
Key Disease Mechanisms Linked to Alzheimer’s Disease in the Entorhinal Cortex


Abstract
:1. Introduction
2. Results
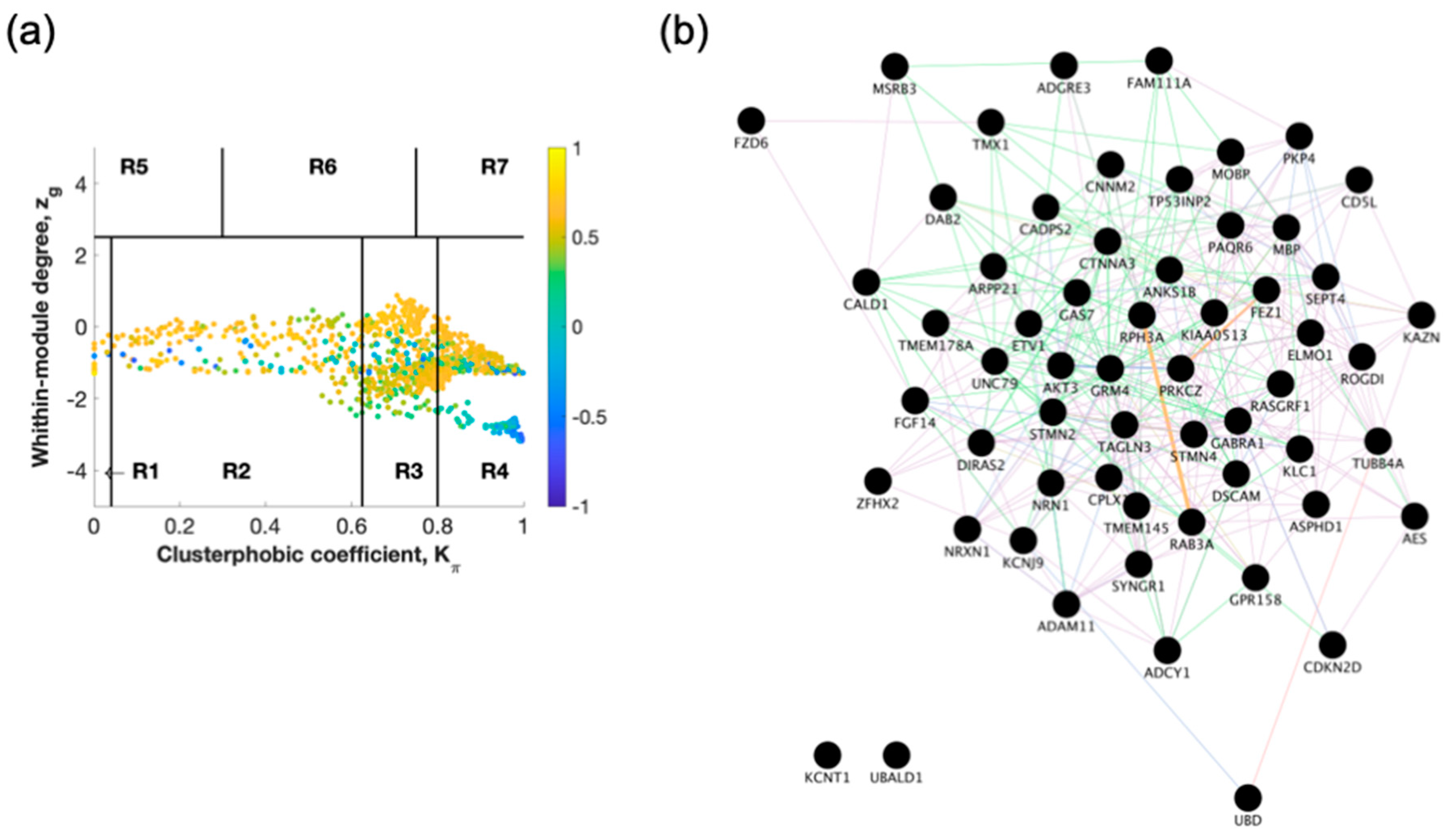
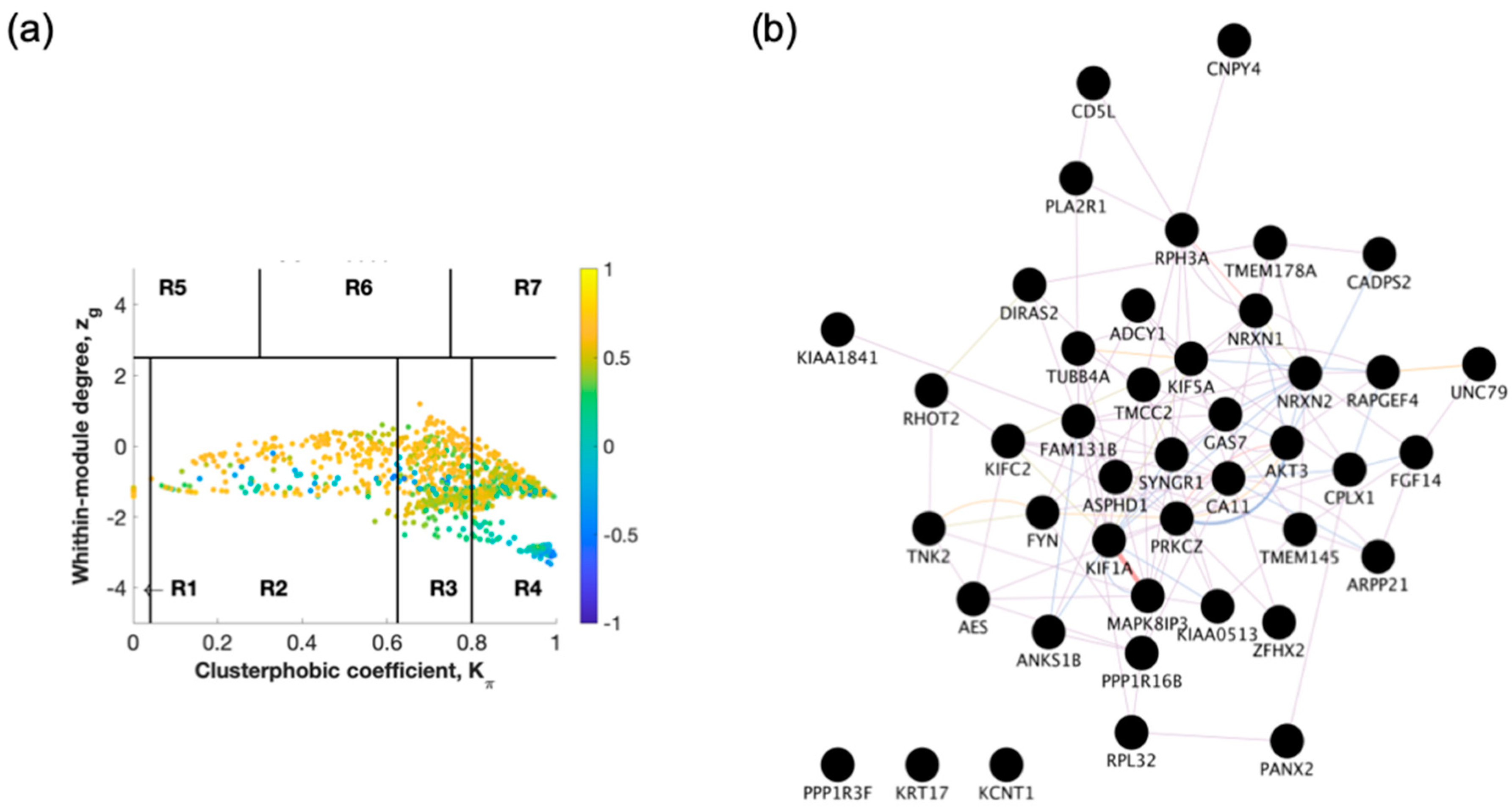
2.1. Identification of Switch Genes for Entorhinal Cortex between AD Versus Healthy or AsymAD
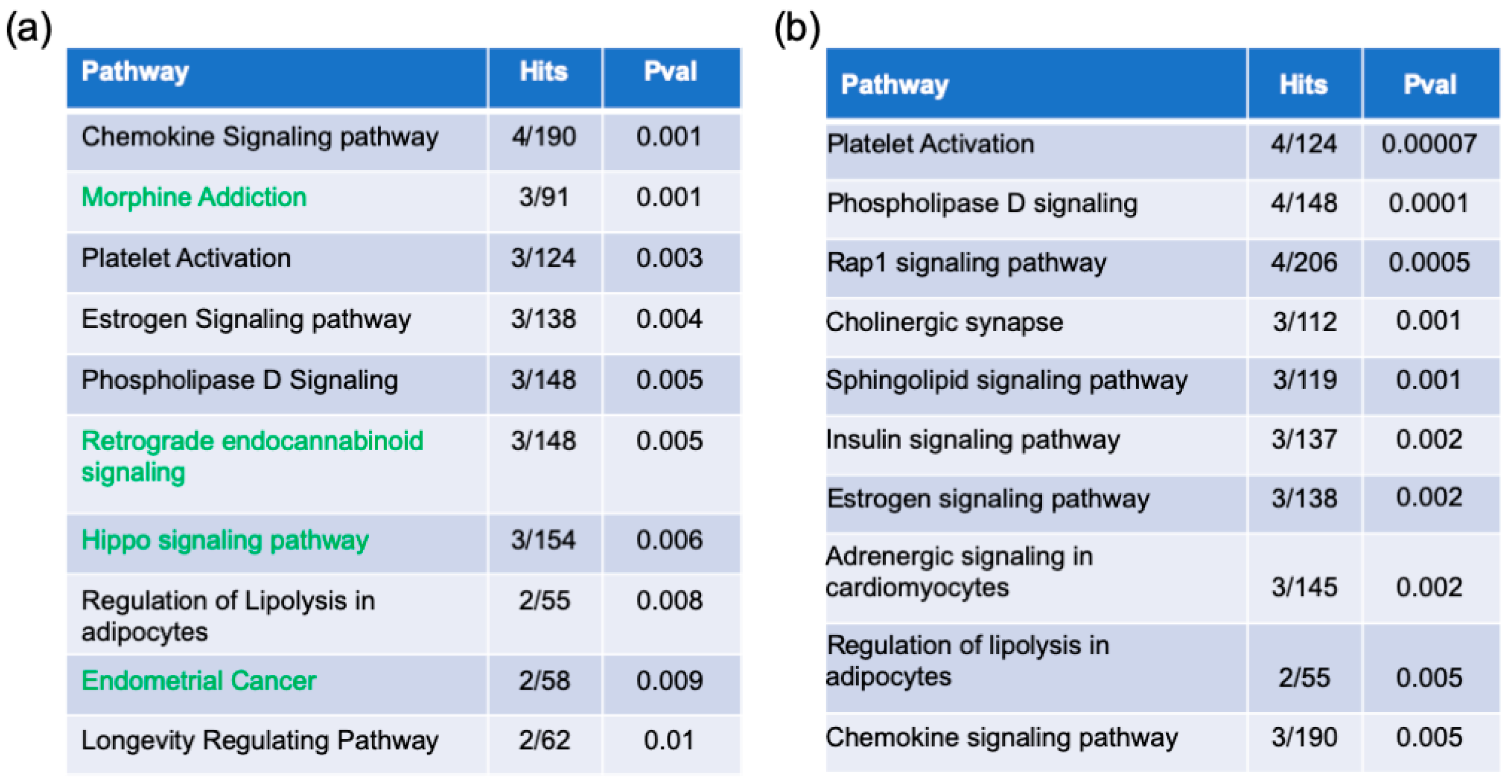
2.2. Pathway Enrichment Analysis
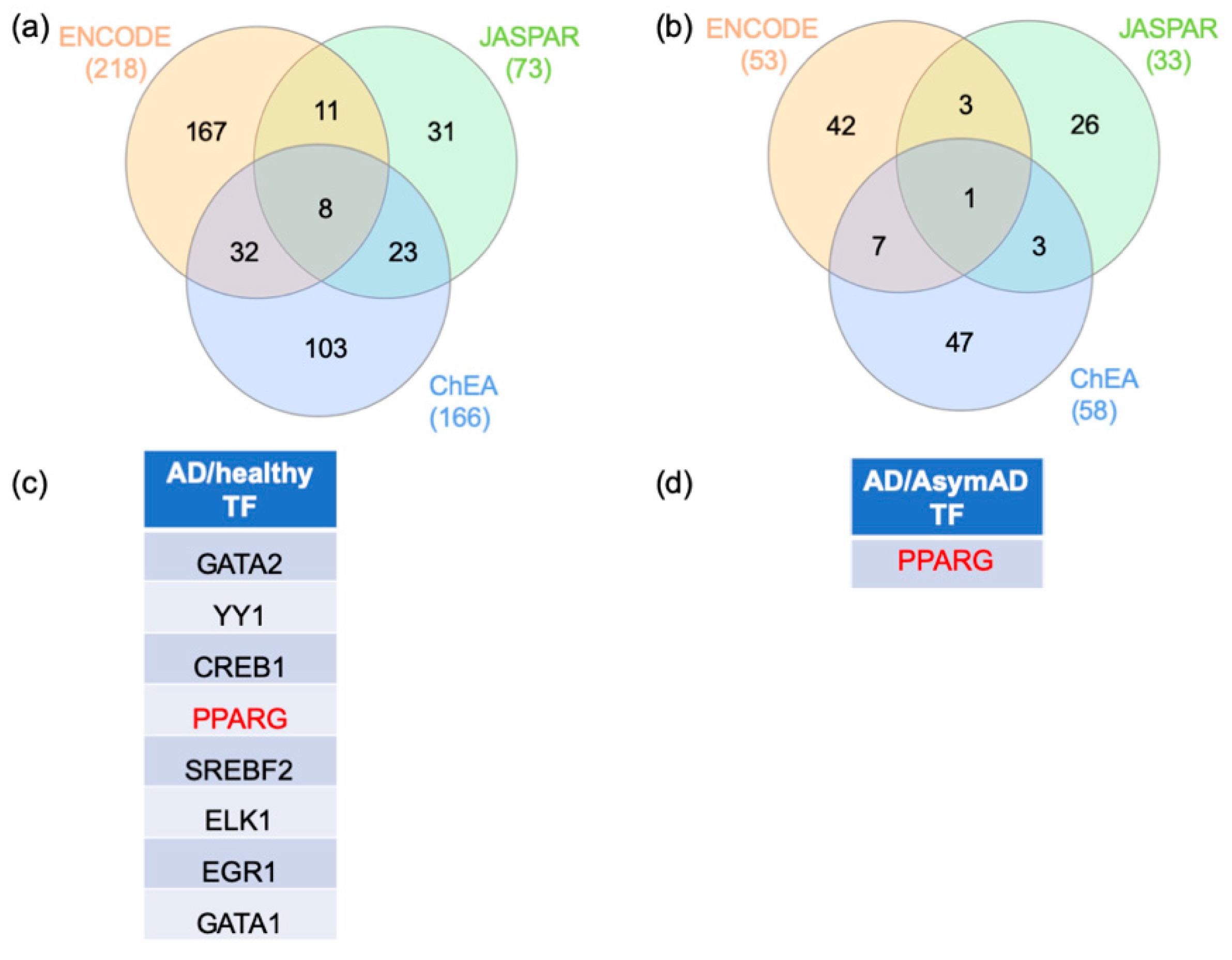
2.3. Gene–Transcription Factor Interaction Analysis
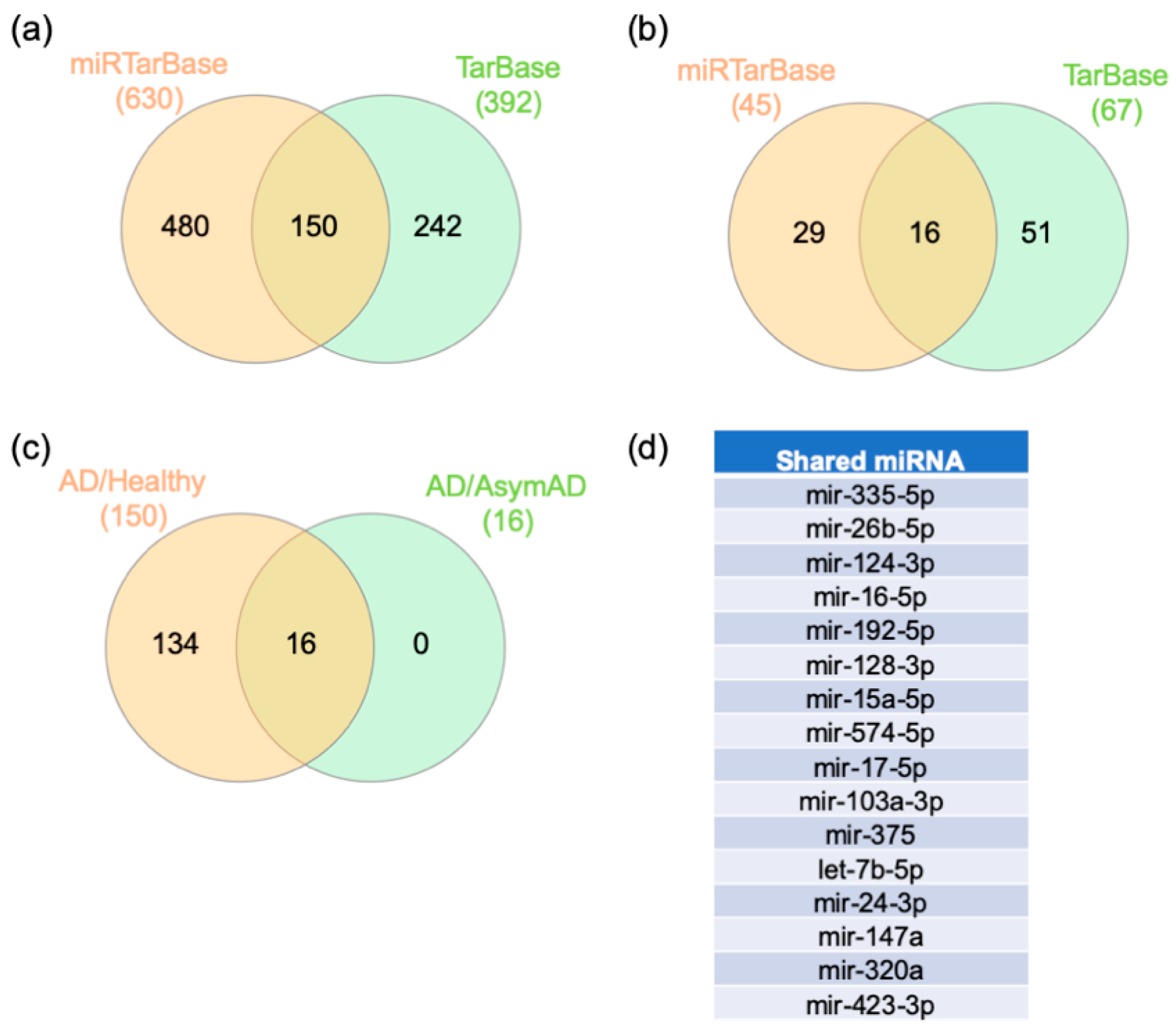
2.4. Gene–miRNA Interaction Analysis
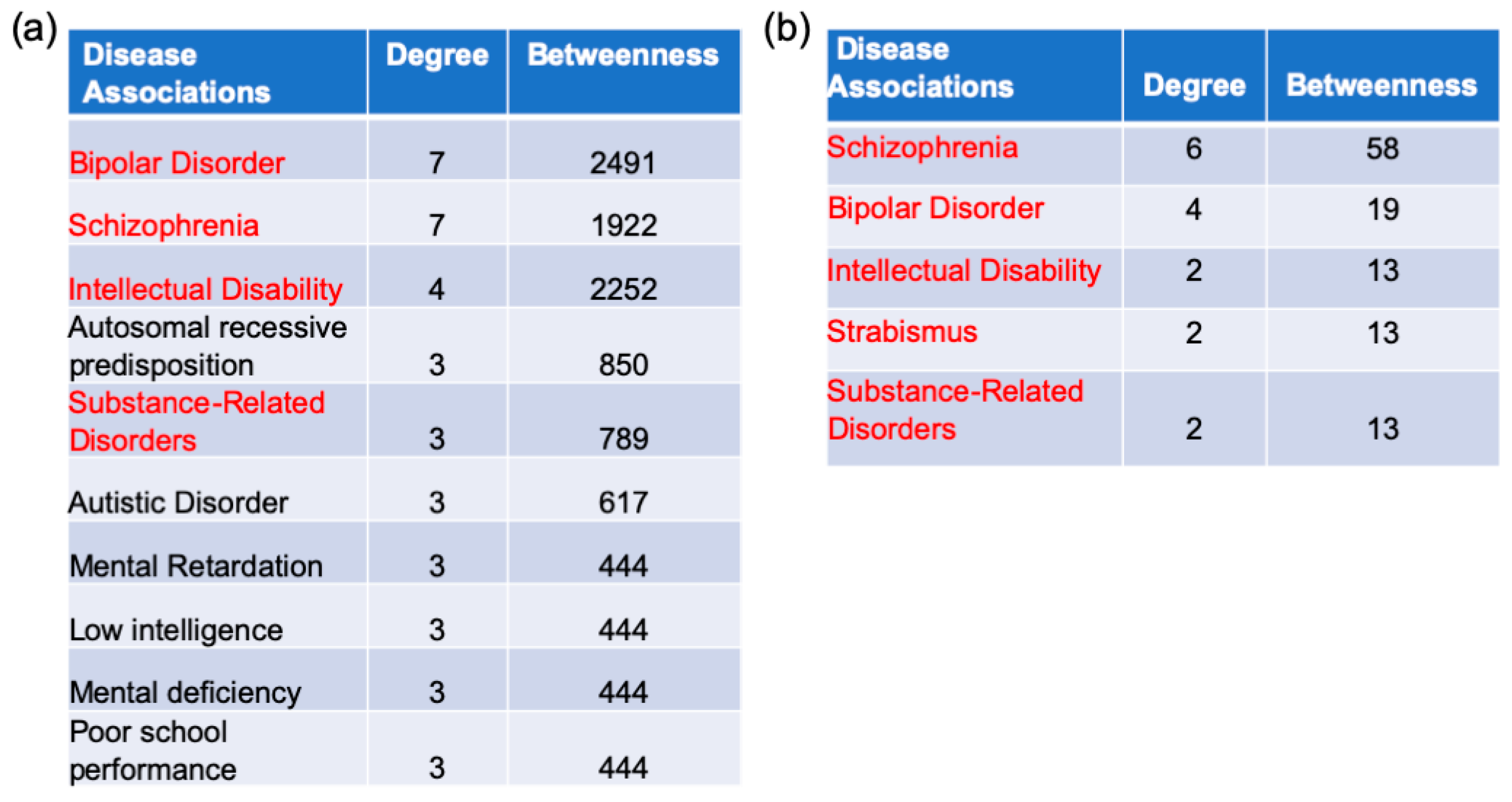
2.5. Gene–Disease Association Analysis
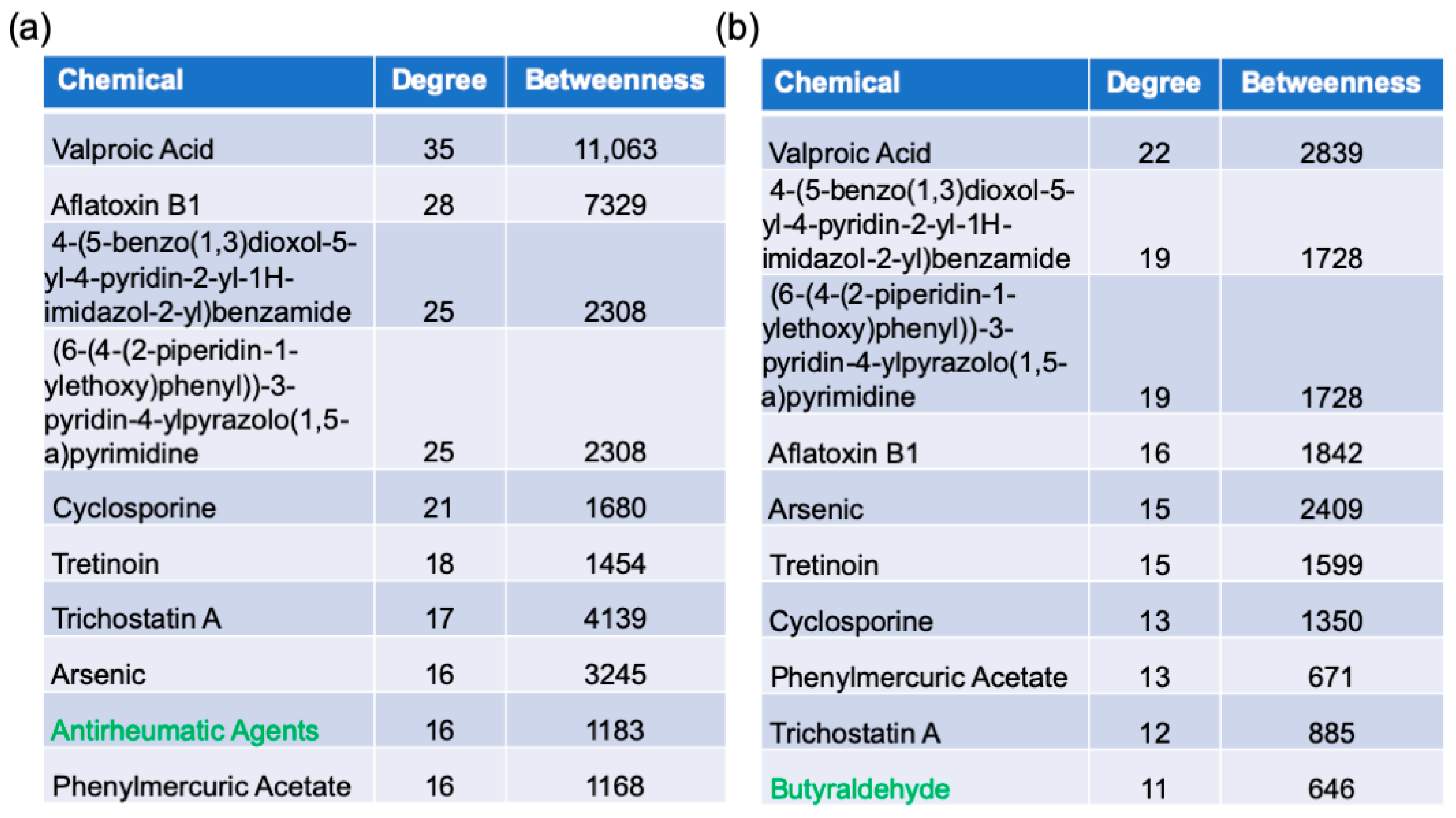
2.6. Protein–Chemical Interaction Analysis
3. Discussion
3.1. Genes
3.2. Pathways
3.3. Transcription Factors
3.4. miRNA
3.5. Disease Association
3.6. Chemicals
3.7. Limitations
4. Materials and Methods
4.1. Data Base Mining, SWIM Analysis to Identify Switch Genes, Switch Gene Analysis
4.2. Pathway Enrichment Analysis
4.3. Gene–Transcription Factor Interaction Analysis
4.4. Gene–miRNA Interaction Analysis
4.5. Gene–Disease Association Analysis
4.6. Gene–Chemical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Contributors, NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Driscoll, I.; Troncoso, J. Asymptomatic Alzheimer’s disease: A prodrome or a state of resilience? Curr. Alzheimer Res. 2011, 8, 330–335. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Kennedy, K.M.; Park, D.C. Beta-amyloid deposition and the aging brain. Neuropsychol. Rev. 2009, 19, 436–450. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Hodges, A.K.; Curtis, C.; Lee, S.H.; Troakes, C.; Dobson, R.J.B.; Newhouse, S.J. Transcriptomic analysis of probable asymptomatic and symptomatic alzheimer brains. Brain Behav. Immun. 2019, 80, 644–656. [Google Scholar] [CrossRef]
- Howett, D.; Castegnaro, A.; Krzywicka, K.; Hagman, J.; Marchment, D.; Henson, R.; Rio, M.; King, J.A.; Burgess, N.; Chan, D. Differentiation of mild cognitive impairment using an entorhinal cortex-based test of virtual reality navigation. Brain 2019, 142, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- Khan, U.A.; Liu, L.; Provenzano, F.A.; Berman, D.E.; Profaci, C.P.; Sloan, R.; Mayeux, R.; Duff, K.E.; Small, S.A. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 2014, 17, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Zhang, F.; Zhao, L.; Qian, J.; Dong, C. Entorhinal cortex: A good biomarker of mild cognitive impairment and mild Alzheimer’s disease. Rev. Neurosci. 2016, 27, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Bevilaqua, L.R.; Rossato, J.I.; Bonini, J.S.; Myskiw, J.C.; Clarke, J.R.; Monteiro, S.; Lima, R.H.; Medina, J.H.; Cammarota, M.; Izquierdo, I. The role of the entorhinal cortex in extinction: Influences of aging. Neural Plast. 2008, 2008, 595282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Walker, D.G.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genom. 2007, 28, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Paci, P.; Colombo, T.; Fiscon, G.; Gurtner, A.; Pavesi, G.; Farina, L. SWIM: A computational tool to unveiling crucial nodes in complex biological networks. Sci. Rep. 2017, 7, 44797. [Google Scholar] [CrossRef] [Green Version]
