
Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study

 ,
,  ,
,  ,
,
Abstract
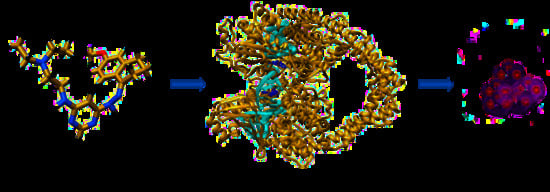
1. Introduction
2. Results and Discussion
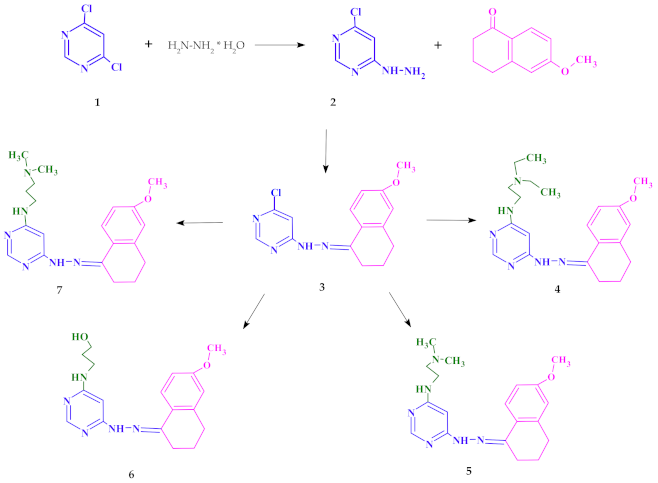
2.1. Chemistry
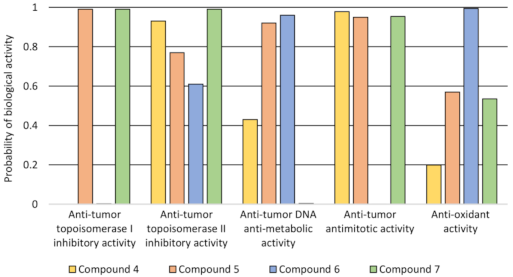
2.2. Lipophilicity and QSAR Study
2.3. Biological Evaluation
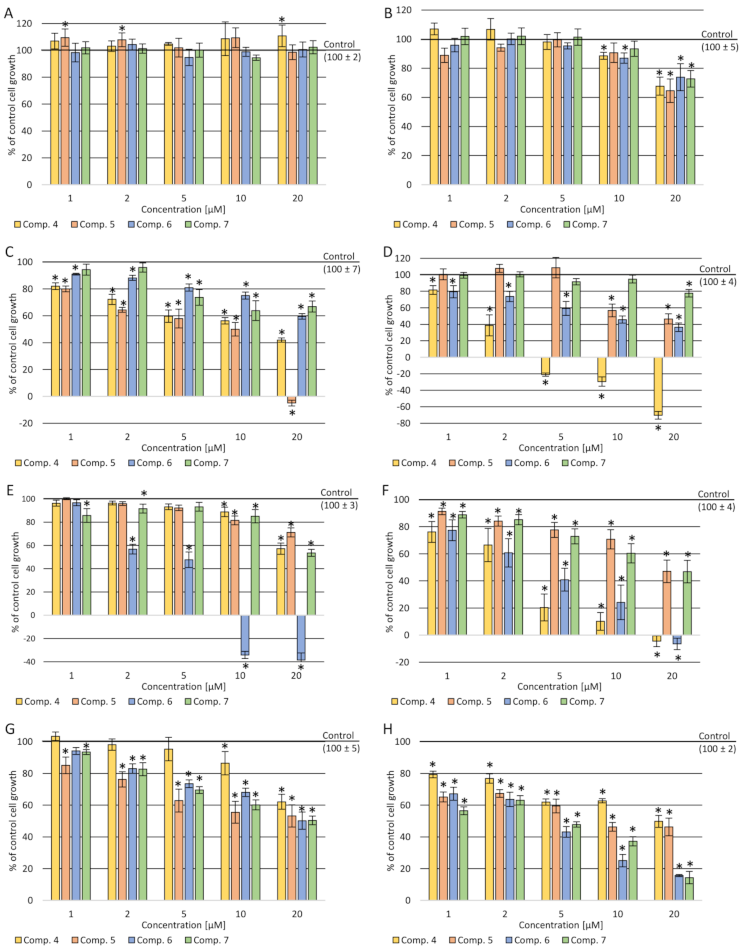
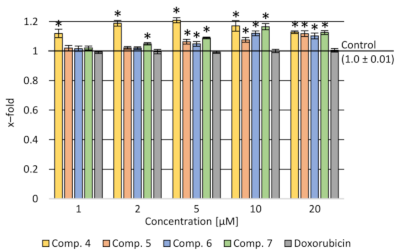
2.3.1. Cytotoxicity Assay
2.3.2. Assessment of the Impact on the Transport Function of P-glycoprotein—Accumulation of Rhodamine 123 (Rod-123) in Cells
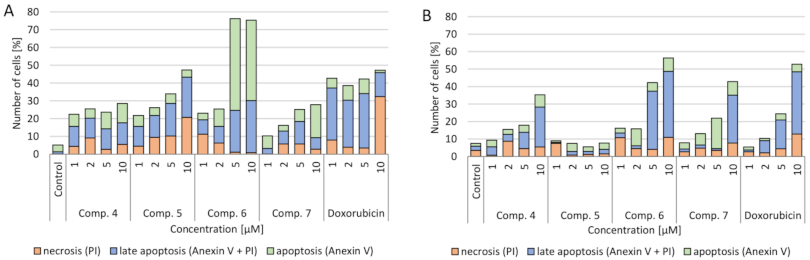
2.3.3. Verification of Apoptotic and Necrotic Cell Death
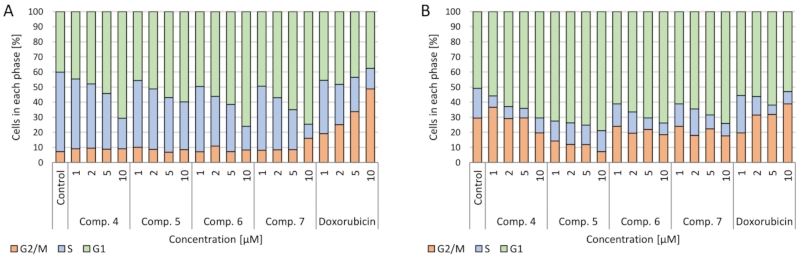
2.3.4. Cell Cycle
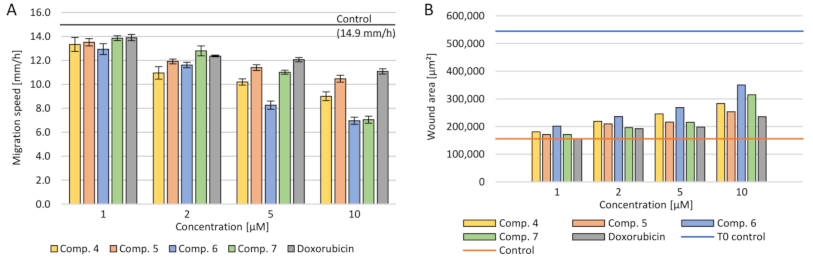
2.3.5. Cell Migration
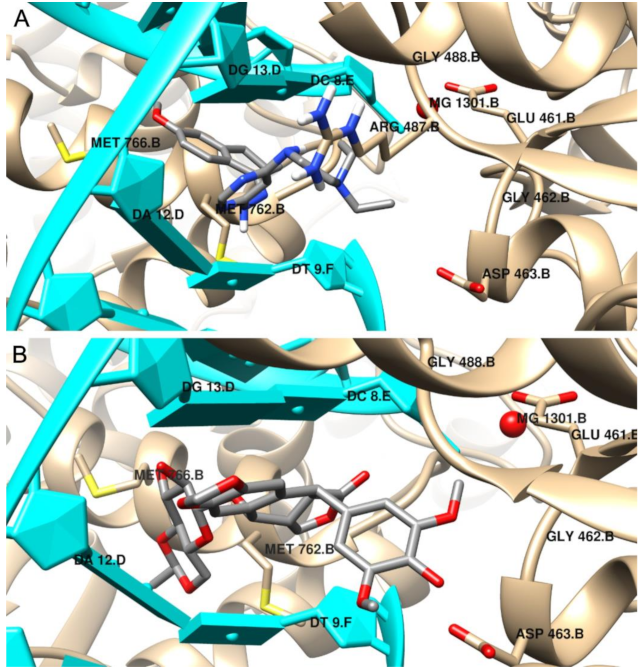
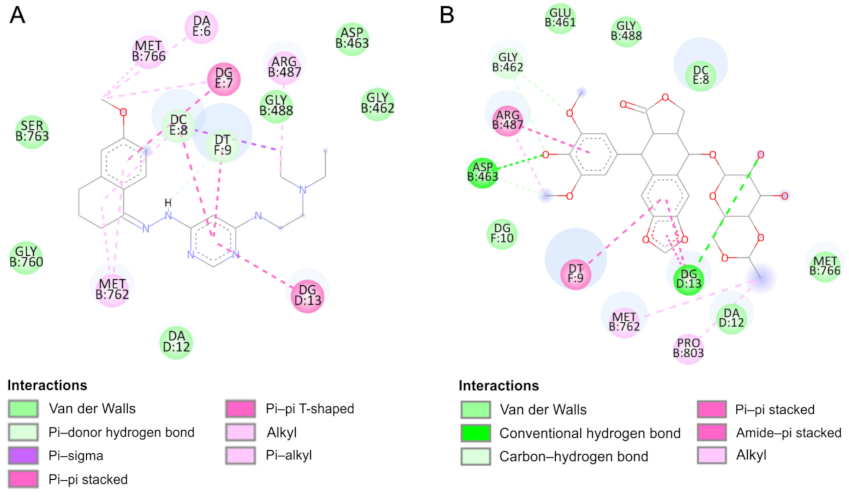
2.4. Molecular Docking
3. Conclusions
4. Materials and Methods
4.1. Instruments and Materials
4.2. Synthesis
4.3. Lipophilicity and QSAR Studies
4.4. Tested Compounds
4.4.1. Cell Lines and Conditions
4.4.2. Viability Assay
4.4.3. Assessment of the Impact on the Transport Function of P-glycoprotein—Accumulation of Rhodamine 123 (Rod-123) in Cells
4.4.4. Verification of Apoptotic and Necrotic Cell Death
4.4.5. Cell Cycle
4.4.6. Cell Migration
4.5. Statistical Analysis
4.6. Molecular docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaur, H.; Machado, M.; de Kock, C.; Smith, P.; Chibale, K.; Prudêncio, M.; Singh, K. Primaquine-pyrimidine hybrids: Synthesis and dual-stage antiplasmodial activity. Eur. J. Med. Chem. 2015, 101, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.C.; Kotadiya, G.M.; Trivedi, A.R. Studies on molecular properties prediction, antitubercular and antimicrobial activities of novel quinoline based pyrimidine motifs. Bioorgan. Med. Chem. Lett. 2014, 24, 3126–3130. