
Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CAA Characterization
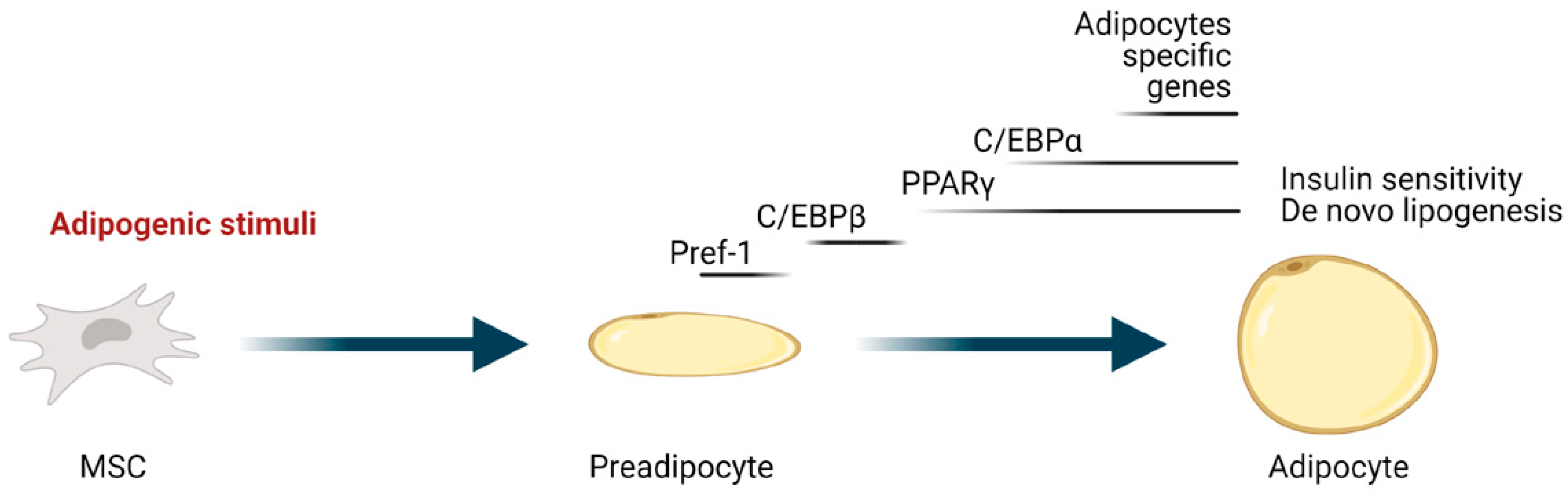
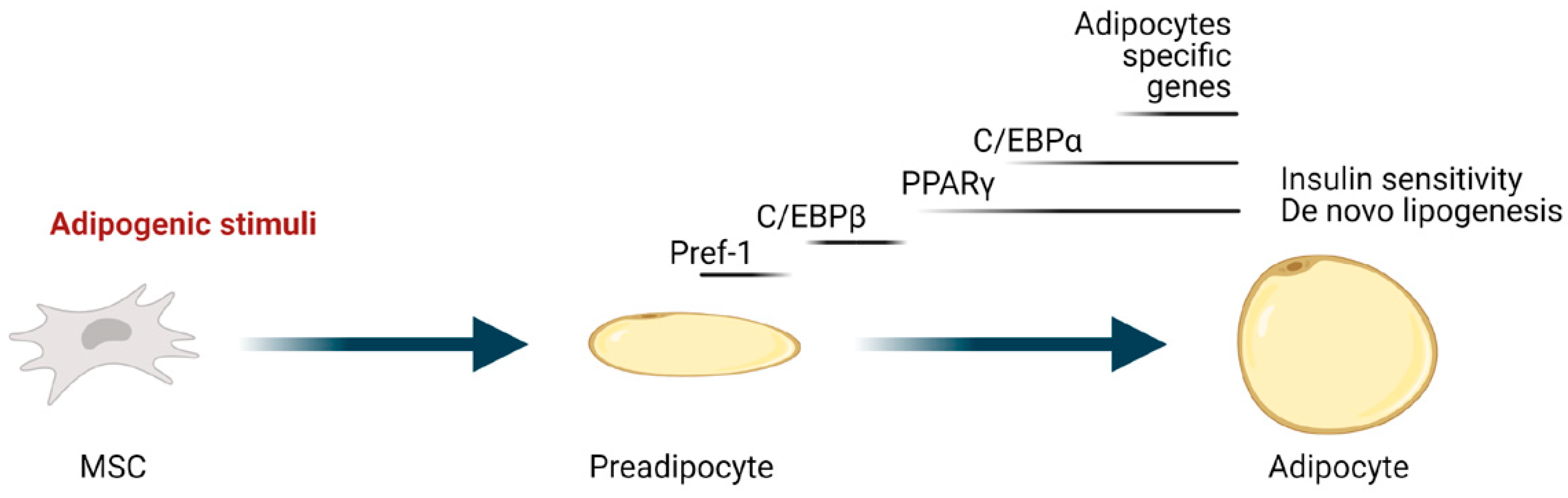
3. Regulation of Adipocyte Differentiation
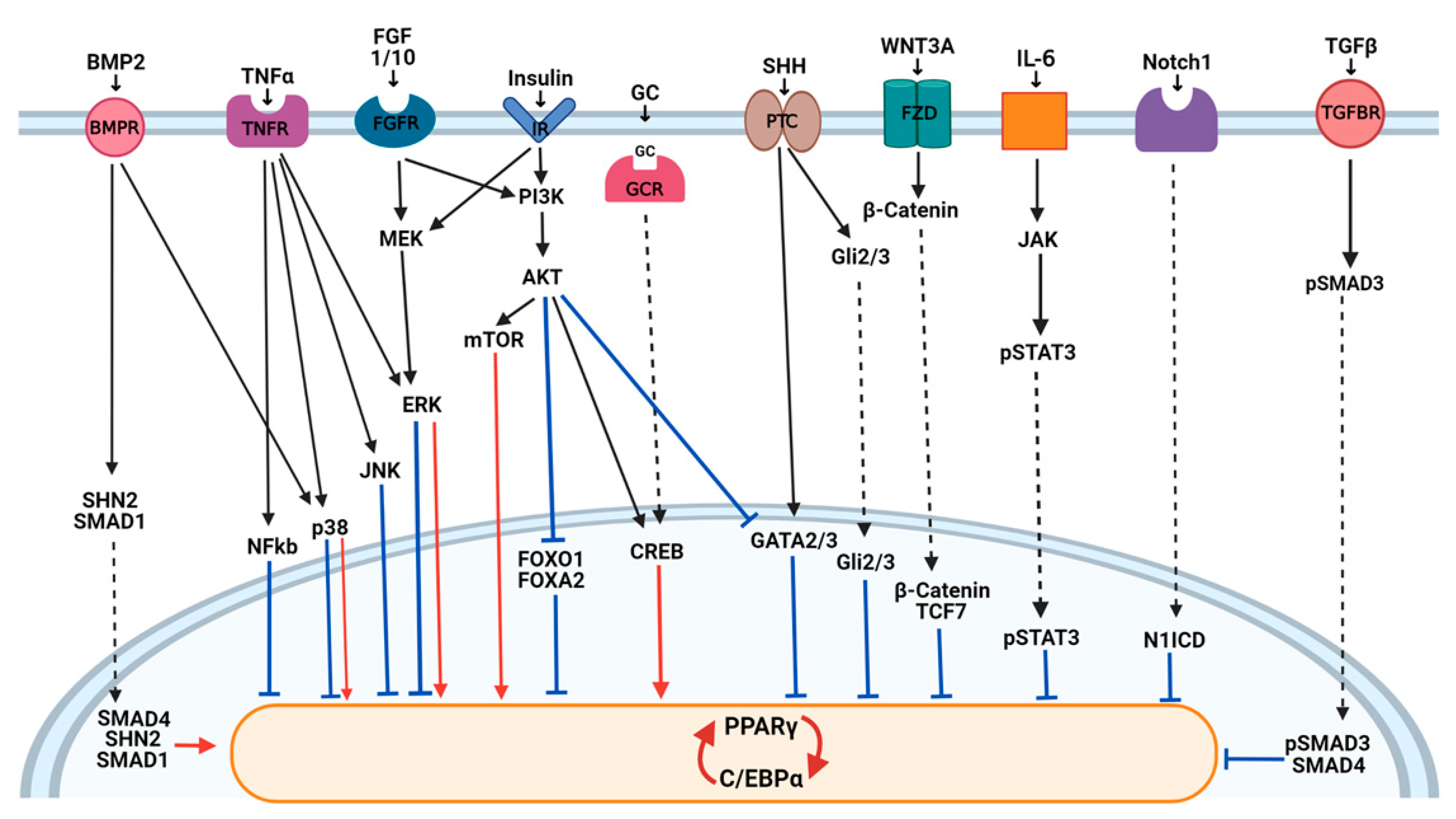
3.1. Wnt Signaling
3.2. Hedgehog Signaling
3.3. Notch Signaling
3.4. TGFβ and BMP Signaling
3.5. Insulin Signaling
3.6. The MAPK Pathway
3.7. FGFs
3.8. Inflammatory Molecules
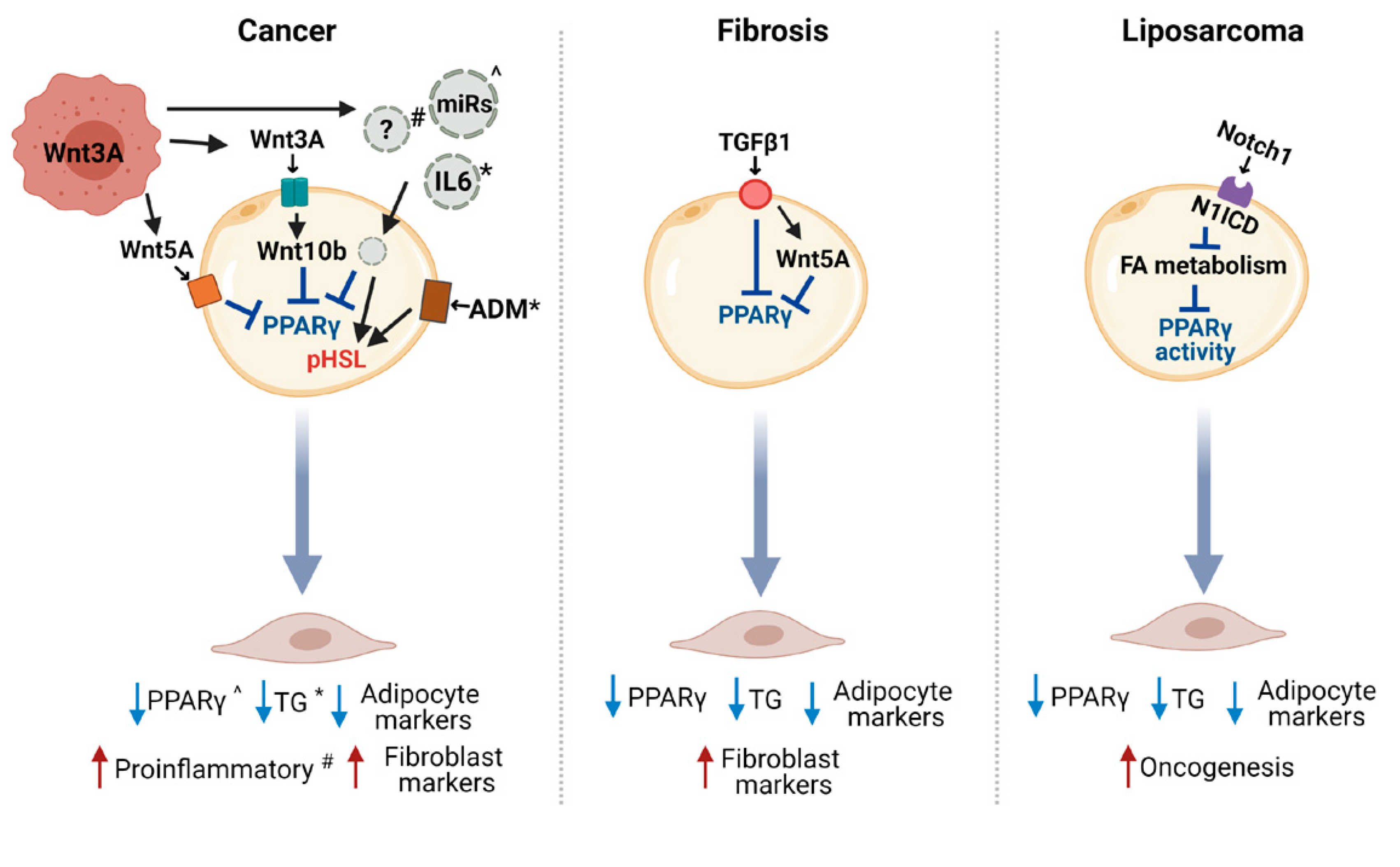
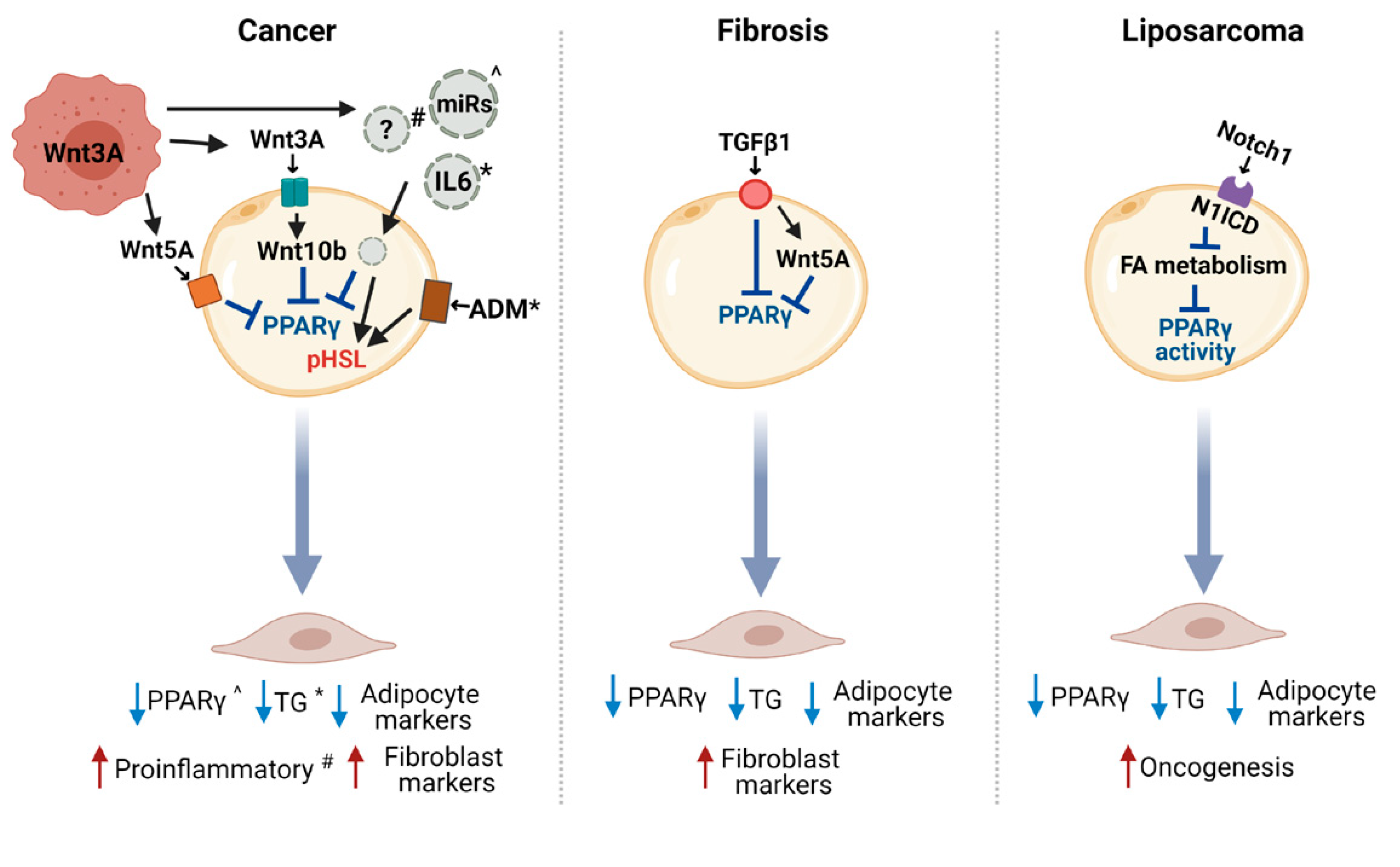
4. Regulation of CAA Induction
4.1. The Wnt Pathway in the Crosstalk between CAA and Cancer Cells
4.2. Other Potential Modulators of Adipocyte Reprogramming in Cancer
4.3. Other Molecular Pathways Involved in Adipocyte Dedifferentiation
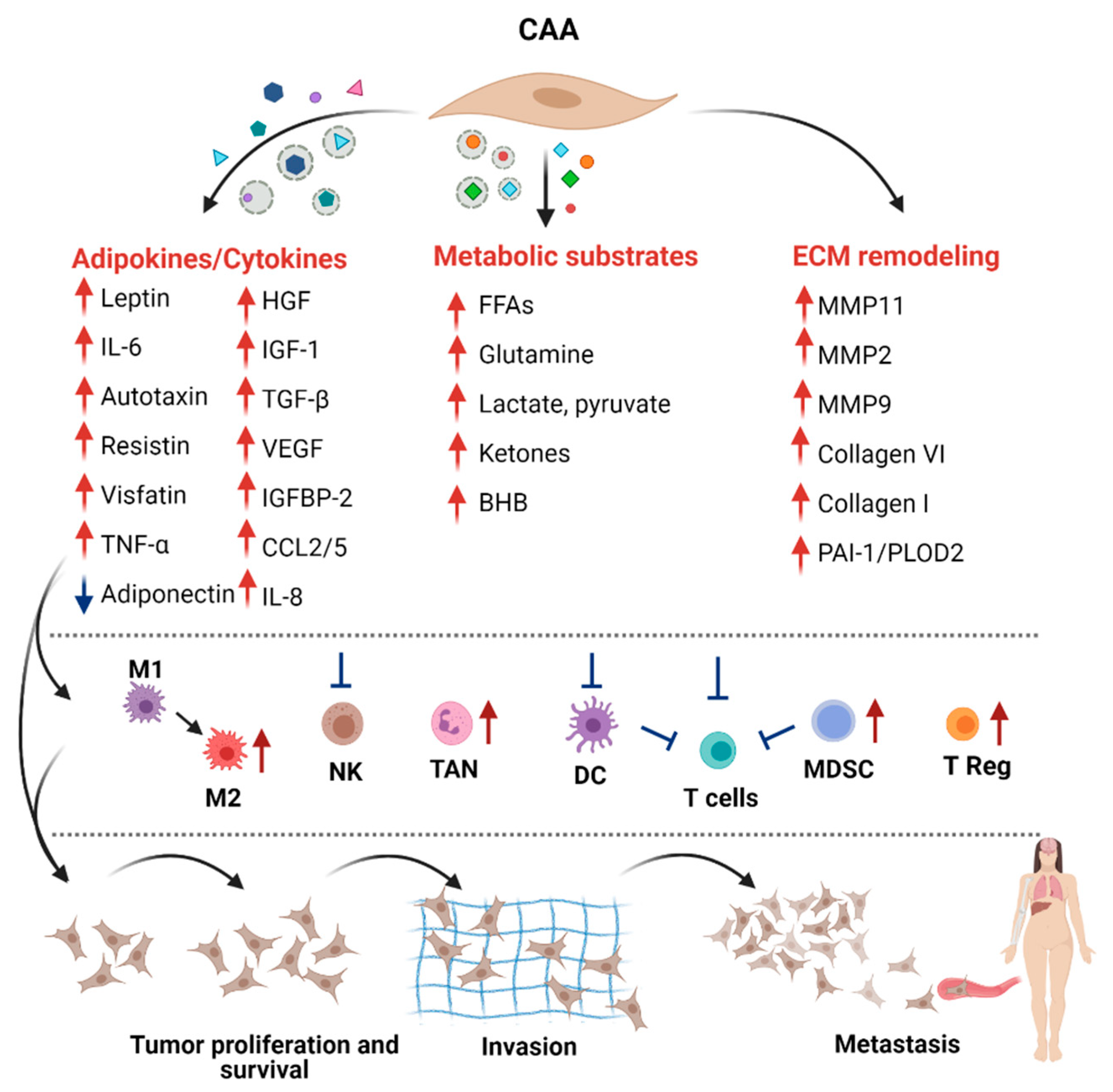
5. CAA-Derived Molecules in Cancer Progression
5.1. Adipokines
5.2. Metabolic Reprogramming
5.3. ECM Remodeling
5.4. Immune Cell Modulation by CAA-Released Molecules
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Deng, C.X. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Song, T.; Kuang, S. Adipocyte dedifferentiation in health and diseases. Clin. Sci. 2019, 133, 2107–2119. [Google Scholar] [CrossRef]
- Corsa, C.A.S.; MacDougald, O.A. Cyclical dedifferentiation and redifferentiation of mammary adipocytes. Cell Metab. 2018, 28, 187–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, S.; Gupta, A.; Beg, M.; Shankar, K.; Srivastava, A.; Varshney, S.; Kumar, D.; Gaikwad, A.N. Adipocyte transdifferentiation and its molecular targets. Differentiation 2014, 87, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid. Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat 2010, 123, 627–635. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in breast cancer, the thick and the thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef] [Green Version]
- Suàrez-Nàjera, L.E.; Chanona-Pérez, J.J.; Valdivia-Flores, A.; Marrero-Rodrìguez, D.; Salcedo-Vargas, M.; Garcìa-Ruiz, D.I.; Castro-Reyes, M.A. Morphometric study of adipocytes on breast cancer by means of photonic microscopy and image analysis. Microsc. Res. Tech. 2018, 81, 240–249. [Google Scholar] [CrossRef]
- Wang, F.; Gao, S.; Chen, F.; Fu, Z.; Yin, H.; Lu, X.; Yu, J.; Lu, C. Mammary fat of breast cancer: Gene expression profiling and functional characterization. PLoS ONE 2014, 9, e109742. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attané, C.; Muller, C. Drilling for Oil: Tumor-surrounding adipocytes fueling cancer. Trends Cancer 2020, 6, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouyafar, A.; Heydarabad, M.Z.; Abdolalizadeh, J.; Zade, J.A.; Rahbarghazi, R.; Talebi, M. Modulation of lipolysis and glycolysis pathways in cancer stem cells changed multipotentiality and differentiation capacity toward endothelial lineage. Cell Biosci. 2019, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, J.F.; Brisson, L. Interaction between adipose tissue and cancer cells: Role for cancer progression. Cancer Metastasis Rev. 2021, 40, 31–46. [Google Scholar] [CrossRef]