- Fiscon, G.; Conte, F.; Licursi, V.; Nasi, S.; Paci, P. Computational identification of specific genes for glioblastoma stem-like cells identity. Sci. Rep. 2018, 8, 7769. [Google Scholar] [CrossRef] [PubMed]
- Potashkin, J.A.; Bottero, V.; Santiago, J.A.; Quinn, J.P. Computational identification of key genes that may regulate gene expression reprogramming in Alzheimer’s patients. PLoS ONE 2019, 14, e0222921. [Google Scholar] [CrossRef]
- Potashkin, J.A.; Bottero, V.; Santiago, J.A.; Quinn, J.P. Bioinformatic Analysis Reveals Phosphodiesterase 4D-Interacting Protein as a Key Frontal Cortex Dementia Switch Gene. Int. J. Mol. Sci. 2020, 21, 3787. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Li, H.; Liu, W.; Zhang, L.; Tian, Q.; Li, H.; Li, M. MiR-485-3p serves as a biomarker and therapeutic target of Alzheimer’s disease via regulating neuronal cell viability and neuroinflammation by targeting AKT3. Mol. Genet. Genom. Med. 2020, e1548. [Google Scholar] [CrossRef]
- Robinson, A.; Lubitz, I.; Atrakchi-Baranes, D.; Licht-Murava, A.; Katsel, P.; Leroith, D.; Liraz-Zaltsman, S.; Haroutunian, V.; Beeri, M.S. Combination of Insulin with a GLP1 Agonist Is Associated with Better Memory and Normal Expression of Insulin Receptor Pathway Genes in a Mouse Model of Alzheimer’s Disease. J. Mol. Neurosci. 2019, 67, 504–510. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, X.; Liang, Y.; Zhu, R. Downregulation of microRNA-15b-5p Targeting the Akt3-Mediated GSK-3beta/beta-Catenin Signaling Pathway Inhibits Cell Apoptosis in Parkinson’s Disease. Biomed. Res. Int. 2021, 2021, 8814862. [Google Scholar] [CrossRef]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. Akt3-Mediated Protection Against Inflammatory Demyelinating Disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, C.J.; Weber, H.; Jungwirth, S.; Danielczyk, W.; Reif, A.; Tragl, K.H.; Fischer, P.; Riederer, P.; Deckert, J.; Grunblatt, E. Explorative results from multistep screening for potential genetic risk loci of Alzheimer’s disease in the longitudinal VITA study cohort. J. Neural Transm. 2018, 125, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, E.; Vito, P.; Lopez, P.; Abdallah, M.; D’Adamio, L. The intracellular localization of amyloid beta protein precursor (AbetaPP) intracellular domain associated protein-1 (AIDA-1) is regulated by AbetaPP and alternative splicing. J. Alzheimers Dis. 2004, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, E.; Noviello, C.; D’Adamio, L. Amyloid-beta protein precursor (AbetaPP) intracellular domain-associated protein-1 proteins bind to AbetaPP and modulate its processing in an isoform-specific manner. J. Biol. Chem. 2004, 279, 49105–49112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, E.; Di Luca, M.; Gardoni, F. Synapse-to-nucleus communication: From developmental disorders to Alzheimer’s disease. Curr. Opin. Neurobiol. 2018, 48, 160–166. [Google Scholar] [CrossRef]
- Drew, C.J.; Kyd, R.J.; Morton, A.J. Complexin 1 knockout mice exhibit marked deficits in social behaviours but appear to be cognitively normal. Hum. Mol. Genet. 2007, 16, 2288–2305. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Tasaki, S.; Schneider, J.A.; Arfanakis, K.; Duong, D.M.; Wingo, A.P.; Wingo, T.S.; Kearns, N.; Thatcher, G.R.J.; Seyfried, N.T.; et al. Cortical Proteins Associated With Cognitive Resilience in Community-Dwelling Older Persons. JAMA Psychiatry 2020. [Google Scholar] [CrossRef]