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, J.; Zhou, Z.; Huang, D.; Zhang, Y.; Pei, H.; Guo, W.; Wu, H.; Wang, X.; Liu, M.; et al. Corrigendum to “Design, synthesis and evaluation of hybrid of tetrahydrocarbazole with 2,4-diaminopyrimidine scaffold as antibacterial agents” [Eur. J. Med. Chem. 162 (162) (2019) 203–211]. Eur. J. Med. Chem. 2019, 168, 385. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Soliman, S.M.; Al-Majid, A.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.K.; Yousuf, S.; Choudhary, M.I.; Wadood, A. Synthesis and structure investigation of novel pyrimidine-2,4,6-trione derivatives of highly potential biological activity as anti-diabetic agent. J. Mol. Struct. 2015, 1098, 365–376. [Google Scholar] [CrossRef]
- Zimmermann, J. Pyrimidine Derivatives and Processes for the Preparation Thereof. U.S. Patent US5521184A, 28 May 1996. [Google Scholar]
- Xie, F.; Zhao, H.; Zhao, L.; Lou, L.; Hu, Y. Synthesis and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity. Bioorgan. Med. Chem. Lett. 2009, 19, 275–278. [Google Scholar] [CrossRef]
- Kaldrikyan, M.A.; Grigoryan, L.A.; Geboyan, V.A.; Arsenyan, F.G.; Stepanyan, G.M.; Garibdzhanyan, B.T. Synthesis and antitumor activity of some disubstituted 5-(3-methyl-4-alkoxybenzyl)pyrimidines. Pharm. Chem. J. 2000, 34, 521–524. [Google Scholar] [CrossRef]
- Tylinska, B.; Jasztold-Howorko, R.; Kowalczewska, K.; Szczaurska-Nowak, K.; Gbarowski, T.; Wietrzyk, J. Design, synthesis and analysis of anticancer activity of new SAR-based S16020 derivatives. Acta Pol. Pharm. Drug Res. 2018. [Google Scholar] [CrossRef]