- Tan, J.; Buache, E.; Chenard, M.P.; Dali-Youcef, N.; Rio, M.C. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int. J. Dev. Biol. 2011, 55, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Andarawewa, K.L.; Motrescu, E.R.; Chenard, M.P.; Gansmuller, A.; Stoll, I.; Tomasetto, C.; Rio, M.C. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res. 2005, 65, 10862–10871. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.A.; Song, A.; Chen, W.; Schwalie, P.C.; Zhang, F.; Vishvanath, L.; Jiang, L.; Ye, R.; Shao, M.; Tao, C.; et al. Reversible de-differentiation of mature white adipocytes into preadipocyte-like precursors during lactation. Cell Metab. 2018, 28, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef]
- Bi, P.; Yue, F.; Karki, A.; Castro, B.; Wirbisky, S.E.; Wang, C.; Durkes, A.; Elzey, B.D.; Andrisani, O.M.; Bidwell, C.A.; et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 2016, 213, 2019–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Lowe, C.E.; O’Rahilly, S.; Rochford, J.J. Adipogenesis at a glance. J. Cell Sci. 2011, 124, 2681–2686. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.S.; Siersbaek, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome proliferator-activated receptor g and C/EBPa synergistically activate key metabolic adipocyte genes by assisted loading. Mol. Cell Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Schmidt, H.; Lai, B.; Ge, K. Transcriptional and epigenomic regulation of adipogenesis. Mol. Cell Biol. 2019, 39, e00601-18. [Google Scholar] [CrossRef] [Green Version]
- Gharanei, S.; Shabir, K.; Brown, J.E.; Weickert, M.O.; Barber, T.M.; Kyrou, I.; Randeva, H.S. Regulatory microRNAs in brown, brite and white adipose tissue. Cells 2020, 9, 2489. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.J.; Choi, H.; Ko, E.H.; Kim, J.W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009, 284, 10601–10609. [Google Scholar] [CrossRef] [Green Version]
- de Winter, T.J.J.; Nusse, R. Running against the wnt: How wnt/b-catenin suppresses adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.N.; Ross, S.E.; Longo, K.A.; Bajnok, L.; Hemati, N.; Johnson, K.W.; Harrison, S.D.; MacDougald, O.A. Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 2002, 277, 30998–31004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizuka, M.; Koyanagi, A.; Osada, S.; Imagawa, M. Wnt4 and Wnt5a promote adipocyte differentiation. FEBS Lett. 2008, 582, 3201–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J. Cell Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef]
- Kanazawa, A.; Tsukada, S.; Kamiyama, M.; Yanagimoto, T.; Nakajima, M.; Maeda, S. Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2005, 330, 505–510. [Google Scholar] [CrossRef]
