
Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016



Abstract
:1. Introduction
2. PDE1 Radioligands
3. PDE2A Radioligands
4. PDE4 Radioligands
4.1. Recent [11C]Rolipram Positron Emission Tomography (PET) Studies
4.2. Novel PDE4 (B, D) Radioligands Apart from [11C]Rolipram
5. PDE5 Radioligands
6. PDE7 Radioligands
7. PDE10A Radioligands
7.1. Recent PET Studies Using Already Known PDE10A Radioligands
7.2. Novel PDE10A Radioligands
8. Summary and Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Scheunemann, M.; Brust, P. Novel radioligands for cyclic nucleotide phosphodiesterase imaging with positron emission tomography: An update on developments since 2012. Molecules 2016, 21, 650. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Reed, T.M.; Repaske, D.R.; Snyder, G.L.; Greengard, P.; Vorhees, C.V. Phosphodiesterase 1B knock-out mice exhibit exaggerated locomotor hyperactivity and DARPP-32 phosphorylation in response to dopamine agonists and display impaired spatial learning. J. Neurosci. 2002, 22, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Blokland, A.; Menniti, F.S.; Prickaerts, J. PDE inhibition and cognition enhancement. Expert Opin. Ther. Pat. 2012, 22, 349–354. [Google Scholar] [CrossRef]
- Gupta, S.; Sharma, B. Protective effects of phosphodiesterase-1 (PDE1) and ATP sensitive potassium (KATP) channel modulators against 3-nitropropionic acid induced behavioral and biochemical toxicities in experimental Huntington’s disease. Eur. J. Pharmacol. 2014, 732, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Wennogle, L.P.; Hoxie, H.; Peng, Y.; Hendrick, J.P. Phosphodiesterase 1: A unique drug target for degenerative diseases and cognitive dysfunction. Adv. Neurobiol. 2017, 17, 349–384. [Google Scholar]
- Li, P.; Wennogle, L.P.; Zhao, J.; Zheng, H. Phosphodiesterase 1-Targeting Tracers and Methods. WO Patent 2011/043816 A1, 14 April 2011. [Google Scholar]
- Andrés, J.I.; De Angelis, M.; Alcazar, J.; Celen, S.; Bormans, G. Recent advances in positron emission tomography (PET) radiotracers for imaging phosphodiesterases. Curr. Top. Med. Chem. 2012, 12, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Kealey, S.; Zhou, X.; Williamson, D.; Kuzhuppilly Ramakrishnan, N.; Aigbirhio, F. In vivo evaluation of a potential PDE1 radiotracer—[11C](±)-PF04822163. In The XII International Symposium of Functional Neuroreceptor Mapping of the Living Brain (NRM18); Authorea: London, UK, 2018. [Google Scholar]
- Humphrey, J.M.; Yang, E.; Ende, C.W.; Arnold, E.P.; Head, J.L.; Jenkinson, S.; Lebel, L.A.; Liras, S.; Pandit, J.; Samas, B.; et al. Small-molecule phosphodiesterase probes: Discovery of potent and selective CNS-penetrable quinazoline inhibitors of PDE1. Med. Chem. Comm. 2014, 5, 1290–1296. [Google Scholar] [CrossRef]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, D.T.; Coskran, T.M.; Kelly, M.P.; Kleiman, R.J.; Morton, D.; O’Neill, S.M.; Schmidt, C.J.; Weinberg, R.J.; Menniti, F.S. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012, 226, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett. 2013, 23, 6522–6527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yu, Y.; Ruan, L.; Wang, C.; Pan, J.; Klabnik, J.; Lueptow, L.; Zhang, H.-T.; O’Donnell, J.M.; Xu, Y. The roles of phosphodiesterase 2 in the central nervous and peripheral systems. Curr. Pharm. Des. 2015, 21, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Ritawidya, R.; Wenzel, B.; Teodoro, R.; Toussaint, M.; Kranz, M.; Deuther-Conrad, W.; Dukic-Stefanovic, S.; Ludwig, F.-A.; Scheunemann, M.; Brust, P. Radiosynthesis and biological investigation of a novel fluorine-18 labeled benzoimidazotriazine-based radioligand for the imaging of phosphodiesterase 2A with positron emission tomography. Molecules 2019, 24, 4149. [Google Scholar] [CrossRef] [Green Version]
- Ritawidya, R.; Ludwig, F.-A.; Briel, D.; Brust, P.; Scheunemann, M. Synthesis and in vitro evaluation of 8-pyridinyl-substituted benzo[e]imidazo[2,1-c][1,2,4]triazines as phosphodiesterase 2A inhibitors. Molecules 2019, 24, 2791. [Google Scholar] [CrossRef] [Green Version]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Kranz, M.; Scheunemann, M.; Egerland, U.; Höfgen, N.; Briel, D.; Steinbach, J.; et al. Investigation of an 18F-labelled imidazopyridotriazine for molecular imaging of cyclic nucleotide phosphodiesterase 2A. Molecules 2018, 23, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef]
- Helal, C.J.; Arnold, E.; Boyden, T.; Chang, C.; Chappie, T.A.; Fisher, E.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Humphrey, J.M.; et al. Identification of a potent, highly selective, and brain penetrant phosphodiesterase 2A inhibitor clinical candidate. J. Med. Chem. 2018, 61, 1001–1018. [Google Scholar] [CrossRef]
- Chen, L.; Nabulsi, N.; Naganawa, M.; Zasadny, K.; Skaddan, M.B.; Zhang, L.; Najafzadeh, S.; Lin, S.-f.; Helal, C.J.; Boyden, T.L.; et al. Preclinical evaluation of 18F-PF-05270430, a novel PET radioligand for the phosphodiesterase 2A enzyme. J. Nucl. Med. 2016, 57, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Naganawa, M.; Nabulsi, N.; Waterhouse, R.; Lin, S.-f.; Zhang, L.; Cass, T.; Ropchan, J.; McCarthy, T.; Huang, Y.; Carson, R. Human PET studies with [18F]PF-05270430, a PET radiotracer for imaging phosphodiesterase-2A. J. Nucl. Med. 2013, 54, 201. [Google Scholar]
- Naganawa, M.; Waterhouse, R.N.; Nabulsi, N.; Lin, S.-F.; Labaree, D.; Ropchan, J.; Tarabar, S.; DeMartinis, N.; Ogden, A.; Banerjee, A.; et al. First-in-human assessment of the novel PDE2A PET radiotracer 18F-PF-05270430. J. Nucl. Med. 2016, 57, 1388–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, T.; Massif, C.; Papin, C.; Carroll, V.; Alagille, D.; Baldwin, R.M.; Tamagnan, G. Improved production of [18F]PF-05270430 for clinical imaging of PDE2A in brain. J. Labelled Compd. Rad. 2015, 58, S199. [Google Scholar]
- Bolger, G.B.; Rodgers, L.; Riggs, M. Differential CNS expression of alternative mRNA isoforms of the mammalian genes encoding cAMP-specific phosphodiesterases. Gene 1994, 149, 237–244. [Google Scholar] [CrossRef]
