
Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
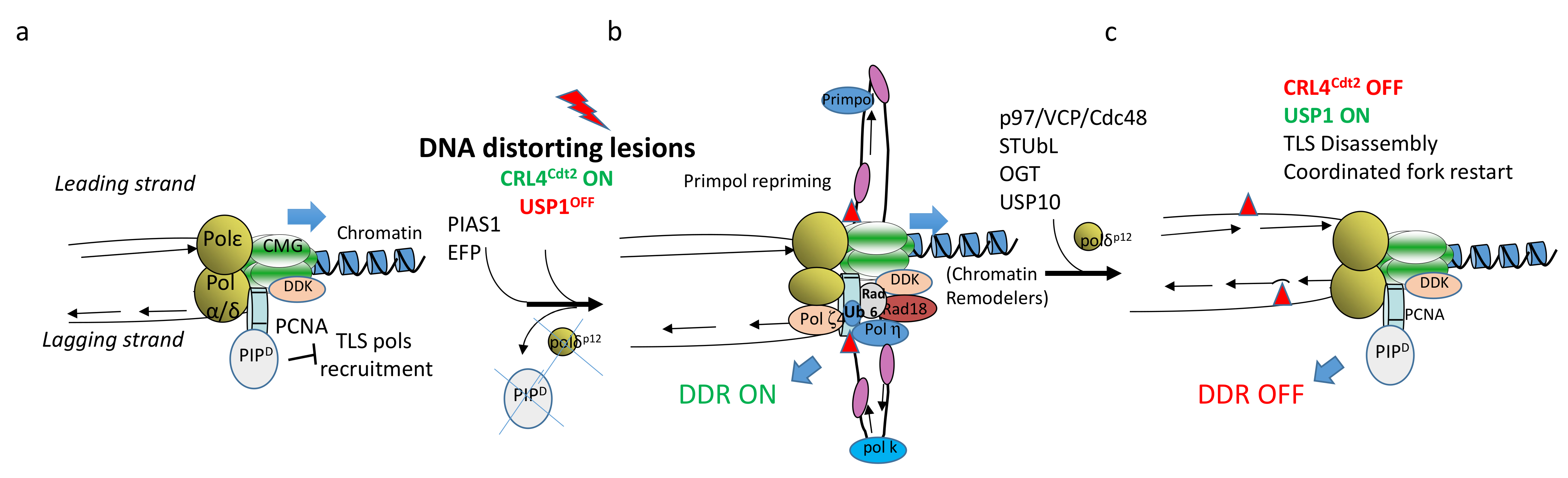
2. The Crucial Role of TLS to Tolerate Structural Impediments during DNA Replication
2.1. TLS at Arrested Forks
2.2. Recruitment and Action of TLS DNA Polymerases
2.3. Rad18 Chromatin Recruitment
2.3.1. Other TLS Pols Regulators
2.3.2. Rad18 and DNA Repair
2.4. TLS Inhibition in Cancer Therapy
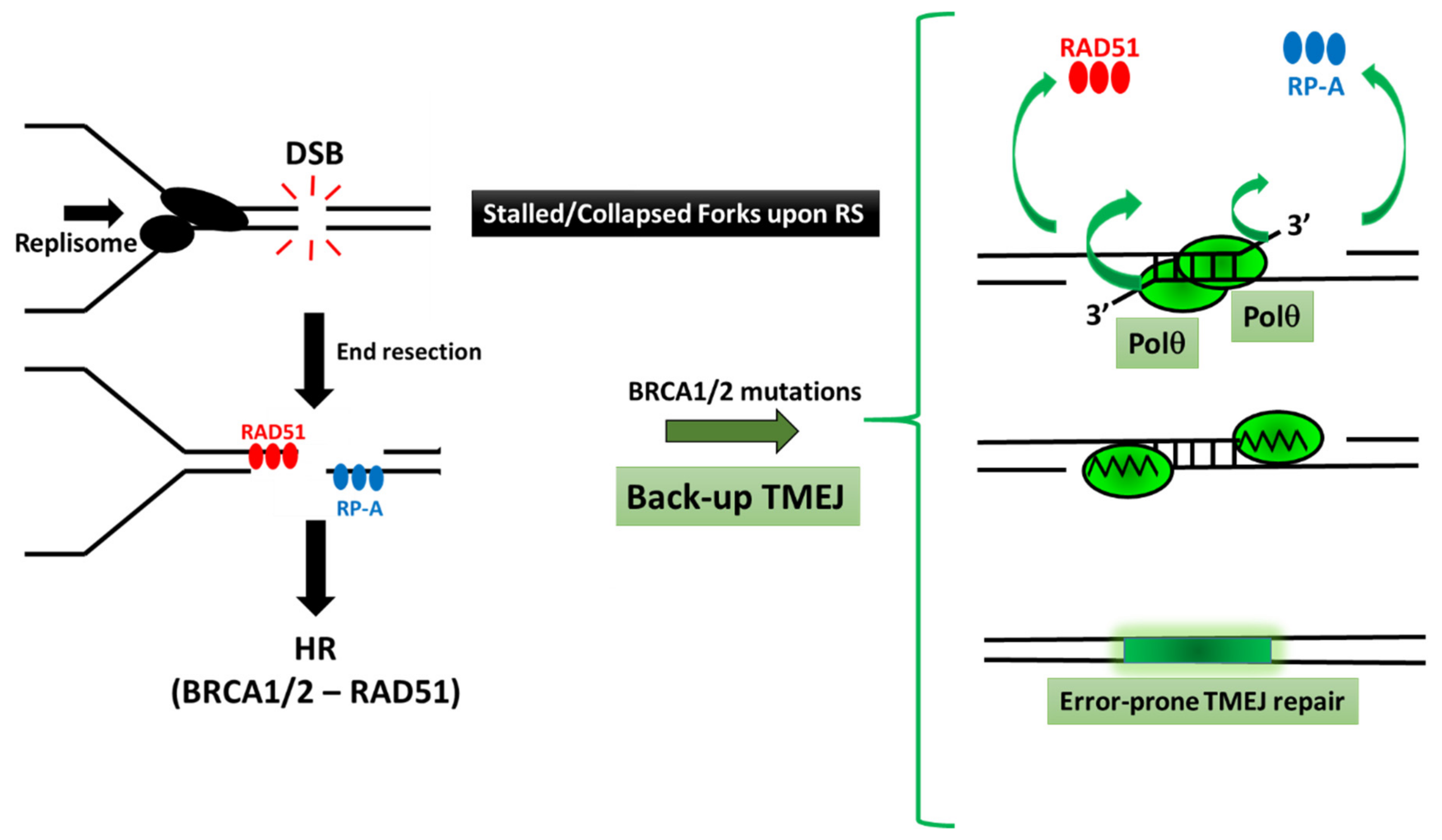
3. The Major Role of Polθ during the Alternative TMEJ Pathway to Repair DSBs
3.1. Several Pathways to Repair DSB
3.2. The Critical Action of Polθ in TMEJ
3.3. Polθ Inhibition in Cancer Therapy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansregret, L.; Patterson, J.O.; Dewhurst, S.; López-García, C.; Koch, A.; McGranahan, N.; Chao, W.C.H.; Barry, D.J.; Rowan, A.; Instrell, R.; et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017, 7, 218–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.-S.; Cazaux, C. Aberrant expression of alternative DNA polymerases: A source of mutator phenotype as well as replicative stress in cancer. Semin. Cancer Biol. 2010, 20, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nat. Cell Biol. 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, T.; Reh, S.; Dunbayev, Y.; Zhong, Y.; Takata, Y.; Shen, J.; McBride, K.M.; Murnane, J.P.; Bhak, J.; Lee, S.; et al. Defining the mutation signatures of DNA polymerase θ in cancer genomes. NAR Cancer 2020, 2, zcaa017. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nat. Cell Biol. 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.; Haipek, C.; Clarkson, J. (6–4)Photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane pyrimidine dimers. Mutat. Res. Lett. 1985, 143, 109–112. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Pasero, P.; Vindigni, A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Powers, K.T.; Washington, M.T. Eukaryotic translesion synthesis: Choosing the right tool for the job. DNA Repair 2018, 71, 127–134. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020, 61, 680–692. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiqui-tinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [Green Version]
- Guilliam, T.A.; Yeeles, J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020, 27, 450–460. [Google Scholar] [CrossRef]
- Tirman, S.; Cybulla, E.; Quinet, A.; Meroni, A.; Vindigni, A. PRIMPOL ready, set, reprime! Crit. Rev. Biochem. Mol. Biol. 2020, 56, 17–30. [Google Scholar] [CrossRef]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple Mechanisms Control Chromosome Integrity after Replication Fork Uncoupling and Restart at Irreparable UV Lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Wong, R.P.; García-Rodríguez, N.; Zilio, N.; Hanulová, M.; Ulrich, H.D. Processing of DNA Polymer-ase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 2020, 77, 3–16.e4. [Google Scholar] [CrossRef]