- Jasztold-Howorko, R.; Tylińska, B.; Biaduń, B.; Gębarowski, T.; Gąsiorowski, K. New pyridocarbazole derivatives. Synthesis and their in vitro anticancer activity. Acta Pol. Pharm. 2013, 70, 823–832. [Google Scholar]
- Nguyen, C.H.; Bisagni, E.; Lhoste, J.M.; Lavelle, F.; Bissery, M.C. Synthesis and Antitumor Activity of l-[[(Dialkylamino)alkyl]amino]-4-methyl-5H-pyrido[4,3-b]benzo[e]- and -benzo[g])indoles. A New Class of Antineoplastic Agents. J. Med. Chem. 1990, 33, 1519–1528. [Google Scholar] [CrossRef]
- Piasny, J.; Wiatrak, B.; Dobosz, A.; Tylińska, B.; Gębarowski, T. Antitumor Activity of New Olivacine Derivatives. Molecules 2020, 25, 2512. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, R.; Cui, Y.; Sheng, L.; Sun, Y.; Tian, W.; Liu, X.; Liang, S. Design, synthesis and biological evaluation of 3,4-dihydronaphthalen-1(2H)-one derivatives as Bcl-2 inhibitors. Res. Chem. Intermed. 2017, 43, 5933–5942. [Google Scholar] [CrossRef]
- Ananth, A.H.; Manikandan, N.; Rajan, R.K.; Elancheran, R.; Lakshmithendral, K.; Ramanathan, M.; Bhattacharjee, A.; Kabilan, S. Design, Synthesis, and Biological Evaluation of 2-(2-Bromo-3-nitrophenyl)-5-phenyl-1,3,4-oxadiazole Derivatives as Possible Anti-Breast Cancer Agents. Chem. Biodivers. 2020, 17. [Google Scholar] [CrossRef]
- Katariya, K.D.; Shah, S.R.; Reddy, D. Anticancer, antimicrobial activities of quinoline based hydrazone analogues: Synthesis, characterization and molecular docking. Bioorgan. Chem. 2020, 94, 103406. [Google Scholar] [CrossRef]
- Horiuchi, T.; Chiba, J.; Uoto, K.; Soga, T. Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: Synthesis, biological evaluation, and structure-activity relationships. Bioorgan. Med. Chem. Lett. 2009, 19, 305–308. [Google Scholar] [CrossRef]