- Horton, Y.M.; Sullivan, M.; Houslay, M.D. Molecular cloning of a novel splice variant of human type IVA (PDE-IVA) cyclic AMP phosphodiesterase and localization of the gene to the p13.2-q12 region of human chromosome 19. Biochem. J. 1995, 308, 683–691. [Google Scholar] [CrossRef]
- Pérez-Torres, S.; Miró, X.; Palacios, J.M.; Cortés, R.; Puigdoménech, P.; Mengod, G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and [3H]rolipram binding autoradiography: Comparison with monkey and rat brain. J. Chem. Neuroanat. 2000, 20, 349–374. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.; Ray, B.; Mahalakshmi, A.M.; Tuladhar, S.; Nandakumar, D.N.; Srinivasan, M.; Essa, M.M.; Chidambaram, S.B.; Guillemin, G.J.; Sakharkar, M.K. Phosphodiesterase-4 enzyme as a therapeutic target in neurological disorders. Pharmacol. Res. 2020, 160, 105078. [Google Scholar] [CrossRef]
- Bodkhe, S.; Nikam, M.; Sherje, A.P.; Khan, T.; Suvarna, V.; Patel, K. Current insights on clinical efficacy of roflumilast for treatment of COPD, asthma and ACOS. Int. Immunopharmacol. 2020, 88, 106906. [Google Scholar] [CrossRef]
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front. Pharmacol. 2018, 9, 1048. [Google Scholar] [CrossRef] [Green Version]
- DaSilva, J.N.; Valente, C.M.; Wilson, A.A.; Warsh, J.J.; Houle, S. Carbon-11 labeling of the selective inhibitors of phosphodiesterase IV RO20-1724 and rolipram. J. Labelled Compd. Rad. 1997, 40, 678–680. [Google Scholar]
- DaSilva, J.N.; Lourenco, C.M.; Wilson, A.A.; Houle, S. Syntheses of the phosphodiesterase-4 inhibitors [11C]RO 20-1724, R-, R/S- and S-[11C]rolipram. J. Labelled Compd. Rad. 2001, 44, 373–384. [Google Scholar] [CrossRef]
- DaSilva, J.N.; Lourenco, C.M.; Meyer, J.H.; Hussey, D.; Potter, W.Z.; Houle, S. Imaging cAMP-specific phosphodiesterase-4 in human brain with R-[11C]rolipram and positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Kenk, M.; Thomas, A.; Lortie, M.; deKemp, R.; Beanlands, R.S.; DaSilva, J.N. PET measurements of cAMP-mediated phosphodiesterase-4 with (R)-[11C]rolipram. Curr. Radiopharm. 2011, 4, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Hines, C.S.; Zoghbi, S.S.; Mallinger, A.G.; Dickstein, L.P.; Liow, J.S.; Zhang, Y.; Pike, V.W.; Drevets, W.C.; Innis, R.B.; et al. Downregulation of brain phosphodiesterase type IV measured with 11C-(R)-rolipram positron emission tomography in major depressive disorder. Biol. Psychiat. 2012, 72, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Lortie, M.; DaSilva, J.N.; Kenk, M.; Thorn, S.; Davis, D.; Birnie, D.; Beanlands, R.S.; deKemp, R.A. Analysis of (R)- and (S)-[11C]rolipram kinetics in canine myocardium for the evaluation of phosphodiesterase-4 with PET. Mol. Imaging Biol. 2012, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Saldou, N.; Obernolte, R.; Huber, A.; Baecker, P.A.; Wilhelm, R.; Alvarez, R.; Li, B.; Xia, L.; Callan, O.; Su, C.; et al. Comparison of recombinant human PDE4 isoforms: Interaction with substrate and inhibitors. Cell. Signal. 1998, 10, 427–440. [Google Scholar] [CrossRef]
- Wang, P.; Myers, J.G.; Wu, P.; Cheewatrakoolpong, B.; Egan, R.W.; Billah, M.M. Expression, purification, and characterization of human cAMP-specific phosphodiesterase (PDE4) subtypes A, B, C, and D. Biochem. Bioph. Res. Commun. 1997, 234, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.D.; Hofer, P.; Spina, D.; Seeds, E.A.; Banner, K.H.; Harrison, S.; Douglas, G.; Matsumoto, T.; Page, C.P.; Wong, R.H.; et al. Pharmacology of a new cyclic nucleotide phosphodiesterase type 4 inhibitor, V11294. Pulm. Pharmacol. Ther. 2003, 16, 97–104. [Google Scholar] [CrossRef]
- Rutter, A.R.; Poffe, A.; Cavallini, P.; Davis, T.G.; Schneck, J.; Negri, M.; Vicentini, E.; Montanari, D.; Arban, R.; Gray, F.A.; et al. GSK356278, a potent, selective, brain-penetrant phosphodiesterase 4 inhibitor that demonstrates anxiolytic and cognition-enhancing effects without inducing side effects in preclinical species. J. Pharmacol. Exp. Ther. 2014, 350, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzelmann, A.; Morcillo, E.J.; Lungarella, G.; Adnot, S.; Sanjar, S.; Beume, R.; Schudt, C.; Tenor, H. The preclinical pharmacology of roflumilast—A selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2010, 23, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Richards, E.M.; Niciu, M.J.; Ionescu, D.F.; Zoghbi, S.S.; Hong, J.; Telu, S.; Hines, C.S.; Pike, V.W.; Zarate, C.A.; et al. CAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol. Psychiatry 2017, 22, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Niccolini, F.; Wilson, H.; Pagano, G.; Coello, C.; Mehta, M.A.; Searle, G.E.; Gunn, R.N.; Rabiner, E.A.; Foltynie, T.; Politis, M. Loss of phosphodiesterase 4 in Parkinson disease. Relev. Cogn. Deficits 2017, 89, 586–593. [Google Scholar] [CrossRef] [Green Version]
- Niccolini, F.; Foltynie, T.; Marques, T.R.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.; Pagano, G.; Niccolini, F.; Muhlert, N.; Mehta, M.A.; Searle, G.; Gunn, R.N.; Rabiner, E.A.; Foltynie, T.; Politis, M. The role of phosphodiesterase 4 in excessive daytime sleepiness in Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 77, 163–169. [Google Scholar] [CrossRef] [Green Version]
- van der Aart, J.; Salinas, C.; Dimber, R.; Pampols-Maso, S.; Weekes, A.A.; Tonkyn, J.; Gray, F.A.; Passchier, J.; Gunn, R.N.; Rabiner, E.A. Quantification of human brain PDE4 occupancy by GSK356278: A [11C](R)-rolipram PET study. J. Cerebr. Blood Flow Met. 2018, 38, 2033–2040. [Google Scholar] [CrossRef] [Green Version]
- Takano, A.; Uz, T.; Garcia-Segovia, J.; Tsai, M.; Lahu, G.; Amini, N.; Nakao, R.; Jia, Z.; Halldin, C. A nonhuman primate PET study: Measurement of brain PDE4 occupancy by roflumilast using (R)-[11C]rolipram. Mol. Imaging Biol. 2018, 20, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Hatzelmann, A.; Schudt, C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J. Pharmacol. Exp. Ther. 2001, 297, 267–279. [Google Scholar] [PubMed]