- Despras, E.; Daboussi, F.; Hyrien, O.; Marheineke, K.; Kannouche, P.L. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum. Mol. Genet. 2010, 19, 1690–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Jimenez, M.I.; García-Gómez, S.; Bebenek, K.; Sastre-Moreno, G.; Calvo, P.A.; Diaz-Talavera, A.; Kunkel, T.A.; Blanco, L. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair 2015, 29, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Pilzecker, B.; Buoninfante, O.A.; Pritchard, C.; Blomberg, O.S.; Huijbers, I.J.; Berk, P.C.M.V.D.; Jacobs, H. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 2016, 44, 4734–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Un-restrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef]
- Genois, M.-M.; Gagné, J.-P.; Yasuhara, T.; Jackson, J.; Saxena, S.; Langelier, M.-F.; Ahel, I.; Bedford, M.T.; Pascal, J.M.; Vindigni, A.; et al. CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol. Cell 2021, 81, 784–800.e8. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome Stability at Defective DNA Replication Forks Is Independent of S Phase Checkpoint Kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Naiman, K.; Thrall, E.S.; Kath, J.E.; Jergic, S.; Dixon, N.E.; Fuchs, R.P.; Loparo, J.J. A gatekeeping function of the replicative polymerase controls pathway choice in the resolution of lesion-stalled replisomes. Proc. Natl. Acad. Sci. USA 2019, 116, 25591–25601. [Google Scholar] [CrossRef]
- Zhao, G.; Gleave, E.S.; Lamers, M.H. Single-molecule studies contrast ordered DNA replication with sto-chastic translesion synthesis. Elife 2017, 6, e32177. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.; Trusa, S.; Meng, X.; Lee, E.Y.C.; Lee, M.Y.W.T. A novel DNA damage response: Rapid degradation of the p12 subunit of dna polymerase delta. J. Biol. Chem. 2007, 282, 15330–15340. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [Green Version]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [Green Version]
- Kermi, C.; Prieto, S.; Van Der Laan, S.; Tsanov, N.; Recolin, B.; Uro-Coste, E.; Delisle, M.-B.; Maiorano, D. RAD18 Is a Maternal Limiting Factor Silencing the UV-Dependent DNA Damage Checkpoint in Xenopus Embryos. Dev. Cell 2015, 34, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2005, 8, 84–90. [Google Scholar] [CrossRef]
- Tsanov, N.; Kermi, C.; Coulombe, P.; Van der Laan, S.; Hodroj, D.; Maiorano, D. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase eta and kappa focus formation on UV damage. Nucleic Acids Res. 2014, 42, 3692–3706. [Google Scholar] [CrossRef]
- Betous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, L.; Bertoletti, F.; Maffia, A.; Liang, C.-C.; Lehmann, A.R.; Cohn, M.A.; Sabbioneda, S. UBR5 interacts with the replication fork and protects DNA replication from DNA polymerase η toxicity. Nucleic Acids Res. 2019, 47, 11268–11283. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nat. Cell Biol. 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Chang, D.J.; Lupardus, P.J.; Cimprich, K.A. Monoubiquitination of proliferating cell nuclear antigen in-duced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J. Biol. Chem. 2006, 281, 32081–32088. [Google Scholar] [CrossRef] [Green Version]