- Cousin, W.; Fontaine, C.; Dani, C.; Peraldi, P. Hedgehog and adipogenesis: Fat and fiction. Biochimie 2007, 89, 1447–1453. [Google Scholar] [CrossRef]
- Suh, J.M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J.M. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006, 3, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Dalgin, G.; Xu, H.; Ting, C.N.; Leiden, J.M.; Hotamisligil, G.S. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 2000, 290, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Ulloa, F.; Itasaki, N.; Briscoe, J. Inhibitory Gli3 activity negatively regulates Wnt/beta-catenin signaling. Curr. Biol. 2007, 20, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Radtke, F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett. 2006, 580, 2860–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, T.; Liu, J.; Wu, W.; Xu, Z.; Wang, Y. Roles of notch signaling in adipocyte progenitor cells and mature adipocytes. J. Cell Physiol. 2017, 232, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- da Silva, C.; Durandt, C.; Kallmeyer, K.; Ambele, M.A.; Pepper, M.S. The Role of Pref-1 during Adipogenic Differentiation: An overview of suggested mechanisms. Int. J. Mol. Sci. 2020, 21, 4104. [Google Scholar] [CrossRef]
- Ross, D.A.; Rao, P.K.; Kadesch, T. Dual roles for the Notch target gene Hes-1 in the differentiation of 3T3-L1 preadipocytes. Mol. Cell Biol. 2004, 24, 3505–3513. [Google Scholar] [CrossRef] [Green Version]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-b receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1160–1171. [Google Scholar] [CrossRef]
- Choy, L.; Derynck, R. Transforming growth factor-b inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 2003, 278, 9609–9619. [Google Scholar] [CrossRef] [Green Version]
- Hata, K.; Nishimura, R.; Ikeda, F.; Yamashita, K.; Matsubara, T.; Nokubi, T.; Yoneda, T. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell 2003, 14, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Takagi, T.; Kanesashi, S.N.; Kurahashi, T.; Nomura, T.; Harada, J.; Ishii, S. Schnurri-2 controls BMP-dependent adipogenesis via interaction with Smad proteins. Dev. Cell 2006, 10, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Choy, L.; Skillington, J.; Derynck, R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 2000, 149, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Li, S.N.; Wu, J.F. TGF-b/SMAD signaling regulation of mesenchymal stem cells in adipocyte commitment. Stem Cell Res. Ther. 2020, 11, 41–1552. [Google Scholar] [CrossRef] [PubMed]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and Insulin receptors in adipose tissue development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, H.; Yamauchi, T.; Suzuki, R.; Komeda, K.; Tsuchida, A.; Kubota, N.; Terauchi, Y.; Kamon, J.; Kaburagi, Y.; Matsui, J.; et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell Biol. 2001, 21, 2521–2532. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Dong, L.Q.; Liu, F. Recent Advances in adipose mTOR Signaling and function: Therapeutic prospects. Trends Pharmacol. Sci. 2016, 37, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Menghini, R.; Marchetti, V.; Cardellini, M.; Hribal, M.L.; Mauriello, A.; Lauro, D.; Sbraccia, P.; Lauro, R.; Federici, M. Phosphorylation of GATA2 by Akt increases adipose tissue differentiation and reduces adipose tissue-related inflammation: A novel pathway linking obesity to atherosclerosis. Circulation 2005, 111, 1946–1953. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Bost, F.; Aouadi, M.; Caron, L.; Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [Green Version]
- Leiva, M.; Matesanz, N.; Pulgarìn-Alfaro, M.; Nikolic, I.; Sabio, G. Uncovering the Role of p38 Family Members in Adipose Tissue Physiology. Front. Endocrinol. (Lausanne) 2020, 11, 572089. [Google Scholar] [CrossRef]
- Rabiee, A.; Schwämmle, V.; Sidoli, S.; Dai, J.; Rogowska-Wrzesinska, A.; Mandrup, S.; Jensen, O.N. Nuclear phosphoproteome analysis of 3T3-L1 preadipocyte differentiation reveals system-wide phosphorylation of transcriptional regulators. Proteomics 2017, 17, 1600248. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Cao, S.; Arhatte, M.; Li, D.; Shi, Y.; Kurz, S.; Hu, J.; Wang, L.; Shao, J.; Atzberger, A.; et al. Adipocyte Piezo1 mediates obesogenic adipogenesis through the FGF1/FGFR1 signaling pathway in mice. Nat. Commun. 2020, 11, 2303. [Google Scholar] [CrossRef]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and inflammation in adipogenesis: An updated review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef]
- Xu, H.; Sethi, J.K.; Hotamisligil, G.S. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J. Biol. Chem. 1999, 274, 26287–26295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, G.N.; Kwak, S.J. NF-kappaB is involved in the TNF-alpha induced inhibition of the differentiation of 3T3-L1 cells by reducing PPARgamma expression. Exp. Mol. Med. 2003, 35, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawthorn, W.P.; Heyd, F.; Hegyi, K.; Sethi, J.K. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007, 14, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zou, T.; Zuo, C.; Zhang, M.; Shi, B.; Jiang, Z.; Cui, H.; Liao, X.; Li, X.; Tang, Y.; et al. IL-1a inhibits proliferation and adipogenic differentiation of human adipose-derived mesenchymal stem cells through NF-kB- and ERK1/2-mediated proinflammatory cytokines. Cell Biol. Int. 2018, 42, 794–803. [Google Scholar] [CrossRef]