- Giembycz, M.A.; Field, S.K. Roflumilast: First phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des. Dev. Ther. 2010, 4, 147–158. [Google Scholar]
- Ooms, M.; Tsujikawa, T.; Lohith, T.G.; Mabins, S.N.; Zoghbi, S.S.; Sumitomo, A.; Jaaro-Peled, H.; Kimura, Y.; Telu, S.; Pike, V.W.; et al. [11C](R)-rolipram positron emission tomography detects DISC1 inhibition of phosphodiesterase type 4 in live DISC1 locus-impaired mice. J. Cerebr. Blood Flow Met. 2019, 39, 1306–1313. [Google Scholar] [CrossRef]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Niwa, M.; Cash-Padgett, T.; Kubo, K.I.; Saito, A.; Ishii, K.; Sumitomo, A.; Taniguchi, Y.; Ishizuka, K.; Jaaro-Peled, H.; Tomoda, T.; et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-more disrupted-in-schizophrenia 1. Mol. Psychiatry 2016, 21, 1488–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, L.D.; Wakabayashi, Y.; Stolz, L.A.; Collins, M.T.; Guthrie, L.; Victorino, M.; Chung, J.; Miller, W.; Zoghbi, S.S.; Pike, V.W.; et al. PET imaging of phosphodiesterase-4 identifies affected dysplastic bone in McCune-Albright syndrome, a genetic mosaic disorder. J. Nucl. Med. 2020, 61, 1672–1677. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Beck, E.M.; Chappie, T.A.; Coelho, R.V.; Doran, S.D.; Fan, K.-H.; Helal, C.J.; Humphrey, J.M.; Hughes, Z.; et al. The discovery of a novel phosphodiesterase (PDE) 4B-preferring radioligand for positron emission tomography (PET) imaging. J. Med. Chem. 2017, 60, 8538–8551. [Google Scholar] [CrossRef]
- Jia, L.; Miao, C.; Dong, F.; Li, W.; Wang, M.; Zheng, Q.-H.; Xu, Z. Facile synthesis of carbon-11-labeled sEH/PDE4 dual inhibitors as new potential PET agents for imaging of sEH/PDE4 enzymes in neuroinflammation. Bioorg. Med. Chem. Lett. 2019, 29, 1654–1659. [Google Scholar] [CrossRef]
- Blöcher, R.; Wagner, K.M.; Gopireddy, R.R.; Harris, T.R.; Wu, H.; Barnych, B.; Hwang, S.H.; Xiang, Y.K.; Proschak, E.; Morisseau, C.; et al. Orally available soluble epoxide hydrolase/phosphodiesterase 4 dual inhibitor treats inflammatory pain. J. Med. Chem. 2018, 61, 3541–3550. [Google Scholar] [CrossRef]
- Wakabayashi, Y.; Telu, S.; Dick, R.M.; Fujita, M.; Ooms, M.; Morse, C.L.; Liow, J.-S.; Hong, J.S.; Gladding, R.L.; Manly, L.S.; et al. Discovery, radiolabeling, and evaluation of subtype-selective inhibitors for positron emission tomography imaging of brain phosphodiesterase-4D. ACS Chem. Neurosci. 2020, 11, 1311–1323. [Google Scholar] [CrossRef]
- Gurney, M.E.; Nugent, R.A.; Mo, X.; Sindac, J.A.; Hagen, T.J.; Fox, D.; O’Donnell, J.M.; Zhang, C.; Xu, Y.; Zhang, H.-T.; et al. Design and synthesis of selective phosphodiesterase 4D (PDE4D) allosteric inhibitors for the treatment of fragile X syndrome and other brain disorders. J. Med. Chem. 2019, 62, 4884–4901. [Google Scholar] [CrossRef]
- Espinoza-Fonseca, L.M. The benefits of the multi-target approach in drug design and discovery. Bioorgan. Med. Chem. 2006, 14, 896–897. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural. Regen. Res. 2019, 14, 437–440. [Google Scholar]
- O’Donnell, J.M.; Zhang, H.-T. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4). Trends Pharmacol. Sci. 2004, 25, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Telu, S.; Fujita, M.; Wakabayashi, Y.; Dick, R.; Liow, J.-S.; Hong, J.; Morse, C.; Ooms, M.; Zoghbi, S.; Mo, X.; et al. Syntheses and evaluation of phosphodiesterase subtype 4D (PDE4D) PET radioligand candidates in monkey. J. Nucl. Med. 2020, 61, 268. [Google Scholar]
- Sopory, S.; Kaur, T.; Visweswariah, S.S. The cGMP-binding, cGMP-specific phosphodiesterase (PDE5): Intestinal cell expression, regulation and role in fluid secretion. Cell. Signal. 2004, 16, 681–692. [Google Scholar] [CrossRef]
- Lin, C.S.; Lin, G.; Xin, Z.C.; Lue, T.F. Expression, distribution and regulation of phosphodiesterase 5. Curr. Pharm. Des. 2006, 12, 3439–3457. [Google Scholar] [CrossRef] [PubMed]
- Teich, A.F.; Sakurai, M.; Patel, M.; Holman, C.; Saeed, F.; Fiorito, J.; Arancio, O. PDE5 exists in human neurons and is a viable therapeutic target for neurologic disease. J. Alzheimer’s Dis. 2016, 52, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Therapeutic potential of phosphodiesterase inhibitors against neurodegeneration: The perspective of the medicinal chemist. ACS Chem. Neurosci. 2020, 11, 1726–1739. [Google Scholar] [CrossRef]
- Serap, G.; Philip, J.K.; Ege Can, S.; Wayne, J.G.H. PDE5 inhibitor treatment options for urologic and non-urologic indications: 2012 Update. Curr. Pharm. Des. 2012, 18, 5590–5606. [Google Scholar]
- Chekol, R.; Gheysens, O.; Ahamed, M.; Cleynhens, J.; Pokreisz, P.; Vanhoof, G.; Janssens, S.; Verbruggen, A.; Bormans, G. Carbon-11 and fluorine-18 radiolabeled pyridopyrazinone derivatives for positron emission tomography (PET) imaging of phosphodiesterase-5 (PDE5). J. Med. Chem. 2017, 60, 486–496. [Google Scholar] [CrossRef]
- Wenzel, B.; Liu, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Teodoro, R.; Ludwig, F.-A.; Chezal, J.-M.; Moreau, E.; Brust, P.; Maisonial-Besset, A. Targeting cyclic nucleotide phosphodiesterase 5 (PDE5) in brain: Toward the development of a PET radioligand labeled with fluorine-18. Bioorganic Chem. 2019, 86, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wenzel, B.; Dukic-Stefanovic, S.; Teodoro, R.; Ludwig, F.-A.; Deuther-Conrad, W.; Schröder, S.; Chezal, J.-M.; Moreau, E.; Brust, P.; et al. Development of a new radiofluorinated quinoline analog for PET imaging of phosphodiesterase 5 (PDE5) in brain. Pharmaceuticals 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Du, J.; Miao, C.; Jia, L.; Li, W.; Wang, M.; Zheng, Q.-H.; Xu, Z. Radiosynthesis of carbon-11 labeled PDE5 inhibitors as new potential PET radiotracers for imaging of Alzheimer’s disease. Appl. Radiat. Isotopes 2019, 154, 108873. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, X.; Zhang, X.; Tian, G.; Yang, R.; Wu, J.; Zou, X.; Liu, Z.; Yang, X.; Wu, C.; et al. Pharmacokinetics-driven optimization of 4(3H)-pyrimidinones as phosphodiesterase type 5 inhibitors leading to TPN171, a clinical candidate for the treatment of pulmonary arterial hypertension. J. Med. Chem. 2019, 62, 4979–4990. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jia, L.; Liu, W.; Li, W.; Song, Y.; Zheng, Q.-H. Radiosynthesis of a carbon-11 labeled PDE5 inhibitor [11C]TPN171 as a new potential PET heart imaging agent. Appl. Radiat. Isotopes 2020, 162, 109190. [Google Scholar] [CrossRef] [PubMed]