- Ramos-Miguel, A.; Jones, A.A.; Sawada, K.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G. Frontotemporal dysregulation of the SNARE protein interactome is associated with faster cognitive decline in old age. Neurobiol. Dis. 2018, 114, 31–44. [Google Scholar] [CrossRef]
- Shimizu, T.; Hirose, K.; Uchida, C.; Uchida, T. Growth arrest specific protein 7 inhibits tau fibrillogenesis. Biochem. Biophys. Res. Commun. 2020, 526, 281–286. [Google Scholar] [CrossRef]
- Hidaka, M.; Koga, T.; Gotoh, A.; Sanada, M.; Hirose, K.; Uchida, T. Alzheimer’s disease-related protein hGas7b interferes with kinesin motility. J. Biochem. 2012, 151, 593–598. [Google Scholar] [CrossRef]
- Akiyama, H.; Gotoh, A.; Shin, R.W.; Koga, T.; Ohashi, T.; Sakamoto, W.; Harada, A.; Arai, H.; Sawa, A.; Uchida, C.; et al. A novel role for hGas7b in microtubular maintenance: Possible implication in tau-associated pathology in Alzheimer disease. J. Biol. Chem. 2009, 284, 32695–32699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Re, J.; Wadsworth, P.A.; Laezza, F. Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front. Cell Neurosci. 2017, 11, 103. [Google Scholar] [CrossRef]
- Sowers, M.L.; Re, J.D.; Wadsworth, P.A.; Shavkunov, A.S.; Lichti, C.; Zhang, K.; Laezza, F. Sex-Specific Proteomic Changes Induced by Genetic Deletion of Fibroblast Growth Factor 14 (FGF14), a Regulator of Neuronal Ion Channels. Proteomes 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, W.J.; Wildburger, N.C.; Haidacher, S.J.; Nenov, M.N.; Folorunso, O.; Singh, A.K.; Chesson, B.C.; Franklin, W.F.; Cortez, I.; Sadygov, R.G.; et al. PPARgamma agonists rescue increased phosphorylation of FGF14 at S226 in the Tg2576 mouse model of Alzheimer’s disease. Exp. Neurol. 2017, 295, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Sindi, I.A.; Tannenberg, R.K.; Dodd, P.R. Role for the neurexin-neuroligin complex in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.K.; Khaled, H.; Chofflet, N.; Takahashi, H. Synaptic Organizers in Alzheimer’s Disease: A Classification Based on Amyloid-beta Sensitivity. Front Cell Neurosci. 2020, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Mir, A.; Gonzalez-Perez, A.; Gayan, J.; Antunez, C.; Marin, J.; Boada, M.; Lopez-Arrieta, J.M.; Fernandez, E.; Ramirez-Lorca, R.; Saez, M.E.; et al. Genetic study of neurexin and neuroligin genes in Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Servian-Morilla, E.; Scholl, F.G. Presenilin/gamma-secretase regulates neurexin processing at synapses. PLoS ONE 2011, 6, e19430. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.; da Cruz, E.S.O.A.B.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.U.; Woodling, N.S.; Wang, Q.; Panchal, M.; Liang, X.; Trueba-Saiz, A.; Brown, H.D.; Mhatre, S.D.; Loui, T.; Andreasson, K.I. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Investig. 2015, 125, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Krauthausen, M.; Kummer, M.P.; Zimmermann, J.; Reyes-Irisarri, E.; Terwel, D.; Bulic, B.; Heneka, M.T.; Muller, M. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. J. Clin. Investig. 2015, 125, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Purandare, N.; Burns, A.; Daly, K.J.; Hardicre, J.; Morris, J.; Macfarlane, G.; McCollum, C. Cerebral emboli as a potential cause of Alzheimer’s disease and vascular dementia: Case-control study. BMJ 2006, 332, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wong, A.; Wang, Z.; Liu, W.; Au, L.; Xiong, Y.; Chu, W.W.; Leung, E.Y.; Chen, S.; Lau, C.; et al. Risk factors for incident dementia after stroke and transient ischemic attack. Alzheimers Dement. 2015, 11, 16–23. [Google Scholar] [CrossRef]