- Gagnon, A.; Foster, C.; Landry, A.; Sorisky, A. The role of interleukin 1β in the anti-adipogenic action of macrophages on human preadipocytes. J. Endocrinol. 2013, 217, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Almuraikhy, S.; Kafienah, W.; Bashah, M.; Diboun, I.; Jaganjac, M.; Al-Khelaifi, F.; Abdesselem, H.; Mazloum, N.A.; Alsayrafi, M.; Mohamed-Ali, V.; et al. Interleukin-6 induces impairment in human subcutaneous adipogenesis in obesity-associated insulin resistance. Diabetologia 2016, 59, 2406–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Wang, Y.; Zhou, G.; Wang, Y.; Li, X. Review: The Roles and mechanisms of glycoprotein 130 cytokines in the regulation of adipocyte biological function. Inflammation 2019, 42, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Fuster, G.; Ametller, E.; Costelli, P.; Pilla, F.; Busquets, S.; Figueras, M.; Argiles, J.M.; Lopez-Soriano, F.J. Interleukin-15 increases calcineurin expression in 3T3-L1 cells: Possible involvement on in vivo adipocyte differentiation. Int. J. Mol. Med. 2009, 24, 453–458. [Google Scholar] [PubMed] [Green Version]
- Tsao, C.H.; Shiau, M.Y.; Chuang, P.H.; Chang, Y.H.; Hwang, J. Interleukin-4 regulates lipid metabolism by inhibiting adipogenesis and promoting lipolysis. J. Lipid Res. 2014, 55, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Gaffen, S.L. IL-17 inhibits adipogenesis in part via C/EBPa, PPARg and Kruppel-like factors. Cytokine 2013, 61, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Um, S.H.; Rhee, D.K.; Pyo, S. Interferon-alpha inhibits adipogenesis via regulation of JAK/STAT1 signaling. Biochim. Biophys. Acta 2016, 1860, 2416–2427. [Google Scholar] [CrossRef]
- Todoric, J.; Strobl, B.; Jais, A.; Boucheron, N.; Bayer, M.; Amann, S.; Lindroos, J.; Teperino, R.; Prager, G.; Bilban, M.; et al. Cross-talk between interferon-g and hedgehog signaling regulates adipogenesis. Diabetes 2011, 60, 1668–1676. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Smith, U. Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases beta-catenin levels and leads to cell dedifferentiation and insulin resistance. J. Biol. Chem. 2010, 285, 14031–14041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef] [Green Version]
- Chirumbolo, S.; Bjørklund, G. Can Wnt5a and Wnt non-canonical pathways really mediate adipocyte de-differentiation in a tumour microenvironment? Eur. J. Cancer 2016, 64, 96–100. [Google Scholar] [CrossRef]
- Li, Y.; Mao, A.S.; Seo, B.R.; Zhao, X.; Gupta, S.K.; Chen, M.; Han, Y.L.; Shih, T.Y.; Mooney, D.J.; Guo, M. Compression-induced dedifferentiation of adipocytes promotes tumor progression. Sci. Adv. 2000, 6, eaax5611. [Google Scholar] [CrossRef] [Green Version]