- Blount, M.A.; Beasley, A.; Zoraghi, R.; Sekhar, K.R.; Bessay, E.P.; Francis, S.H.; Corbin, J.D. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol. Pharmacol. 2004, 66, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.-C.; Coste, H.; Linget, J.M.; Kirilovsky, J.; Hyafil, F.; Labaudinière, R. The discovery of tadalafil: A novel and highly selective PDE5 inhibitor. 2: 2,3,6,7,12,12a-hexahydropyrazino[1‘,2‘:1,6]pyrido[3,4-b]indole-1,4-dione analogues. J. Med. Chem. 2003, 46, 4533–4542. [Google Scholar] [CrossRef]
- Saenz de Tejada, I.; Angulo, J.; Cuevas, P.; Fernández, A.; Moncada, I.; Allona, A.; Lledó, E.; Körschen, H.G.; Niewöhner, U.; Haning, H.; et al. The phosphodiesterase inhibitory selectivity and the in vitro and in vivo potency of the new PDE5 inhibitor vardenafil. Int. J. Impot. Res. 2001, 13, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Maisonial-Besset, A.; Wenzel, B.; Canitrot, D.; Baufond, A.; Chezal, J.-M.; Brust, P.; Moreau, E. Synthesis and in vitro evaluation of new fluorinated quinoline derivatives with high affinity for PDE5: Towards the development of new PET neuroimaging probes. Eur. J. Med. Chem. 2017, 136, 548–560. [Google Scholar] [CrossRef]
- Fiorito, J.; Vendome, J.; Saeed, F.; Staniszewski, A.; Zhang, H.; Yan, S.; Deng, S.-X.; Arancio, O.; Landry, D.W. Identification of a novel 1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridine analogue as a potent phosphodiesterase 5 inhibitor with improved aqueous solubility for the treatment of Alzheimer’s disease. J. Med. Chem. 2017, 60, 8858–8875. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Brit. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro, X.; Perez-Torres, S.; Palacios, J.M.; Puigdomenech, P.; Mengod, G. Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in rat brain and peripheral organs. Synapse 2001, 40, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.M.; Reyes-Irisarri, E.; Mengod, G. Comparison of cAMP-specific phosphodiesterase mRNAs distribution in mouse and rat brain. Neurosci. Lett. 2012, 525, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Irisarri, E.; Perez-Torres, S.; Mengod, G. Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience 2005, 132, 1173–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gortari, P.; Mengod, G. Dopamine D1, D2 and mu-opioid receptors are co-expressed with adenylyl cyclase 5 and phosphodiesterase 7B mRNAs in striatal rat cells. Brain Res. 2010, 1310, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.P.; Adamowicz, W.; Bove, S.; Hartman, A.J.; Mariga, A.; Pathak, G.; Reinhart, V.; Romegialli, A.; Kleiman, R.J. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell. Signal. 2014, 26, 383–397. [Google Scholar] [CrossRef]
- Nakata, A.; Ogawa, K.; Sasaki, T.; Koyama, N.; Wada, K.; Kotera, J.; Kikkawa, H.; Omori, K.; Kaminuma, O. Potential role of phosphodiesterase 7 in human T cell function: Comparative effects of two phosphodiesterase inhibitors. Clin. Exp. Immunol. 2002, 128, 460–466. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Perez, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE 2011, 6, e17240. [Google Scholar] [CrossRef] [Green Version]
- Demopulos, G.A.; Gaitanaris, G.A. Treatment of Addiction and Impulse-Control Disorders Using PDE7 Inhibitors. WO Patent 2012/064667 A3, 14 November 2013. [Google Scholar]
- Gil, C.; Campillo, N.E.; Perez, D.I.; Martinez, A. PDE7 inhibitors as new drugs for neurological and inflammatory disorders. Expert Opin. Ther. Pat. 2008, 18, 1127–1139. [Google Scholar] [CrossRef]
- Szczypka, M. Role of phosphodiesterase 7 (PDE7) in T cell activity. Effects of selective PDE7 inhibitors and dual PDE4/7 inhibitors on T cell functions. Int. J. Mol. Sci. 2020, 21, 6118. [Google Scholar] [CrossRef]
- Jankowska, A.; Swierczek, A.; Chlon-Rzepa, G.; Pawlowski, M.; Wyska, E. PDE7-selective and dual inhibitors: Advances in chemical and biological research. Curr. Med. Chem. 2017, 24, 673–700. [Google Scholar] [CrossRef] [PubMed]
- McQuown, S.; Paes, D.; Baumgärtel, K.; Prickaerts, J.; Peters, M. Pharmacological inhibition of phosphodiesterase 7 enhances consolidation processes of spatial memory. Neurobiol. Learn. Mem. 2021, 177, 107357. [Google Scholar] [CrossRef] [PubMed]
- Thomae, D.; Servaes, S.; Vazquez, N.; Wyffels, L.; Dedeurwaerdere, S.; Van der Veken, P.; Joossens, J.; Augustyns, K.; Stroobants, S.; Staelens, S. Synthesis and preclinical evaluation of an 18F labeled PDE7 inhibitor for PET neuroimaging. Nucl. Med. Biol. 2015, 42, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Thomae, D.; Servaes, S.; Vazquez, N.; Wyffels, L.; Dedeurwaerdere, S.; Van der Veken, P.; Joossens, J.; Augustyns, K.; Stroobants, S.; Staelens, S. Synthesis and preclinical evaluation of two novel radioligands for PDE7 imaging in the brain. J. Labelled Compd. Rad. 2015, 58, S295. [Google Scholar]
- Chen, J.; Gu, G.; Chen, M.; Scott, T.; Heger, L.; Zook, D.; Chung, D.; Keenan, T.; Renick, J.; Santora, V.J.; et al. Rapid identification of a novel phosphodiesterase 7B tracer for receptor occupancy studies using LC-MS/MS. Neurochem. Int. 2020, 137, 104735. [Google Scholar] [CrossRef]
- Obokata, N.; Seki, C.; Hirata, T.; Maeda, J.; Ishii, H.; Nagai, Y.; Matsumura, T.; Takakuwa, M.; Fukuda, H.; Minamimoto, T.; et al. Synthesis and preclinical evaluation of [11C]MTP38 as a novel PET ligand for phosphodiesterase 7 in the brain. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kubota, M.; Seki, C.; Kimura, Y.; Takahata, K.; Shimada, H.; Takado, Y.; Matsuoka, K.; Tagai, K.; Sano, Y.; Yamamoto, Y.; et al. A first-in-human study of 11C-MTP38, a novel PET ligand for phosphodiesterase 7. Eur. J. Nucl. Med. Mol. Imaging 2021, in press. [Google Scholar] [CrossRef]
