cytokines and chemokines within lacrimal glands leading to dysregulation of normal immune tolerance mechanism through complex phenotypic changes in innate and adaptive immune cells. Further studies are needed to more completely define the downstream consequences of type I IFN signaling and to specifically identify the lymphocytes and innate immune cells directly affected by the type I IFN signals. Moreover, the upstream signals driving the type I IFN response are also not fully elucidated, though our recent findings of a large common set of genes similarly upregulated in lacrimal glands in a TLR7-dependent manner suggest that TLR7 is one of those upstream type I IFN-inducing signals [
60]. Ultimately, understanding the specific molecular mechanisms of type I IFN in SD-like autoimmunity may provide targets for novel therapeutics or for diagnostic or prognostic testing in SD and, potentially, other type I IFN-associated systemic autoimmune diseases.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}