- Fonar, Y.; Gutkovich, Y.E.; Root, H.; Malyarova, A.; Aamar, E.; Golubovskaya, V.M.; Elias, S.; Elkouby, Y.M.; Frank, D. Focal adhesion kinase protein regulates Wnt3a gene expression to control cell fate specification in the developing neural plate. Mol. Biol. Cell 2011, 22, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Giussani, M.; Merlino, G.; Cappelletti, V.; Tagliabue, E.; Daidone, M.G. Tumor-Extracellular matrix interactions: Identification of tools associated with breast cancer progression. Semin. Cancer Biol. 2015, 35, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Zuo, Y.; Farmer, S.R. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol. Cell Biol. 2006, 26, 5827–5837. [Google Scholar] [CrossRef] [Green Version]
- Schupp, M.; Cristancho, A.G.; Lefterova, M.I.; Hanniman, E.A.; Briggs, E.R.; Steger, D.J.; Qatanani, M.; Curtin, J.C.; Schug, J.; Ochsner, S.A.; et al. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. J. Biol. Chem. 2009, 284, 9458–9464. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xu, M.; Li, X.; Su, X.; Xiao, X.; Keating, A.; Zhao, R.C. Exosomes released by hepatocarcinoma cells endow adipocytes with tumor-promoting properties. J. Hematol. Oncol. 2018, 11, 82–0625. [Google Scholar] [CrossRef]
- Hu, W.; Ru, Z.; Zhou, Y.; Xiao, W.; Sun, R.; Zhang, S.; Gao, Y.; Li, X.; Zhang, X.; Yang, H. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1091–1102. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Wang, C.; et al. Breast cancer-released exosomes trigger cancer-associated cachexia to promote tumor progression. Adipocyte 2019, 8, 31–45. [Google Scholar] [PubMed]
- Parè, M.; Darini, C.Y.; Yao, X.; Chignon-Sicard, B.; Rekima, S.; Lachambre, S.; Virolle, V.; Guilar-Mahecha, A.; Basik, M.; Dani, C.; et al. Breast cancer mammospheres secrete Adrenomedullin to induce lipolysis and browning of adjacent adipocytes. BMC Cancer 2020, 20, 784. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Jha, N.K.; Jha, S.K.; Sharma, A.; Dholpuria, S.; Asthana, N.; Chaurasiya, K.; Singh, V.K.; Burgee, S.; Nand, P. A NOTCH deeper into the epithelial-to-mesenchymal transition (EMT) program in breast cancer. Genes 2019, 10, 961. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Arteaga, C.L. Transforming growth factor-beta and breast cancer: Tumor promoting effects of transforming growth factor-beta. Breast Cancer Res. 2000, 2, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Khoo, N.K.; Schoiswohl, G.; O’Doherty, R.M.; Kensler, T.W. Notch intracellular domain overexpression in adipocytes confers lipodystrophy in mice. Mol. Metab. 2015, 4, 543–550. [Google Scholar] [CrossRef]
- Wei, J.; Melichian, D.; Komura, K.; Hinchcliff, M.; Lam, A.P.; Lafyatis, R.; Gottardi, C.J.; MacDougald, O.A.; Varga, J. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: A novel mouse model for scleroderma? Arthritis Rheum. 2011, 63, 1707–1717. [Google Scholar] [CrossRef] [Green Version]