- Coskran, T.M.; Morton, D.; Menniti, F.S.; Adamowicz, W.O.; Kleiman, R.J.; Ryan, A.M.; Strick, C.A.; Schmidt, C.J.; Stephenson, D.T. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J. Histochem. Cytochem. 2006, 54, 1205–1213. [Google Scholar] [CrossRef]
- Xie, Z.; Adamowicz, W.O.; Eldred, W.D.; Jakowski, A.B.; Kleiman, R.J.; Morton, D.G.; Stephenson, D.T.; Strick, C.A.; Williams, R.D.; Menniti, F.S. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience 2006, 139, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Seeger, T.F.; Bartlett, B.; Coskran, T.M.; Culp, J.S.; James, L.C.; Krull, D.L.; Lanfear, J.; Ryan, A.M.; Schmidt, C.J.; Strick, C.A.; et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003, 985, 113–126. [Google Scholar] [CrossRef]
- Kehler, J. Phosphodiesterase 10A inhibitors: A 2009–2012 patent update. Expert Opin. Ther. Pat. 2013, 23, 31–45. [Google Scholar] [CrossRef]
- Agnieszka, Z.; Anna, P.; Adam, B.; Alicja, G.; Anna, C.; Maciej, P. Phosphodiesterase 10 inhibitors—Novel perspectives for psychiatric and neurodegenerative drug discovery. Curr. Med. Chem. 2018, 25, 3455–3481. [Google Scholar]
- Agnieszka, J.; Artur, Ś.; Elżbieta, W.; Alicja, G.; Adam, B.; Maciej, P.; Grażyna, C.-R. Advances in discovery of PDE10A inhibitors for CNS-related disorders. Part 1: Overview of the chemical and biological research. Curr. Drug Targets 2019, 20, 122–143. [Google Scholar]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life 2012, 64, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Chappie, T.A.; Helal, C.J.; Hou, X. Current landscape of phosphodiesterase 10A (PDE10A) inhibition. J. Med. Chem. 2012, 55, 7299–7331. [Google Scholar] [CrossRef]
- Artur, Ś.; Agnieszka, J.; Grażyna, C.-R.; Maciej, P.; Elżbieta, W. Advances in the discovery of PDE10A inhibitors for CNS-related disorders. Part 2: Focus on schizophrenia. Curr. Drug Targets 2019, 20, 1652–1669. [Google Scholar]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-(4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl)-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Attili, B.; Celen, S.; Koole, M.; Verbruggen, A.; Van Laere, K.; Bormans, G. [18F]JNJ42259152 binding to phosphodiesterase 10A, a key regulator of medium spiny neuron excitability, is altered in the presence of cyclic AMP. J. Neurochem. 2016, 139, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Celen, S.; Koole, M.; Ooms, M.; De Angelis, M.; Sannen, I.; Cornelis, J.; Alcazar, J.; Schmidt, M.; Verbruggen, A.; Langlois, X.; et al. Preclinical evaluation of [18F]JNJ42259152 as a PET tracer for PDE10A. Neuroimage 2013, 82, 13–22. [Google Scholar] [CrossRef]
- Koole, M.; Van Laere, K.; Ahmad, R.; Ceccarini, J.; Bormans, G.; Vandenberghe, W. Brain PET imaging of phosphodiesterase 10A in progressive supranuclear palsy and Parkinson’s disease. Mov. Disord. 2017, 32, 943–945. [Google Scholar] [CrossRef]
- Grauer, S.M.; Pulito, V.L.; Navarra, R.L.; Kelly, M.P.; Kelley, C.; Graf, R.; Langen, B.; Logue, S.; Brennan, J.; Jiang, L.; et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J. Pharmacol. Exp. Ther. 2009, 331, 574–590. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Fan, J.; Li, S.; Jones, L.A.; Cui, J.; Padakanti, P.K.; Xu, J.; Zeng, D.; Shoghi, K.I.; Perlmutter, J.S.; et al. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a PET probe for imaging PDE10A in rodent and non-human primate brain. Bioorg. Med. Chem. 2011, 19, 1666–1673. [Google Scholar] [CrossRef] [Green Version]
- Plisson, C.; Weinzimmer, D.; Jakobsen, S.; Natesan, S.; Salinas, C.; Lin, S.F.; Labaree, D.; Zheng, M.Q.; Nabulsi, N.; Marques, T.R.; et al. Phosphodiesterase 10A PET radioligand development program: From pig to human. J. Nucl. Med. 2014, 55, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, M.; Celen, S.; Koole, M.; Langlois, X.; Schmidt, M.; De Angelis, M.; Andrés, J.I.; Verbruggen, A.; Van Laere, K.; Bormans, G. Synthesis and biological evaluation of carbon-11 and fluorine-18 labeled tracers for in vivo visualization of PDE10A. Nucl. Med. Biol. 2014, 41, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, K.; Ahmad, R.U.; Hudyana, H.; Dubois, K.; Schmidt, M.E.; Celen, S.; Bormans, G.; Koole, M. Quantification of [18F]JNJ42259152, a novel phosphodiesterase 10A PET tracer: Kinetic modeling and test–retest study in human brain. J. Nucl. Med. 2013, 54, 1285–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, X.; Jin, H.; Fan, J.; Flores, H.; Perlmutter, J.S.; Tu, Z. Synthesis of fluorine-containing phosphodiesterase 10A (PDE10A) inhibitors and the in vivo evaluation of F-18 labeled PDE10A PET tracers in rodent and nonhuman primate. J. Med. Chem. 2015, 58, 8584–8600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jin, H.; Zhou, H.; Rothfuss, J.; Tu, Z. Synthesis and in vitro biological evaluation of pyrazole group-containing analogues for PDE10A. Med. Chem. Comm. 2013, 4, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jin, H.; Yue, X.; Zhang, X.; Yang, H.; Li, J.; Flores, H.; Su, Y.; Perlmutter, J.S.; Tu, Z. Preclinical evaluation of a promising C-11 labeled PET tracer for imaging phosphodiesterase 10A in the brain of living subject. Neuroimage 2015, 121, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barret, O.; Thomae, D.; Tavares, A.; Alagille, D.; Papin, C.; Waterhouse, R.; McCarthy, T.; Jennings, D.; Marek, K.; Russell, D.; et al. In vivo assessment and dosimetry of 2 novel PDE10A PET radiotracers in humans: 18F-MNI-659 and 18F-MNI-654. J. Nucl. Med. 2014, 55, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Mori, W.; Yamasaki, T.; Fujinaga, M.; Ogawa, M.; Zhang, Y.; Hatori, A.; Xie, L.; Kumata, K.; Wakizaka, H.; Kurihara, Y.; et al. Development of 2-(2-(3-(4-([18f]fluoromethoxy-d2)phenyl)-7-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)ethyl)-4-isopropoxyisoindoline-1,3-dione for positron-emission-tomography imaging of phosphodiesterase 10A in the brain. J. Med. Chem. 2019, 62, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J.; Kilburn, J.P.; Estrada, S.; Christensen, S.R.; Wall, A.; Thibblin, A.; Lubberink, M.; Bundgaard, C.; Brennum, L.T.; Steiniger-Brach, B.; et al. Discovery and development of 11C-LU AE92686 as a radioligand for PET imaging of phosphodiesterase 10A in the brain. J. Nucl. Med. 2014, 55, 1513–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang-Andersen, B.; Kehler, J. Radiolabelled Phenylimidazole-Based Ligands. WO Patent 2012/062319 A1, 18 May 2012. [Google Scholar]