- Bailly, V.; Lauder, S.; Prakash, S.; Prakash, L. Yeast DNA Repair Proteins Rad6 and Rad18 Form a Heterodimer That Has Ubiquitin Conjugating, DNA Binding, and ATP Hydrolytic Activities. J. Biol. Chem. 1997, 272, 23360–23365. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef]
- Hedglin, M.; Aitha, M.; Pedley, A.; Benkovic, S.J. Replication protein A dynamically regulates mon-oubiquitination of proliferating cell nuclear antigen. J. Biol. Chem. 2019, 294, 5157–5168. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Sengupta, B.; Benkovic, S.J.; Lee, T.H.; Hedglin, M. PCNA Monoubiquitination Is Regulated by Diffusion of Rad6/Rad18 Complexes along RPA Filaments. Biochemistry 2020, 59, 4694–4702. [Google Scholar] [CrossRef]
- Yang, X.H.; Shiotani, B.; Classon, M.; Zou, L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008, 22, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zhou, B.; Gong, J.; Xing, L.; Ma, X.; Wang, F.; Wu, W.; Shen, H.; Sun, C.; Zhu, X.; et al. RNA-splicing factor SART3 regulates translesion DNA synthesis. Nucleic Acids Res. 2018, 46, 4560–4574. [Google Scholar] [CrossRef] [Green Version]
- Cano-Linares, M.I.; Yáñez-Vilches, A.; García-Rodríguez, N.; Barrientos-Moreno, M.; González-Prieto, R.; San-Segundo, P.; Ulrich, H.D.; Prado, F. Non-recombinogenic roles for Rad52 in translesion synthesis during DNA damage tolerance. EMBO Rep. 2021, 22, e50410. [Google Scholar] [CrossRef]
- Durando, M.; Tateishi, S.; Vaziri, C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013, 41, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Pentz, K.; Zhu, Q.; Wang, Q.; He, J.; Srivastava, A.K.; Wani, A.A. USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene 2014, 34, 4791–4796. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Barkley, L.R.; Slater, D.M.; Tateishi, S.; Yamaizumi, M.; Ohmori, H.; Vaziri, C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell Biol. 2006, 26, 3527–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagihara, H.; Kobayashi, J.; Tateishi, S.; Kato, A.; Matsuura, S.; Tauchi, H.; Yamada, K.; Takezawa, J.; Sugasawa, K.; Masutani, C.; et al. NBS1 recruits RAD18 via a RAD6-like domain and regulates Pol eta-dependent translesion DNA synthesis. Mol. Cell 2011, 43, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Liu, T.; Huen, M.S.; Hu, L.; Chen, Z.; Huang, J. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J. Cell Biol. 2014, 205, 811–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Watanabe, K.; Mistrik, M.; Vesela, E.; Protivankova, I.; Mailand, N.; Lee, M.; Masai, H.; Lukas, J.; Bartek, J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev. 2013, 27, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.Y.