- Koc, E.R.; Uzar, E.; Cirak, Y.; Parlak Demir, Y.; Ilhan, A. The increase of mean platelet volume in patients with Alzheimer disease. Turk. J. Med. Sci. 2014, 44, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.C.; Jin, D.; Li, Y.; Wang, R.T. Mean platelet volume and platelet distribution width in vascular dementia and Alzheimer’s disease. Platelets 2014, 25, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.T.; Jin, D.; Li, Y.; Liang, Q.C. Decreased mean platelet volume and platelet distribution width are associated with mild cognitive impairment and Alzheimer’s disease. J. Psychiatr. Res. 2013, 47, 644–649. [Google Scholar] [CrossRef]
- Inyushin, M.; Zayas-Santiago, A.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Platelet-generated amyloid beta peptides in Alzheimer’s disease and glaucoma. Histol. Histopathol. 2019, 34, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Li, Q.X. Platelets and Alzheimer’s disease: Potential of APP as a biomarker. World J. Psychiatry 2012, 2, 102–113. [Google Scholar] [CrossRef]
- Rosenberg, R.N.; Baskin, F.; Fosmire, J.A.; Risser, R.; Adams, P.; Svetlik, D.; Honig, L.S.; Cullum, C.M.; Weiner, M.F. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol. 1997, 54, 139–144. [Google Scholar] [CrossRef]
- Baskin, F.; Rosenberg, R.N.; Iyer, L.; Hynan, L.; Cullum, C.M. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology 2000, 54, 1907–1909. [Google Scholar] [CrossRef]
- Bruntz, R.C.; Lindsley, C.W.; Brown, H.A. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol. Rev. 2014, 66, 1033–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohman, M.A. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol. Sci. 2015, 36, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Cruchaga, C.; Karch, C.M.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.; Bertelsen, S.; et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, T.G.; Di Paolo, G. Phospholipase D in brain function and Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, B.; Kayed, R.; Taglialatela, G. Elevated phospholipase D isoform 1 in Alzheimer’s disease patients’ hippocampus: Relevance to synaptic dysfunction and memory deficits. Alzheimers Dement. 2018, 4, 89–102. [Google Scholar] [CrossRef]
- Bourne, K.Z.; Natarajan, C.; Perez, C.X.M.; Tumurbaatar, B.; Taglialatela, G.; Krishnan, B. Suppressing aberrant phospholipase D1 signaling in 3xTg Alzheimer’s disease mouse model promotes synaptic resilience. Sci. Rep. 2019, 9, 18342. [Google Scholar] [CrossRef]