- Zeve, D.; Seo, J.; Suh, J.M.; Stenesen, D.; Tang, W.; Berglund, E.D.; Wan, Y.; Williams, L.J.; Lim, A.; Martinez, M.J.; et al. Wnt signaling activation in adipose progenitors promotes insulin-independent muscle glucose uptake. Cell Metab. 2012, 15, 492–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariman, E.C.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene 2008, 27, 6347–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. 2019, 75-76, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Pelletier, M.; Biertho, L.; Biron, S.; Marceau, S.; Hould, F.S.; Lebel, S.; Moustarah, F.; Lescelleur, O.; Marceau, P.; et al. Characterization of dedifferentiating human mature adipocytes from the visceral and subcutaneous fat compartments: Fibroblast-activation protein alpha and dipeptidyl peptidase 4 as major components of matrix remodeling. PLoS ONE 2015, 10, e0122065. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef] [PubMed]
- Sànchez-Jiménez, F.; Pérez-Pérez, A.; de la Cruz-Merino, L.; Sànchez-Margalet, V. Obesity and breast cancer: Role of leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Deng, L.L.; Cui, J.Q.; Shi, L.; Yang, Y.C.; Luo, J.H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine (Baltimore) 2018, 97, e11345. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; de Graffenried, L.A.; Hursting, S.D. Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Naimo, G.D.; Gelsomino, L.; Catalano, S.; Mauro, L.; Andò, S. Interfering Role of ERa on Adiponectin Action in Breast Cancer. Front. Endocrinol. (Lausanne) 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Højfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat 2013, 138, 657–664. [Google Scholar] [CrossRef]
- Brindley, D.N.; Tang, X.; Meng, G.; Benesch, M.G.K. Role of adipose tissue-derived autotaxin, lysophosphatidate signaling, and inflammation in the progression and treatment of breast cancer. Int. J. Mol. Sci. 2020, 21, 5938. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Hung, A.C.; Lo, S.; Yuan, S.F. Adipocytokines visfatin and resistin in breast cancer: Clinical relevance. Cancer Lett. 2021, 498, 229–239. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, C.; Meng, K.; Qiao, H.; Wang, Y. Human adipocytes stimulate invasion of breast cancer MCF-7 cells by secreting IGFBP-2. PLoS ONE 2015, 10, e0119348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wu, Y.; Zhang, C.; Zhou, C.; Li, Y.; Zeng, Y.; Zhang, C.; Li, R.; Luo, D.; Wang, L.; et al. Cancer-associated adipocyte-derived G-CSF promotes breast cancer malignancy via Stat3 signaling. J. Mol. Cell Biol. 2020, 12, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Hoy, A.J.; Balaban, S.; Saunders, D.N. Adipocyte-Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Enciu, A.M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How far from translation into clinical practice? Biomed Res. Int. 2018, 2018, 7801202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le, G.S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: A novel mechanism linking obesity and cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, M.; Wang, M.; Yao, L.; Li, X.; Dong, H.; Li, M.; Sun, T.; Liu, X.; Liu, Y.; et al. Role of proton-coupled monocarboxylate transporters in cancer: From metabolic crosstalk to therapeutic potential. Front. Cell Dev. Biol. 2020, 8, 651. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Q.; Sun, S.; Wu, J.; Li, J.; Zhang, Y.; Wang, C.; Yuan, J.; Sun, S. Monocarboxylate transporters in breast cancer and adipose tissue are novel biomarkers and potential therapeutic targets. Biochem. Biophys. Res. Commun. 2018, 501, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.K.; Chang, P.H.; Kuo, W.H.; Chen, C.L.; Jeng, Y.M.; Chang, K.J.; Shew, J.Y.; Hu, C.M.; Lee, W.H. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via b-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 2005, 115, 1163–1176. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Investig. 2012, 22, 4243–4256. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Li, S.; He, J.; Du, H.; Liu, Y.; Yu, W.; Hu, H.; Han, L.; Wang, C.; Li, H.; et al. Tumor-secreted PAI-1 promotes breast cancer metastasis via the induction of adipocyte-derived collagen remodeling. Cell Commun. Signal 2019, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.Ã.; Ortuño-Pineda, C.; Moreno-Godínez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src-dependent manner in breast cancer cells. Endocr. Connect 2019, 8, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, J.; Sun, S.; Yuan, J.; Sun, S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Larabee, C.M.; Neely, O.C.; Domingos, A.I. Obesity: A neuroimmunometabolic perspective. Nat. Rev. Endocrinol. 2020, 16, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Vazquez Rodriguez, G.; Abrahamsson, A.; Jensen, L.D.E.; Dabrosin, C. Adipocytes promote early steps of breast cancer cell dissemination via interleukin-8. Front. Immunol. 2018, 9, 1767. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.C.; Ostrand-Rosenberg, S. Frontline science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. https://doi.org/10.3390/ijms22073775
Rybinska I, Mangano N, Tagliabue E, Triulzi T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. International Journal of Molecular Sciences. 2021; 22(7):3775. https://doi.org/10.3390/ijms22073775
Chicago/Turabian StyleRybinska, Ilona, Nunzia Mangano, Elda Tagliabue, and Tiziana Triulzi. 2021. "Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences" International Journal of Molecular Sciences 22, no. 7: 3775. https://doi.org/10.3390/ijms22073775
APA StyleRybinska, I., Mangano, N., Tagliabue, E., & Triulzi, T. (2021). Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. International Journal of Molecular Sciences, 22(7), 3775. https://doi.org/10.3390/ijms22073775