-L.; Chiba, T.; Truong, L.N.; Cheng, A.N.; Do, J.; Cho, M.J.; Chen, L.; Wu, X. Dbf4 Is Direct Downstream Target of Ataxia Telangiectasia Mutated (ATM) and Ataxia Telangiectasia and Rad3-related (ATR) Protein to Regulate Intra-S-phase Checkpoint. J. Biol. Chem. 2012, 287, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Mutter-Rottmayer, E.; Greenwalt, A.M.; Goldfarb, D.; Yan, F.; Yang, Y.; Martinez-Chacin, R.C.; Pearce, K.H.; Tateishi, S.; Major, M.B.; et al. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 2016, 7, 12105. [Google Scholar] [CrossRef] [Green Version]
- Centore, R.C.; Yazinski, S.A.; Tse, A.; Zou, L. Spartan/C1orf124, a Reader of PCNA Ubiquitylation and a Regulator of UV-Induced DNA Damage Response. Mol. Cell 2012, 46, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Juhasz, S.; Balogh, D.; Hajdu, I.; Burkovics, P.; Villamil, M.A.; Zhuang, Z.; Haracska, L. Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance. Nucleic Acids Res. 2012, 40, 10795–10808. [Google Scholar] [CrossRef] [Green Version]
- Machida, Y.; Kim, M.S.; Machida, Y.J. Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle 2012, 11, 3395–3402. [Google Scholar] [CrossRef] [Green Version]
- Mosbech, A.; Gibbs-Seymour, I.; Kagias, K.; Thorslund, T.; Beli, P.; Povlsen, L.K.; Nielsen, S.V.; Smedegaard, S.; Sedgwick, G.; Lukas, C.; et al. DVC1 (C1orf124) is a DNA damage–targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012, 19, 1084–1092. [Google Scholar] [CrossRef]
- Davis, E.J.; Lachaud, C.; Appleton, P.L.; Macartney, T.J.; Nathke, I.S.; Rouse, J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 2012, 19, 1093–1100. [Google Scholar] [CrossRef]
- Ghosal, G.; Leung, J.W.-C.; Nair, B.C.; Fong, K.-W.; Chen, J. Proliferating Cell Nuclear Antigen (PCNA)-binding Protein C1orf124 Is a Regulator of Translesion Synthesis. J. Biol. Chem. 2012, 287, 34225–34233. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 341–347. [Google Scholar] [CrossRef]
- Gallina, I.; Hendriks, I.A.; Hoffmann, S.; Larsen, N.B.; Johansen, J.; Colding-Christensen, C.S.; Schubert, L.; Sellés-Baiget, S.; Fábián, Z.; Kühbacher, U.; et al. The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Mol. Cell 2021, 81, 442–458.e9. [Google Scholar] [CrossRef]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [Green Version]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [Green Version]
- Guérillon, C.; Smedegaard, S.; Hendriks, I.A.; Nielsen, M.L.; Mailand, N. Multisite SUMOylation restrains DNA polymerase η interactions with DNA damage sites. J. Biol. Chem. 2020, 295, 8350–8362. [Google Scholar] [CrossRef]
- Moquin, D.M.; Genois, M.-M.; Zhang, J.-M.; Ouyang, J.; Yadav, T.; Buisson, R.; Yazinski, S.A.; Tan, J.; Boukhali, M.; Gagné, J.-P.; et al. Localized protein biotinylation at DNA damage sites identifies ZPET, a repressor of homologous recombination. Genes Dev. 2018, 33, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Ting, L.; Jun, H.; Junjie, C. RAD18 lives a double life: Its implication in DNA double-strand break repair. DNA Repair 2010, 9, 1241–1248. [Google Scholar] [CrossRef]