- Oliveira, T.G.; Chan, R.B.; Tian, H.; Laredo, M.; Shui, G.; Staniszewski, A.; Zhang, H.; Wang, L.; Kim, T.W.; Duff, K.E.; et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J. Neurosci. 2010, 30, 16419–16428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, A.I.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Brain insulin signalling, glucose metabolism and females’ reproductive aging: A dangerous triad in Alzheimer’s disease. Neuropharmacology 2018, 136, 223–242. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasoohi, S.; Parveen, K.; Ishrat, T. Metabolic Syndrome, Brain Insulin Resistance, and Alzheimer’s Disease: Thioredoxin Interacting Protein (TXNIP) and Inflammasome as Core Amplifiers. J. Alzheimers Dis. 2018, 66, 857–885. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.J.; Chen, Y.M.; Hewson, E.G.; Richter, J.E.; Dalton, C.B.; Gelfand, D.W.; Wu, W.C. Esophageal motility: Assessment with synchronous video tape fluoroscopy and manometry. Radiology 1989, 173, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sarruf, D.A.; Talukdar, S.; Sharma, S.; Li, P.; Bandyopadhyay, G.; Nalbandian, S.; Fan, W.; Gayen, J.R.; Mahata, S.K.; et al. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat. Med. 2011, 17, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other Demen. 2009, 24, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guan, L.; Luo, D.; Liu, J.; Lin, H.; Li, X.; Liu, X. Gene- Gene Interaction between PPARG and APOE Gene on Late-Onset Alzheimer’s Disease: A Case- Control Study in Chinese Han Population. J. Nutr. Health Aging 2017, 21, 397–403. [Google Scholar] [CrossRef] [PubMed]
- West, N.A.; Tingle, J.V.; Simino, J.; Selvin, E.; Bressler, J.; Mosley, T.H., Jr. The PPARG Pro12Ala Polymorphism and 20-year Cognitive Decline: Race and Sex Heterogeneity. Alzheimer Dis. Assoc. Disord. 2018, 32, 131–136. [Google Scholar] [CrossRef]
- Helisalmi, S.; Tarvainen, T.; Vepsalainen, S.; Koivisto, A.M.; Hiltunen, M.; Soininen, H. Lack of genetic association between PPARG gene polymorphisms and Finnish late-onset Alzheimer’s disease. Neurosci. Lett. 2008, 441, 233–236. [Google Scholar] [CrossRef]
- Chen, Z.S.; Chan, H.Y.E. Transcriptional dysregulation in neurodegenerative diseases: Who tipped the balance of Yin Yang 1 in the brain? Neural Regen. Res. 2019, 14, 1148–1151. [Google Scholar] [CrossRef]
- Nowak, K.; Lange-Dohna, C.; Zeitschel, U.; Gunther, A.; Luscher, B.; Robitzki, A.; Perez-Polo, R.; Rossner, S. The transcription factor Yin Yang 1 is an activator of BACE1 expression. J. Neurochem. 2006, 96, 1696–1707. [Google Scholar] [CrossRef]
- Chen, Z.S.; Li, L.; Peng, S.; Chen, F.M.; Zhang, Q.; An, Y.; Lin, X.; Li, W.; Koon, A.C.; Chan, T.F.; et al. Planar cell polarity gene Fuz triggers apoptosis in neurodegenerative disease models. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Qin, W.; Jia, L.; Zhou, A.; Zuo, X.; Cheng, Z.; Wang, F.; Shi, F.; Jia, J. The -980C/G polymorphism in APH-1A promoter confers risk of Alzheimer’s disease. Aging Cell 2011, 10, 711–719. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, C.; Julien, C.; Frappier, J.; Miron, J.; Theroux, L.; Dea, D.; United Kingdom Brain Expression, C.; for the Alzheimer’s Disease Neuroimaging, I.; Breitner, J.C.S.; Poirier, J. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 66, 180.e181–180.e189. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, F.; Shen, K.; Wang, W.; Siedlak, S.L.; Lee, H.G.; Phelix, C.F.; Perry, G.; Shen, L.; Tang, B.; et al. The sterol regulatory element-binding protein 2 is dysregulated by tau alterations in Alzheimer disease. Brain Pathol. 2019, 29, 530–543. [Google Scholar] [CrossRef]