- Ritzén, A.; Kehler, J.; Langgârd, M.; Nielsen, J.; Kilburn, J.P.; Farah, M.M. Novel Phenylimidazole Derivatives as PDE10A Enzyme Inhibitors. WO Patent 2009/152825 A1, 23 December 2009. [Google Scholar]
- Stepanov, V.; Miura, S.; Takano, A.; Amini, N.; Nakao, R.; Hasui, T.; Nakashima, K.; Taniguchi, T.; Kimura, H.; Kuroita, T.; et al. Development of a series of novel carbon-11 labeled PDE10A inhibitors. J. Labelled Compd. Rad. 2015, 58, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Suzuki, K.; Miura, S.; Hasui, T.; Kamiguchi, N.; Ishii, T.; Taniguchi, T.; Kuroita, T.; Takano, A.; Stepanov, V.; et al. Characterization of the binding properties of T-773 as a PET radioligand for phosphodiesterase 10A. Nucl. Med. Biol. 2015, 42, 146–154. [Google Scholar] [CrossRef]
- Kunitomo, J.; Yoshikawa, M.; Fushimi, M.; Kawada, A.; Quinn, J.F.; Oki, H.; Kokubo, H.; Kondo, M.; Nakashima, K.; Kamiguchi, N.; et al. Discovery of 1-[2-fluoro-4-(1H-pyrazol-1-yl)phenyl]-5-methoxy-3-(1-phenyl-1H-pyrazol-5-yl)pyridazin-4(1H)-one (TAK-063), a highly potent, selective, and orally active phosphodiesterase 10A (PDE10A) inhibitor. J. Med. Chem. 2014, 57, 9627–9643. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Scheunemann, M.; Dipper, K.; Egerland, U.; Hoefgen, N.; Steinbach, J.; Brust, P. Development of highly potent phosphodiesterase 10A (PDE10A) inhibitors: Synthesis and in vitro evaluation of 1,8-dipyridinyl- and 1-pyridinyl-substituted imidazo[1,5-a]quinoxalines. Eur. J. Med. Chem. 2016, 107, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jin, H.; Luo, Z.; Yue, X.; Zhang, X.; Flores, H.; Su, Y.; Perlmutter, J.S.; Tu, Z. In vivo characterization of two 18F-labeled PDE10A PET radioligands in nonhuman primate brains. ACS Chem. Neurosci. 2018, 9, 1066–1073. [Google Scholar] [CrossRef]
- Threlfell, S.; West, A.R. Modulation of striatal neuron activity by cyclic nucleotide signalling and phosphodiesterase inhibition. Basal Ganglia 2013, 3, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Jin, H.; Yue, X.; Han, J.; Yang, H.; Flores, H.; Su, Y.; Alagille, D.; Perlmutter, J.S.; Tamagnan, G.; et al. Comparison of [11C]TZ1964B and [18F]MNI659 for PET imaging brain PDE10A in nonhuman primates. Pharmacol. Res. Perspect. 2016, 4, e00253. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, X.; Li, J.; Jin, H.; Padakanti, P.K.; Jones, L.A.; Flores, H.P.; Su, Y.; Perlmutter, J.S.; Tu, Z. Radiosyntheses and in vivo evaluation of carbon-11 PET tracers for PDE10A in the brain of rodent and nonhuman primate. Bioorgan. Med. Chem. 2014, 22, 2648–2654. [Google Scholar] [CrossRef] [Green Version]
- Barret, O.; Thomae, D.; Alagille, D.; Lee, H.; Papin, C.; Baldwin, R.; Jennings, D.; Marek, K.; Seibyl, J.; Tamagnan, G. First in vivo assessment of two PDE10 tracers [18F]MNI654 and [18F]MNI659. J. Nucl. Med. 2012, 53, 361. [Google Scholar]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early Huntington disease. J. Am. Med. Assoc. Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caille, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P.; et al. Change in PDE10 across early Huntington disease assessed by [18F]MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Mori, W.; Takei, M.; Furutsuka, K.; Fujinaga, M.; Kumata, K.; Muto, M.; Ohkubo, T.; Hashimoto, H.; Tamagnan, G.; Higuchi, M.; et al. Comparison between [18F]fluorination and [18F]fluoroethylation reactions for the synthesis of the PDE10A PET radiotracer [18F]MNI-659. Nucl. Med. Biol. 2017, 55, 12–18. [Google Scholar] [CrossRef]
- Zhang, M.-R.; Tsuchiyama, A.; Haradahira, T.; Yoshida, Y.; Furutsuka, K.; Suzuki, K. Development of an automated system for synthesizing 18F-labeled compounds using [18F]fluoroethyl bromide as a synthetic precursor. Appl. Radiat. Isotopes 2002, 57, 335–342. [Google Scholar] [CrossRef]
- Fazio, P.; Schain, M.; Mrzljak, L.; Amini, N.; Nag, S.; Al-Tawil, N.; Fitzer-Attas, C.J.; Bronzova, J.; Landwehrmeyer, B.; Sampaio, C.; et al. Patterns of age related changes for phosphodiesterase type-10A in comparison with dopamine D2/3 receptors and sub-cortical volumes in the human basal ganglia: A PET study with 18F-MNI-659 and 11C-raclopride with correction for partial volume effect. NeuroImage 2017, 152, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svedberg, M.M.; Varnäs, K.; Varrone, A.; Mitsios, N.; Mulder, J.; Gulyás, B.; Beaumont, V.; Munoz-Sanjuan, I.; Zaleska, M.M.; Schmidt, C.J.; et al. In vitro phosphodiesterase 10A (PDE10A) binding in whole hemisphere human brain using the PET radioligand [18F]MNI-659. Brain Res. 2019, 1711, 140–145. [Google Scholar] [CrossRef]
- Delnomdedieu, M.; Forsberg, A.; Ogden, A.; Fazio, P.; Yu, C.-R.; Stenkrona, P.; Duvvuri, S.; David, W.; Al-Tawil, N.; Vitolo, O.V.; et al. In vivo measurement of PDE10A enzyme occupancy by positron emission tomography (PET) following single oral dose administration of PF-02545920 in healthy male subjects. Neuropharmacology 2017, 117, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, V.; Zhong, S.; Lin, H.; Xu, W.; Bradaia, A.; Steidl, E.; Gleyzes, M.; Wadel, K.; Buisson, B.; Padovan-Neto, F.E.; et al. Phosphodiesterase 10A inhibition improves cortico-basal ganglia function in Huntington’s disease models. Neuron 2016, 92, 1220–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoglio, D.; Verhaeghe, J.; Kosten, L.; Thomae, D.; Van der Linden, A.; Stroobants, S.; Wityak, J.; Dominguez, C.; Mrzljak, L.; Staelens, S. MR-based spatial normalization improves [18F]MNI-659 PET regional quantification and detectability of disease effect in the Q175 mouse model of Huntington’s disease. PLoS ONE 2018, 13, e0206613. [Google Scholar] [CrossRef]