- Huang, J.; Huen, M.S.Y.; Kim, H.; Leung, C.C.Y.; Glover, J.N.M.; Yu, X.; Chen, J. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 2009, 11, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Iwabuchi, K.; Sun, J.; Tsuji, Y.; Tani, T.; Tokunaga, K.; Date, T.; Hashimoto, M.; Yamaizumi, M.; Tateishi, S. RAD18 promotes DNA double-strand break repair during G1 phase through chromatin retention of 53BP1. Nucleic Acids Res. 2009, 37, 2176–2193. [Google Scholar] [CrossRef] [Green Version]
- Lo Furno, E.; Busseau, I.; Lorenzi, C.; Saghira, C.; Zuchner, S. Translesion DNA synthesis-driven mutagenesis in very early embryogenesis of fast cleaving embryos. BioRxiv 2020. [Google Scholar] [CrossRef]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Räschle, M.; Smeenk, G.; Hansen, R.K.; Temu, T.; Oka, Y.; Hein, M.Y.; Nagaraj, N.; Long, D.T.; Walter, J.C.; Hofmann, K.; et al. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 2015, 348, 1253671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, N.B.; Gao, A.O.; Sparks, J.L.; Gallina, I.; Wu, R.A.; Mann, M.; Räschle, M.; Walter, J.C.; Duxin, J.P. Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 2019, 73, 574–588.e7. [Google Scholar] [CrossRef] [Green Version]
- Sparks, J.L.; Chistol, G.; Gao, A.O.; Räschle, M.; Larsen, N.B.; Mann, M.; Duxin, J.P.; Walter, J.C. The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell 2019, 176, 167–181.e21. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Gao, Y.; Mutter-Rottmayer, L.; Zlatanou, A.; Durando, M.; Ding, W.; Wyatt, D.; Ramsden, D.; Tanoue, Y.; Tateishi, S.; et al. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 2017, 216, 3097–3115. [Google Scholar] [CrossRef] [Green Version]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020, 6, eaaz7808. [Google Scholar] [CrossRef]
- Poveda, A.; Méndez, M.Á.; Armijos-Jaramillo, V. Analysis of DNA Polymerases Reveals Specific Genes Expansion in Leishmania and Trypanosoma spp. Front. Cell. Infect. Microbiol. 2020, 10, 570493. [Google Scholar] [CrossRef] [PubMed]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair 2005, 4, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Rocha, C.R.R.; Martins, D.J.; Fiore, A.P.Z.P.; Kinker, G.S.; Bruni-Cardoso, A.; Menck, C.F.M. ATR mediates cisplatin resistance in 3D-cultured breast cancer cells via translesion DNA synthesis modulation. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wang, H.; Cheng, H.; Li, J.; Wang, Z.; Yue, W. RAD18 mediates resistance to ionizing radiation in human glioma cells. Biochem. Biophys. Res. Commun. 2014, 445, 263–268. [Google Scholar] [CrossRef]