- Barbero-Camps, E.; Fernandez, A.; Martinez, L.; Fernandez-Checa, J.C.; Colell, A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; de Dios, C.; Fernandez-Checa, J.C.; Mari, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 2018, 14, 1129–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duclot, F.; Kabbaj, M. The Role of Early Growth Response 1 (EGR1) in Brain Plasticity and Neuropsychiatric Disorders. Front. Behav. Neurosci. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanke, V.; Moolamalla, S.T.R.; Roy, D.; Vinod, P.K. Integrative Analysis of Hippocampus Gene Expression Profiles Identifies Network Alterations in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.T.; Chen, X.L.; Huang, S.H.; Zhu, Q.B.; Yu, S.Y.; Shen, Y.; Sluiter, A.; Verhaagen, J.; Zhao, J.; Swaab, D.; et al. Early growth response-1 regulates acetylcholinesterase and its relation with the course of Alzheimer’s disease. Brain Pathol. 2019, 29, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.B.; Unmehopa, U.; Bossers, K.; Hu, Y.T.; Verwer, R.; Balesar, R.; Zhao, J.; Bao, A.M.; Swaab, D. MicroRNA-132 and early growth response-1 in nucleus basalis of Meynert during the course of Alzheimer’s disease. Brain 2016, 139, 908–921. [Google Scholar] [CrossRef]
- Chang, W.S.; Wang, Y.H.; Zhu, X.T.; Wu, C.J. Genome-Wide Profiling of miRNA and mRNA Expression in Alzheimer’s Disease. Med. Sci. Monit 2017, 23, 2721–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Qi, Y.; Liu, C.F.; Lu, J.M.; Shi, J.; Shi, Y. MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer’s disease. J. Gene Med. 2018, 20, e3014. [Google Scholar] [CrossRef]
- Dursun, E.; Candas, E.; Yilmazer, S.; Gezen-Ak, D. Amyloid Beta 1-42 Alters the Expression of miRNAs in Cortical Neurons. J. Mol. Neurosci. 2019, 67, 181–192. [Google Scholar] [CrossRef]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3beta pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [Google Scholar] [CrossRef]
- Zhou, Y.; Deng, J.; Chu, X.; Zhao, Y.; Guo, Y. Role of Post-Transcriptional Control of Calpain by miR-124-3p in the Development of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Guo, M.; Hu, T.; Li, W.; Huang, S.; Yin, Z.; Li, Y.; Chen, F.; Zhu, L.; Kang, C.; et al. Increased Microglial Exosomal miR-124-3p Alleviates Neurodegeneration and Improves Cognitive Outcome after rmTBI. Mol. Ther. 2020, 28, 503–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, S.H.; Park, Y.; Park, J.; Lee, J.H.; Kim, B.C.; Song, W.K. miR-16-5p is upregulated by amyloid beta deposition in Alzheimer’s disease models and induces neuronal cell apoptosis through direct targeting and suppression of BCL-2. Exp. Gerontol. 2020, 136, 110954. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, C.; Venturin, M.; Sojic, A.; Jesuthasan, N.; Orro, A.; Spinelli, R.; Musicco, M.; De Bellis, G.; Adorni, F. Candidate Genes and MiRNAs Linked to the Inverse Relationship Between Cancer and Alzheimer’s Disease: Insights From Data Mining and Enrichment Analysis. Front. Genet. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Hilty, D.M.; Leamon, M.H.; Lim, R.F.; Kelly, R.H.; Hales, R.E. A review of bipolar disorder in adults. Psychiatry 2006, 3, 43–55. [Google Scholar] [PubMed]
- Karabulut, S.; Tasdemir, I.; Akcan, U.; Kucukali, C.I.; Tuzun, E.; Cakir, S. Inflammation and Neurodegeneration in Patients with Early-Stageand Chronic Bipolar Disorder. Turk. Psikiyatri Derg. 2019, 30, 75–81. [Google Scholar] [PubMed]