- Pandey, J.; Pal, R.; Dwivedi, A.; Hajela, K. Synthesis of some new diaryl and triaryl hydrazone derivatives as possible estrogen receptor modulators. Arzneimittelforschung Drug Res. 2002, 52, 39–44. [Google Scholar] [CrossRef]
- Kaplancıklı, Z.A.; Yurttaa, L.; Turan-Zitouni, G.; Özdemir, A.; Göger, G.; Demirci, F.; Mohsen, U.A. Synthesis and Antimicrobial Activity of New Pyrimidine-Hydrazones. Lett. Drug Des. Discov. 2014, 11, 76–81. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Rashed, H.M.; Fayad, W.; Youns, M.; Sakr, T.M. Novel hydrazide-hydrazone and amide substituted coumarin derivatives: Synthesis, cytotoxicity screening, microarray, radiolabeling and in vivo pharmacokinetic studies. Eur. J. Med. Chem. 2018, 151, 723–739. [Google Scholar] [CrossRef]
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.S.; Abdel-Aziz, H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280. [Google Scholar] [CrossRef]
- Park, S.-E.; Chang, I.-H.; Jun, K.-Y.; Lee, E.; Lee, E.-S.; Na, Y.; Kwon, Y. 3-(3-Butylamino-2-hydroxy-propoxy)-1-hydroxy-xanthen-9-one acts as a topoisomerase IIα catalytic inhibitor with low DNA damage. Eur. J. Med. Chem. 2013, 69, 139–145. [Google Scholar] [CrossRef]
- Byl, J.A.W.; Cline, S.D.; Utsugi, T.; Kobunai, T.; Yamada, Y.; Osheroff, N. DNA topoisomerase II as the target for the anticancer drug TOP-53: Mechanistic basis for drug action. Biochemistry 2001, 40, 712–718. [Google Scholar] [CrossRef]
- Chikamori, K.; Grozav, A.; Kozuki, T.; Grabowski, D.; Ganapathi, R.; K Ganapathi, M. DNA Topoisomerase II Enzymes as Molecular Targets for Cancer Chemotherapy. Curr. Cancer Drug Targets 2010, 10, 758–771. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Wilkinson, B. Drug discovery beyond the “rule-of-five”. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- OECD. Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method. In OECD Guidelines for the Testing of Chemicals, Section 1; OECD Publishing: Paris, France, 1995. [Google Scholar] [CrossRef]
- Boucek, R.J.; Olson, R.D.; Brenner, D.E.; Ogunbunmi, E.M.; Inui, M.; Fleischer, S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J. Biol. Chem. 1987, 262, 15851–15856. [Google Scholar] [CrossRef]
- Vekariya, M.K.; Vekariya, R.H.; Brahmkshatriya, P.S.; Shah, N.K. Pyrimidine-based pyrazoles as cyclin-dependent kinase 2 inhibitors: Design, synthesis, and biological evaluation. Chem. Biol. Drug Des. 2018, 92, 1683–1691. [Google Scholar] [CrossRef]
- Malik, A.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Zahoor, A.F.; Ahmad, G.; Altaf, A.A.; Shah, S.A.A.; Sultan, S.; Zakaria, Z.A. Suzuki–Miyaura Reactions of (4-bromophenyl)-4,6-dichloropyrimidine through Commercially Available Palladium Catalyst: Synthesis, Optimization and Their Structural Aspects Identification through Computational Studies. Processes 2020, 8, 1342. [Google Scholar] [CrossRef]
- Kirby, A.J.; Le Lain, R.; Maharlouie, F.; Mason, P.; Nicholls, P.J.; John Smith, H.; Simons, C. Inhibition of Retinoic Acid Metabolising Enzymes by 2-(4-aminophenylmethyl)-6-hydroxy-3,4-dihydronaphthalen-1(2H)-one and Related Compounds. J. Enzyme Inhib. Med. Chem. 2003, 18, 27–33. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, Y.-Q.; Liu, Y.-K.; Zhang, H.-Q.; Hou, G.-G.; Meng, Q.-G.; Hou, Y. Potential anti-neuroinflammatory NF-кB inhibitors based on 3,4-dihydronaphthalen-1(2H)-one derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 1631–1640. [Google Scholar] [CrossRef]