- Fazio, P.; Fitzer-Attas, C.J.; Mrzljak, L.; Bronzova, J.; Nag, S.; Warner, J.H.; Landwehrmeyer, B.; Al-Tawil, N.; Halldin, C.; Forsberg, A.; et al. PET molecular imaging of phosphodiesterase 10A: An early biomarker of Huntington’s disease progression. Mov. Disord. 2020, 35, 606–615. [Google Scholar] [CrossRef]
- Ward, K.M.; Chen, L.; Zhang, L.; Smith, D.; Chappie, T.; Schmidt, C.; Grimwood, S.; O’Connor, R.; Rizzo, S.; Schildknegt, K. Development of novel PET ligands (PF-04831704 and PF-06327104) for PDE10A. Schizophr. Res. 2014, 153, S104–S105. [Google Scholar] [CrossRef]
- Giampà, C.; Laurenti, D.; Anzilotti, S.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS ONE 2010, 5, e13417. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Carretón, O.; Arumí, H.; Tyebji, S.; Alberch, J.; Pérez-Navarro, E. PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus 2013, 23, 684–695. [Google Scholar] [CrossRef]
- Saavedra, A.; Giralt, A.; Arumí, H.; Alberch, J.; Pérez-Navarro, E. Regulation of hippocampal cGMP levels as a candidate to treat cognitive deficits in Huntington’s disease. PLoS ONE 2013, 8, e73664. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.; Niccolini, F.; Haider, S.; Marques, T.R.; Pagano, G.; Coello, C.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of extra-striatal phosphodiesterase 10A expression in early premanifest Huntington’s disease gene carriers. J. Neurol. Sci. 2016, 368, 243–248. [Google Scholar] [CrossRef]
- Niccolini, F.; Haider, S.; Marques, T.R.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Piccini, P.; Kapur, S.; Rabiner, E.A.; et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington’s disease. Brain 2015, 138, 3016–3029. [Google Scholar] [CrossRef] [Green Version]
- Marques, T.R.; Natesan, S.; Niccolini, F.; Politis, M.; Gunn, R.N.; Searle, G.E.; Howes, O.; Rabiner, E.A.; Kapur, S. Phosphodiesterase 10A in schizophrenia: A PET study using [11C]IMA107. Am. J. Psychiat. 2016, 173, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Diggle, C.P.; Sukoff Rizzo, S.J.; Popiolek, M.; Hinttala, R.; Schülke, J.-P.; Kurian, M.A.; Carr, I.M.; Markham, A.F.; Bonthron, D.T.; Watson, C.; et al. Biallelic mutations in PDE10A lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am. J. Hum. Genet. 2016, 98, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccolini, F.; Mencacci, N.E.; Yousaf, T.; Rabiner, E.A.; Salpietro, V.; Pagano, G.; Balint, B.; Efthymiou, S.; Houlden, H.; Gunn, R.N.; et al. PDE10A and ADCY5 mutations linked to molecular and microstructural basal ganglia pathology. Mov. Disord. 2018, 33, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Niccolini, F.; Wilson, H.; Yousaf, T.; Khan, N.L.; Martino, D.; Plisson, C.; Gunn, R.N.; Rabiner, E.A.; Piccini, P.; et al. Comparison of phosphodiesterase 10A and dopamine transporter levels as markers of disease burden in early Parkinson’s disease. Mov. Disord. 2019, 34, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, S.; Gertler, J.; Himes, M.L.; Paris, J.; Kendro, S.; Lopresti, B.; Scott Mason, N.; Narendran, R. Imaging phosphodiesterase-10A availability in cocaine use disorder with [11C]IMA107 and PET. Synapse 2019, 73, e22070. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-W.; Kim, Y.; Kim, A.M.; Helmin, K.; Nairn, A.C.; Greengard, P. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc. Natl. Acad. Sci. USA 2006, 103, 3399–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, R.M.; Smith, Y. Differential striatal spine pathology in Parkinson’s disease and cocaine addiction: A key role of dopamine? Neuroscience 2013, 251, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.M.; Self, D.W. It’s only a matter of time: Longevity of cocaine-induced changes in dendritic spine density in the nucleus accumbens. Curr. Opin. Behav. Sci. 2017, 13, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-C.; Stepanov, V.; Amini, N.; Martinsson, S.; Takano, A.; Nielsen, J.; Bundgaard, C.; Bang-Andersen, B.; Grimwood, S.; Halldin, C.; et al. Characterization of [11C]LU AE92686 as a PET radioligand for phosphodiesterase 10A in the nonhuman primate brain. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 308–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodén, R.; Persson, J.; Wall, A.; Lubberink, M.; Ekselius, L.; Larsson, E.M.; Antoni, G. Striatal phosphodiesterase 10A and medial prefrontal cortical thickness in patients with schizophrenia: A PET and MRI study. Transl. Psychiatry 2017, 7, e1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlenius, S. Clozapine: Dopamine D1 receptor agonism in the prefrontal cortex as the code to decipher a rosetta stone of antipsychotic drugs. Pharmacol. Toxicol. 1999, 84, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Persson, J.; Szalisznyó, K.; Antoni, G.; Wall, A.; Fällmar, D.; Zora, H.; Bodén, R. Phosphodiesterase 10A levels are related to striatal function in schizophrenia: A combined positron emission tomography and functional magnetic resonance imaging study. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 451–459. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Deng, W.; Li, M.; Chen, Z.; Jiang, L.; Wang, Q.; Huang, C.; Collier, D.A.; Gong, Q.; Ma, X.; et al. Aberrant intrinsic brain activity and cognitive deficit in first-episode treatment-naive patients with schizophrenia. Psychol. Med. 2012, 43, 769–780. [Google Scholar] [CrossRef]
- Huang, X.-Q.; Lui, S.; Deng, W.; Chan, R.C.K.; Wu, Q.-Z.; Jiang, L.-J.; Zhang, J.-R.; Jia, Z.-Y.; Li, X.-L.; Li, F.; et al. Localization of cerebral functional deficits in treatment-naive, first-episode schizophrenia using resting-state fMRI. NeuroImage 2010, 49, 2901–2906. [Google Scholar] [CrossRef]