- Naserkhaki, R.; Zamanzadeh, S.; Baharvand, H.; Nabavi, S.M.; Pakdaman, H.; Shahbazi, S.; Vosough, M.; Ghaedi, G.; Barzegar, A.; Mirtorabi, D.; et al. cis pT231-Tau Drives Neurodegeneration in Bipolar Disorder. ACS Chem. Neurosci. 2019, 10, 1214–1221. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Appukuttan, A.T.; Moon, S.; Duce, J.A.; Volitakis, I.; Cherny, R.; Wood, S.J.; Greenough, M.; Berger, G.; et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry 2017, 22, 396–406. [Google Scholar] [CrossRef]
- Prestia, A.; Boccardi, M.; Galluzzi, S.; Cavedo, E.; Adorni, A.; Soricelli, A.; Bonetti, M.; Geroldi, C.; Giannakopoulos, P.; Thompson, P.; et al. Hippocampal and amygdalar volume changes in elderly patients with Alzheimer’s disease and schizophrenia. Psychiatry Res. 2011, 192, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Religa, D.; Laudon, H.; Styczynska, M.; Winblad, B.; Naslund, J.; Haroutunian, V. Amyloid beta pathology in Alzheimer’s disease and schizophrenia. Am. J. Psychiatry 2003, 160, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt signaling: Role in Alzheimer disease and schizophrenia. J. Neuroimmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Ruthirakuhan, M.T.; Herrmann, N.; Abraham, E.H.; Chan, S.; Lanctot, K.L. Pharmacological interventions for apathy in Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 5, CD012197. [Google Scholar] [CrossRef]
- Diniz, B.S.; Teixeira, A.L.; Cao, F.; Gildengers, A.; Soares, J.C.; Butters, M.A.; Reynolds, C.F., 3rd. History of Bipolar Disorder and the Risk of Dementia: A Systematic Review and Meta-Analysis. Am. J. Geriatr. Psychiatry 2017, 25, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Venkataramani, V.; Rossner, C.; Iffland, L.; Schweyer, S.; Tamboli, I.Y.; Walter, J.; Wirths, O.; Bayer, T.A. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J. Biol. Chem. 2010, 285, 10678–10689. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhu, L.; Guo, X. Valproic acid attenuates Abeta25-35-induced neurotoxicity in PC12 cells through suppression of mitochondria-mediated apoptotic pathway. Biomed. Pharmacother. 2018, 106, 77–82. [Google Scholar] [CrossRef]
- Xuan, A.G.; Pan, X.B.; Wei, P.; Ji, W.D.; Zhang, W.J.; Liu, J.H.; Hong, L.P.; Chen, W.L.; Long, D.H. Valproic acid alleviates memory deficits and attenuates amyloid-beta deposition in transgenic mouse model of Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Liu, Y.J.; Zhang, W.; Luo, S.F.; Zhou, X.; He, G.Q. Combined treatment with valproic acid and estrogen has neuroprotective effects in ovariectomized mice with Alzheimer’s disease. Neural Regen. Res. 2021, 16, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Truran, D.; Mai, J.T.; Langbaum, J.B.; Aisen, P.S.; Cummings, J.L.; Jack, C.R., Jr.; Weiner, M.W.; Thomas, R.G.; Schneider, L.S.; et al. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 2011, 77, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Chou, R.C.; Kane, M.; Ghimire, S.; Gautam, S.; Gui, J. Treatment for Rheumatoid Arthritis and Risk of Alzheimer’s Disease: A Nested Case-Control Analysis. CNS Drugs 2016, 30, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimers Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Han, S.B.; Nam, S.Y.; Oh, K.W.; Hong, J.T. Inflammation and Alzheimer’s disease. Arch Pharm. Res. 2010, 33, 1539–1556. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Jana, M.; Pahan, K. Aspirin Induces Lysosomal Biogenesis and Attenuates Amyloid Plaque Pathology in a Mouse Model of Alzheimer’s Disease via PPARalpha. J. Neurosci. 2018, 38, 6682–6699. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimers Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Ronowska, A.; Pawelczyk, T. Retinoic acid as a therapeutic option in Alzheimer’s disease: A focus on cholinergic restoration. Expert Rev. Neurother. 2015, 15, 239–249. [Google Scholar] [CrossRef]
- Yang, Y.W.; Liou, S.H.; Hsueh, Y.M.; Lyu, W.S.; Liu, C.S.; Liu, H.J.; Chung, M.C.; Hung, P.H.; Chung, C.J. Risk of Alzheimer’s disease with metal concentrations in whole blood and urine: A case-control study using propensity score matching. Toxicol. Appl. Pharmacol. 2018, 356, 8–14. [Google Scholar] [CrossRef]
- Gong, G.; O’Bryant, S.E. The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 311–316. [Google Scholar] [CrossRef]
- Nino, S.A.; Martel-Gallegos, G.; Castro-Zavala, A.; Ortega-Berlanga, B.; Delgado, J.M.; Hernandez-Mendoza, H.; Romero-Guzman, E.; Rios-Lugo, J.; Rosales-Mendoza, S.; Jimenez-Capdeville, M.E.; et al. Chronic Arsenic Exposure Increases Abeta(1-42) Production and Receptor for Advanced Glycation End Products Expression in Rat Brain. Chem. Res. Toxicol. 2018, 31, 13–21. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, H.; Ma, J.; Zang, C.; Wang, C.; Wang, J.; Tang, Q.; Meyer, C.A.; Zhang, Y.; Liu, X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013, 8, 2502–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranasic, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Pinero, J.; Ramirez-Anguita, J.M.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattingly, C.J.; Colby, G.T.; Forrest, J.N.; Boyer, J.L. The Comparative Toxicogenomics Database (CTD). Environ. Health Perspect. 2003, 111, 793–795. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | AsymAD | AD | |
|---|---|---|---|
| Sample number | 16 | 28 | 34 |
| Age (±SD) | 71.9 (±15.6) | 85.4 (±9.5) | 83.9 (±9.7) |
| Sex (M/F) | 9/7 | 8/20 | 13/21 |
| BRAAK (±SD) | 0 | 2.2 (±1.2) | 4.9 (±1) |
| Disease duration (y) | 0 | 0 | 11.8 (5.2) |
| Post-mortem delay | 33.8 (17.8) | 52.5 (15.9) | 39.5 (21.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottero, V.; Powers, D.; Yalamanchi, A.; Quinn, J.P.; Potashkin, J.A. Key Disease Mechanisms Linked to Alzheimer’s Disease in the Entorhinal Cortex. Int. J. Mol. Sci. 2021, 22, 3915. https://doi.org/10.3390/ijms22083915
Bottero V, Powers D, Yalamanchi A, Quinn JP, Potashkin JA. Key Disease Mechanisms Linked to Alzheimer’s Disease in the Entorhinal Cortex. International Journal of Molecular Sciences. 2021; 22(8):3915. https://doi.org/10.3390/ijms22083915
Chicago/Turabian StyleBottero, Virginie, Dallen Powers, Ashna Yalamanchi, James P. Quinn, and Judith A. Potashkin. 2021. "Key Disease Mechanisms Linked to Alzheimer’s Disease in the Entorhinal Cortex" International Journal of Molecular Sciences 22, no. 8: 3915. https://doi.org/10.3390/ijms22083915
APA StyleBottero, V., Powers, D., Yalamanchi, A., Quinn, J. P., & Potashkin, J. A. (2021). Key Disease Mechanisms Linked to Alzheimer’s Disease in the Entorhinal Cortex. International Journal of Molecular Sciences, 22(8), 3915. https://doi.org/10.3390/ijms22083915