- Rath, S.K.; Singh, S.; Kumar, S.; Wani, N.A.; Rai, R.; Koul, S.; Khan, I.A.; Sangwan, P.L. Synthesis of amides from (E)-3-(1-chloro-3,4-dihydronaphthalen-2-yl)acrylic acid and substituted amino acid esters as NorA efflux pump inhibitors of Staphylococcus aureus. Bioorgan. Med. Chem. 2019, 27, 343–353. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Potemkin, V.A.; Grishina, M.A.; Bartashevich, E.V. Modeling of drug molecule orientation within a receptor cavity in the BiS algorithm framework. J. Struct. Chem. 2007, 48, 155–160. [Google Scholar] [CrossRef][Green Version]
- Potemkin, V.; Grishina, M. Principles for 3D/4D QSAR classification of drugs. Drug Discov. Today 2008, 13, 952–959. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Wang, Y.R.; Chen, S.F.; Wu, C.C.; Liao, Y.W.; Lin, T.S.; Liu, K.T.; Chen, Y.S.; Li, T.K.; Chien, T.C.; Chan, N.L. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. [Google Scholar] [CrossRef]
- Chen, D.; Menche, G.; Power, T.D.; Sower, L.; Peterson, J.W.; Schein, C.H. Accounting for ligand-bound metal ions in docking small molecules on adenylyl cyclase toxins. Proteins Struct. Funct. Bioinform. 2007, 67, 593–605. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]










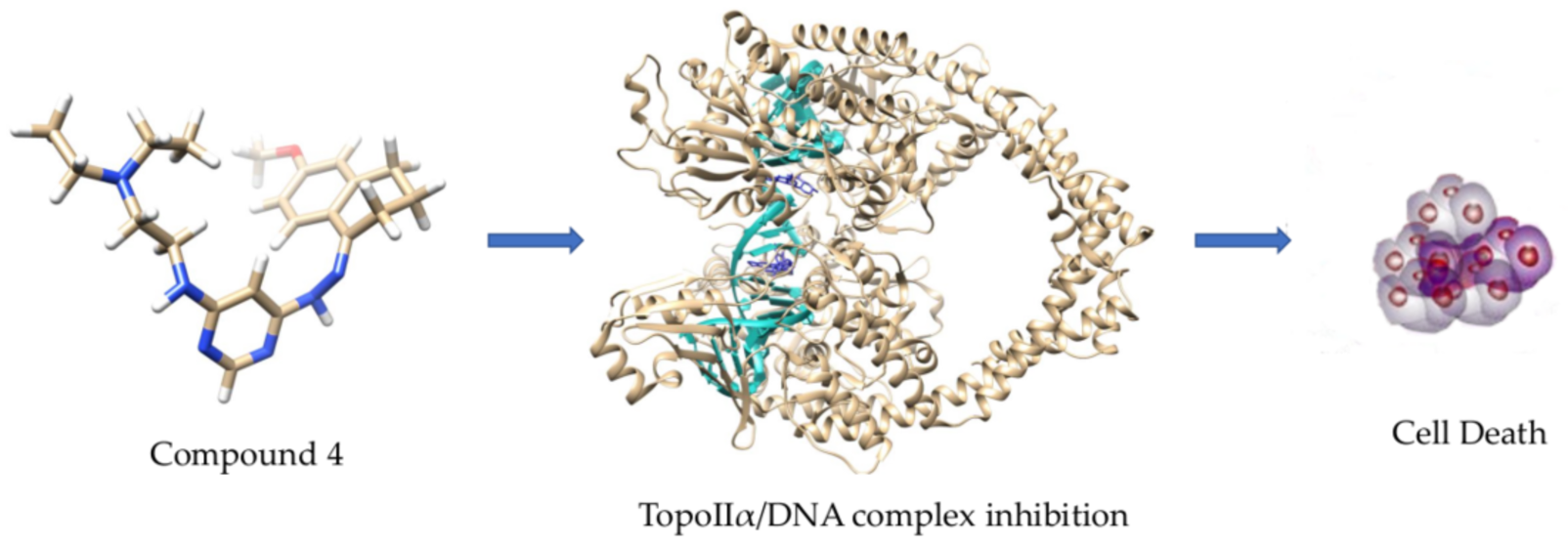{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lipinski’s Rules | Veber’s Rules | |||||
|---|---|---|---|---|---|---|---|
| MW ≤ 500 | Log P ≤ 5 | NHD ≤ 5 | NHA ≤ 10 | Violations of Rules | NBR ≤ 10 | TPSA ≤ 140 | |
| Doxorubicin | 543.52 | 0.52 | 6 | 12 | 3 | 5 | 206.07 |
| 4 | 382.50 | 3.14 | 2 | 5 | 0 | 9 | 74.67 |
| 5 | 354.45 | 2.47 | 2 | 5 | 0 | 7 | 74.67 |
| 6 | 341.41 | 2.45 | 3 | 5 | 0 | 7 | 91.66 |
| 7 | 368.48 | 2.77 | 2 | 5 | 0 | 8 | 74.67 |