- Li, Z.; Lei, W.; Deng, W.; Zheng, Z.; Li, M.; Ma, X.; Wang, Q.; Huang, C.; Li, N.; Collier, D.A.; et al. Aberrant spontaneous neural activity and correlation with evoked-brain potentials in first-episode, treatment-naïve patients with deficit and non-deficit schizophrenia. Psychiatry Res. Neuroimaging 2017, 261, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A.; Chapin, D.S.; Harms, J.F.; Lebel, L.A.; McCarthy, S.A.; Chambers, L.; Shrikhande, A.; Wong, S.; Menniti, F.S.; Schmidt, C.J. Inhibition of the striatum-enriched phosphodiesterase PDE10A: A novel approach to the treatment of psychosis. Neuropharmacology 2006, 51, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miura, S.; Hasui, T.; Halldin, C.; Stepanov, V.; Takano, A. Radiolabeled Compounds and Their Use as Radiotracers for Quantitative Imaging of Phosphodiesterase (PDE10A) in Mammals. WO Patent 2013/027845 A1, 28 February 2013. [Google Scholar]
- Takano, A.; Stenkrona, P.; Stepanov, V.; Amini, N.; Martinsson, S.; Tsai, M.; Goldsmith, P.; Xie, J.; Wu, J.; Uz, T.; et al. A human [11C]T-773 PET study of PDE10A binding after oral administration of TAK-063, a PDE10A inhibitor. NeuroImage 2016, 141, 10–17. [Google Scholar] [CrossRef]
- Takano, A.; Stepanov, V.; Gulyás, B.; Nakao, R.; Amini, N.; Miura, S.; Kimura, H.; Taniguchi, T.; Halldin, C. Evaluation of a novel PDE10A PET radioligand, [11C]T-773, in nonhuman primates: Brain and whole body PET and brain autoradiography. Synapse 2015, 69, 345–355. [Google Scholar] [CrossRef]
- Takano, A.; Stepanov, V.; Nakao, R.; Amini, N.; Gulyás, B.; Kimura, H.; Halldin, C. Brain PET measurement of PDE10A occupancy by TAK-063, a new PDE10A inhibitor, using [11C]T-773 in nonhuman primates. Synapse 2016, 70, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, A.R.; Rodriguez, C.G.; Toolan, D.M.; Price, O.; Henry, M.; Forrest, G.; Szeto, D.; Keohane, C.A.; Pan, Y.; Smith, K.M.; et al. Genetic deletion and pharmacological inhibition of phosphodiesterase 10A protects mice from diet-induced obesity and insulin resistance. Diabetes 2014, 63, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Kranz, M.; Hankir, M.; Deuther-Conrad, W.; Scheunemann, M.; Teodoro, R.; Wenzel, B.; Fischer, S.; Egerland, U.; Fenske, W.K.; et al. Evaluation of the new radioligand [18F]AQ-28A by small animal PET/MR demonstrates increse of PDE10A expression in striatum and brown adipose tissue (BAT) of obese mice. J. Labelled Compd. Rad. 2015, 58, S52. [Google Scholar]
- Wagner, S.; Teodoro, R.; Deuther-Conrad, W.; Kranz, M.; Scheunemann, M.; Fischer, S.; Wenzel, B.; Egerland, U.; Hoefgen, N.; Steinbach, J.; et al. Radiosynthesis and biological evaluation of the new PDE10A radioligand [18F]AQ28A. J. Labelled Compd. Rad. 2017, 60, 36–48. [Google Scholar] [CrossRef]
- Hankir, M.K.; Kranz, M.; Gnad, T.; Weiner, J.; Wagner, S.; Deuther-Conrad, W.; Bronisch, F.; Steinhoff, K.; Luthardt, J.; Klöting, N.; et al. A novel thermoregulatory role for PDE10A in mouse and human adipocytes. EMBO Mol. Med. 2016, 8, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Kranz, M.; Hankir, M.; Deuther-Conrad, W.; Wagner, S.; Teodoro, R.; Wenzel, B.; Fenske, W.; Brust, P. Preclinical PET/MR: Defining novel roles for phosphodiesterase 10A in brain and brown adipose tissue (BAT) in the regulation of energy homeostasis. J. Nucl. Med. 2016, 57, 405. [Google Scholar]
- Stepanov, V.; Takano, A.; Nakao, R.; Amini, N.; Miura, S.; Hasui, T.; Kimura, H.; Taniguchi, T.; Halldin, C. Development of two fluorine-18 labeled PET radioligands targeting PDE10A and in vivo PET evaluation in nonhuman primates. Nucl. Med. Biol. 2018, 57, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Hou, L.; Xiao, Z.; Che, C.; Li, G.; Zhang, L.; Xu, H.; Liang, S.; Wang, L. Development and preliminary evaluation of an aryl 18F-labeled PET tracer for imaging PDE10A. J. Nucl. Med. 2020, 61, 1040. [Google Scholar]
- Schröder, S.; Lai, T.H.; Toussaint, M.; Kranz, M.; Chovsepian, A.; Shang, Q.; Dukić-Stefanović, S.; Deuther-Conrad, W.; Teodoro, R.; Wenzel, B.; et al. PET imaging of the adenosine A2A receptor in the rotenone-based mouse model of Parkinson’s disease with [18F]FESCH synthesized by a simplified two-step one-pot radiolabeling strategy. Molecules 2020, 25, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phosphodiesterase | Radioligands Review Compound No. (Code No. Given) | References |
|---|---|---|
| PDE1 | [11C]1 ((±)-[11C]PF-04822163) | [10] |
| PDE2A | [18F]2 ([18F]BIT1) | [16,17] |
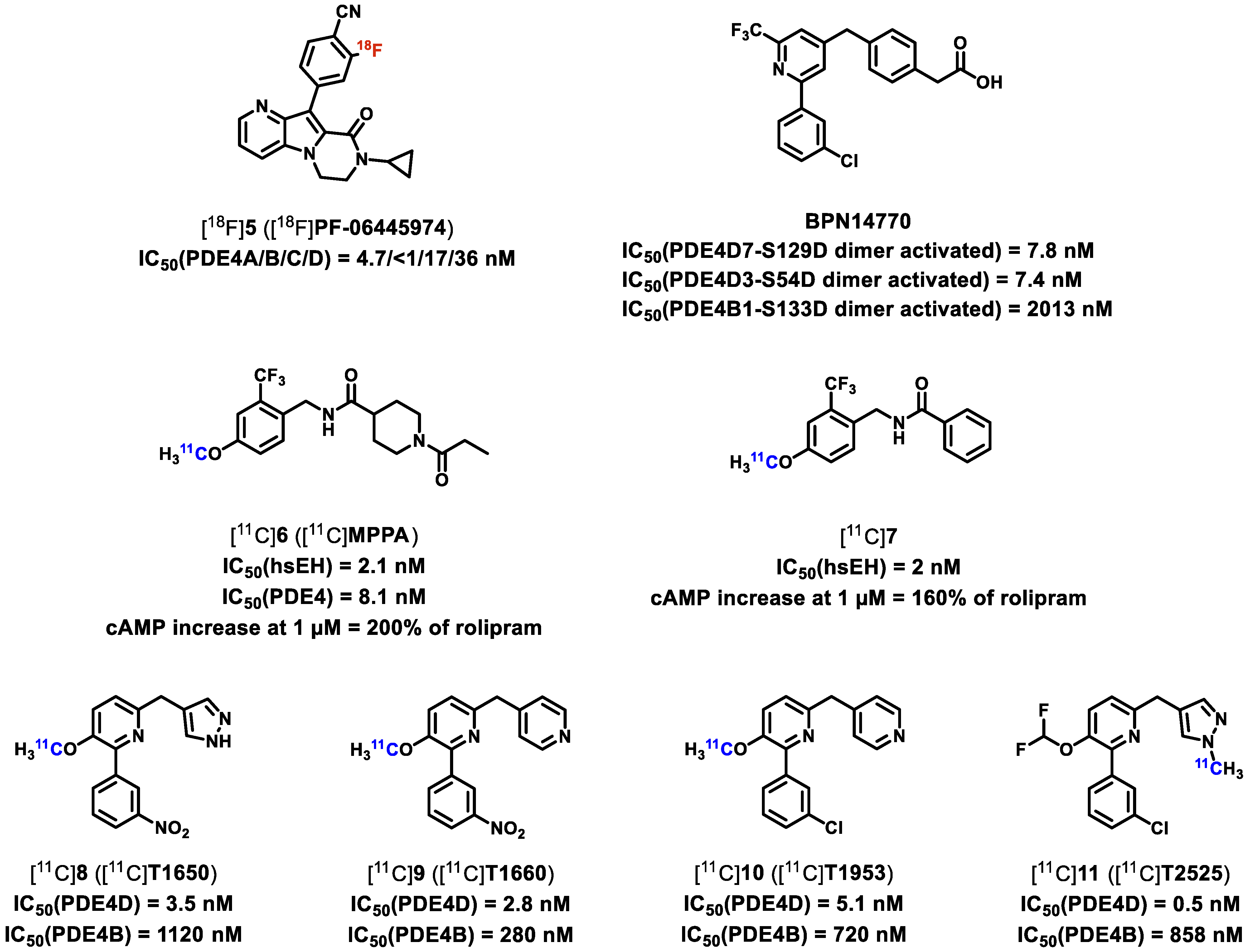
| PDE4B | [18F]5 ([18F]PF-06445974) | [55] |
| PDE4/sEH | [11C]6, [11C]7 | [56] |
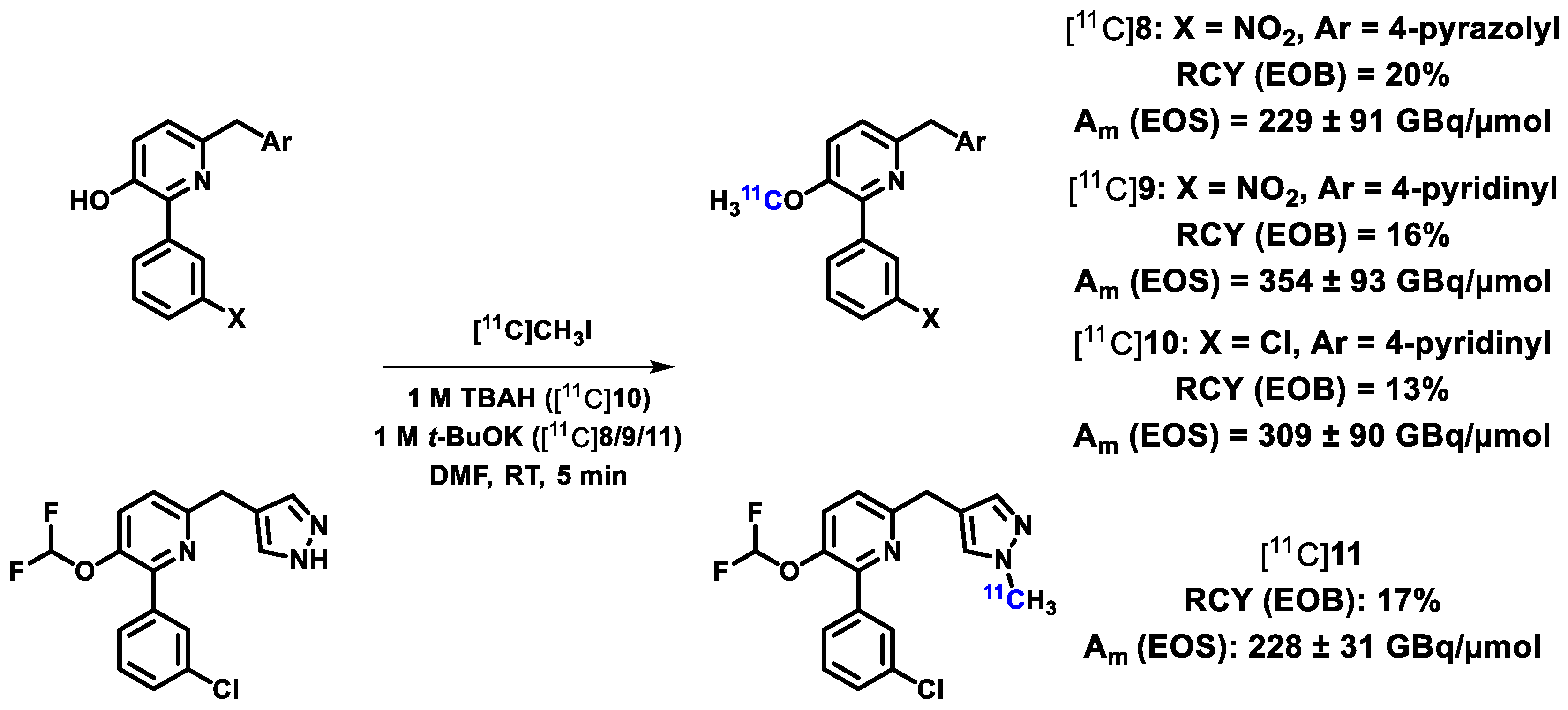
| PDE4D | [11C]8, [11C]9, [11C]10, [11C]11 ([11C]T1650, [11C]T1660, [11C]T1953, [11C]T2525) | [58] |
| [11C]12, [11C]13, [18F]14 (and four more 11C-labeled radioligands) | [66] | |
| PDE5 | [11C]15, [18F]16 | [72] |
| [18F]17 | [73,81] | |
| [11C]19, [11C]20 | [75] | |
| [11C]21 ([11C]TPN171) | [76,77] | |
| PDE7B | [3H]24 | [98] |
| PDE7A/B | [11C]25 ([11C]MTP38) | [99] |
| PDE10A | [18F]35, [18F]36 ([18F]FM-T-773-d2, [18F]FE-T-773-d4) | [177] |
| [18F]37, [18F]38, [18F]39, [18F]40 | [123] | |
| [18F]41 ([18F]P10A-1910) | [178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schröder, S.; Scheunemann, M.; Wenzel, B.; Brust, P. Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016. Int. J. Mol. Sci. 2021, 22, 3832. https://doi.org/10.3390/ijms22083832
Schröder S, Scheunemann M, Wenzel B, Brust P. Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016. International Journal of Molecular Sciences. 2021; 22(8):3832. https://doi.org/10.3390/ijms22083832
Chicago/Turabian StyleSchröder, Susann, Matthias Scheunemann, Barbara Wenzel, and Peter Brust. 2021. "Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016" International Journal of Molecular Sciences 22, no. 8: 3832. https://doi.org/10.3390/ijms22083832
APA StyleSchröder, S., Scheunemann, M., Wenzel, B., & Brust, P. (2021). Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016. International Journal of Molecular Sciences, 22(8), 3832. https://doi.org/10.3390/ijms22083832