- Lou, J.; Yang, Y.; Gu, Q.; Price, B.A.; Qiu, Y.; Fedoriw, Y.; Desai, S.; Mose, L.E.; Chen, B.; Tateishi, S.; et al. Rad18 mediates specific mutational signatures and shapes the genomic landscape of carcinogen-induced tumors in vivo. NAR Cancer 2021, 3, zcaa037. [Google Scholar] [CrossRef]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Temprine, K.; Campbell, N.R.; Huang, R.; Langdon, E.M.; Simon-Vermot, T.; Mehta, K.; Clapp, A.; Chipman, M.; White, R.M. Regulation of the error-prone DNA polymerase Polκ by oncogenic signaling and its contribution to drug resistance. Sci. Signal. 2020, 13, eaau1453. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Z.; Wang, S.; Wang, H.W.; Qiu, W.; Zhao, L.; Xu, R.; Luo, H.; Chen, Y.; Chen, D.; et al. The Er-ror-Prone DNA Polymerase kappa Promotes Temozolomide Resistance in Glioblastoma through Rad17-Dependent Activation of ATR-Chk1 Signaling. Cancer Res. 2016, 76, 2340–2353. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, M.; Gorgun, F.M.; Tyler, D.S.; Englander, E.W. Transient activation of tumoral DNA damage tolerance pathway coupled with immune checkpoint blockade exerts durable tumor regression in mouse melanoma. Pigment Cell Melanoma Res. 2020. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Waters, C.A.; Strande, N.T.; Wyatt, D.W.; Pryor, J.M.; Ramsden, D.A. Nonhomologous end joining: A good solution for bad ends. DNA Repair 2014, 17, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microho-mology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi1, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [Green Version]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: What is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, H.; Jehi, S.; Li, J.; Liu, S.; Wang, Z.; Truong, L.; Chiba, T.; Wang, Z.; Wu, X. PIF1 helicase promotes break- induced replication in mammalian cells. EMBO J. 2021, e104509. [Google Scholar] [CrossRef]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.F.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alter-native Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Mentegari, E.; Bertoletti, F.; Kissova, M.; Zucca, E.; Galli, S.; Tagliavini, G.; Garbelli, A.; Maffia, A.; Bione, S.; Ferrari, E.; et al. A Role for Human DNA Polymerase λ in Alternative Lengthening of Telomeres. Int. J. Mol. Sci. 2021, 22, 2365. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA polymerase θ (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Koole, W.; Van Schendel, R.; Karambelas, A.E.; Van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [Green Version]
- Roerink, S.F.; van Schendel, R.; Tijsterman, M. Polymerase theta-mediated end joining of replica-tion-associated DNA breaks in C. elegans. Genome Res. 2014, 24, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929.e6. [Google Scholar] [CrossRef] [Green Version]
- Arana, M.E.; Seki, M.; Wood, R.D.; Rogozin, I.B.; Kunkel, T.A. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008, 36, 3847–3856. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Averill, A.M.; Aller, P.; Wood, R.D.; Doublié, S. Human DNA polymerase θ grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 2015, 22, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Jensen, R.B.; Wood, R.D.; Doublié, S. Human DNA polymerase θ harbors DNA end-trimming activity critical for DNA repair. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Yoshimura, M.; Kohzaki, M.; Nakamura, J.; Asagoshi, K.; Sonoda, E.; Hou, E.; Prasad, R.; Wilson, S.H.; Keizo, T.; Yasui, A.; et al. Vertebrate POLQ and POLβ Cooperate in Base Excision Repair of Oxidative DNA Damage. Mol. Cell 2006, 24, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.-H.; McArthur, M.J.; Park, J.; Basu, D.; Wakamiya, M.; Prakash, L.; Prakash, S. Error-Prone Replication through UV Lesions by DNA Polymerase θ Protects against Skin Cancers. Cell 2019, 176, 1295–1309.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Prodhomme, M.K.; Pommier, R.M.; Franchet, C.; Fauvet, F.; Bergoglio, V.; Brousset, P.; Morel, A.-P.; Brunac, A.-C.; Devouassoux-Shisheboran, M.; Petrilli, V.; et al. EMT Transcription Factor ZEB1 Represses the Mutagenic POLθ-Mediated End-Joining Pathway in Breast Cancers. Cancer Res. 2021, 81, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Garcia, J.; Cho, J.-E.; Carvajal-Garcia, P.; Feng, W.; Wood, R.D.; Sekelsky, J.; Gupta, G.P.; Roberts, S.A.; Ramsden, D.A. Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc. Natl. Acad. Sci. USA 2020, 117, 8476–8485. [Google Scholar] [CrossRef] [Green Version]
- Allera-Moreau, C.; Rouquette, I.; Lepage, B.; Oumouhou, N.; Walschaerts, M.; Leconte, E.; Schilling, V.; Gordien, K.; Brouchet, L.; Delisle, M.B.; et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis 2012, 1, e30. [Google Scholar] [CrossRef]
- Lemée, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.-J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.-L.; Leduc, C.; et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [Green Version]
- Pillaire, M.J.; Selves, J.; Gordien, K.; Gourraud, P.A.; Gentil, C.; Danjoux, M.; Do, C.; Negre, V.; Bieth, A.; Guimbaud, R.; et al. A “DNA replication” signature of progression and negative outcome in colorectal cancer. Oncogene 2010, 29, 876–887. [Google Scholar] [CrossRef]
- Cong, K.; Kousholt, A.N.; Peng, M.; Panzarino, N.J.; Ting, L., W.T.C.; Nayak, S.; Krais, J.; Calvo, J.; Bere, M.; Rothenberg, E.; et al. PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorano, D.; El Etri, J.; Franchet, C.; Hoffmann, J.-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 2021, 22, 3924. https://doi.org/10.3390/ijms22083924
Maiorano D, El Etri J, Franchet C, Hoffmann J-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. International Journal of Molecular Sciences. 2021; 22(8):3924. https://doi.org/10.3390/ijms22083924
Chicago/Turabian StyleMaiorano, Domenico, Jana El Etri, Camille Franchet, and Jean-Sébastien Hoffmann. 2021. "Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress" International Journal of Molecular Sciences 22, no. 8: 3924. https://doi.org/10.3390/ijms22083924
APA StyleMaiorano, D., El Etri, J., Franchet, C., & Hoffmann, J.-S. (2021). Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. International Journal of Molecular Sciences, 22(8), 3924. https://doi.org/10.3390/ijms22083924