| Compound | Log Po/w | Consensus log Po/w |
|---|---|---|
| Doxorubicin | –0.18 | 0.52 |
| 4 | 1.05 | 3.14 |
| 5 | 1.53 | 2.47 |
| 7 | 1.25 | 2.77 |
| Compound | ΔGbinding (kJ/mol) | Ki (µM) | ΔGint (kJ/mol) | ΔGvdw + ΔGhbond + ΔGdesolv (kJ/mol) | ΔGel (kJ/mol) |
|---|---|---|---|---|---|
| 4 | −30.8 | 4.12 | −42.0 | −41.6 | −0.4 |
| 5 | −29.0 | 8.56 | −37.7 | −37.5 | −0.2 |
| 6 | −26.3 | 24.36 | −36.3 | −35.5 | −0.8 |
| 7 | −27.0 | 15.77 | −37.0 | −36.8 | −0.2 |
| Etoposide | −44.6 | 0.02 | −50.9 | −50.6 | −0.3 |
| Compound | λmax (nm) | ε (M−1cm−1) |
|---|---|---|
| Doxorubicin | 539 | 4459 |
| 4 | 345 | 15,326 |
| 5 | 328 | 50,465 |
| 7 | 328 | 20,430 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tylińska, B.; Wiatrak, B.; Czyżnikowska, Ż.; Cieśla-Niechwiadowicz, A.; Gębarowska, E.; Janicka-Kłos, A. Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 3825. https://doi.org/10.3390/ijms22083825
Tylińska B, Wiatrak B, Czyżnikowska Ż, Cieśla-Niechwiadowicz A, Gębarowska E, Janicka-Kłos A. Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. International Journal of Molecular Sciences. 2021; 22(8):3825. https://doi.org/10.3390/ijms22083825
Chicago/Turabian StyleTylińska, Beata, Benita Wiatrak, Żaneta Czyżnikowska, Aneta Cieśla-Niechwiadowicz, Elżbieta Gębarowska, and Anna Janicka-Kłos. 2021. "Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study" International Journal of Molecular Sciences 22, no. 8: 3825. https://doi.org/10.3390/ijms22083825
APA StyleTylińska, B., Wiatrak, B., Czyżnikowska, Ż., Cieśla-Niechwiadowicz, A., Gębarowska, E., & Janicka-Kłos, A. (2021). Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. International Journal of Molecular Sciences, 22(8), 3825. https://doi.org/10.3390/ijms22083825