
An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
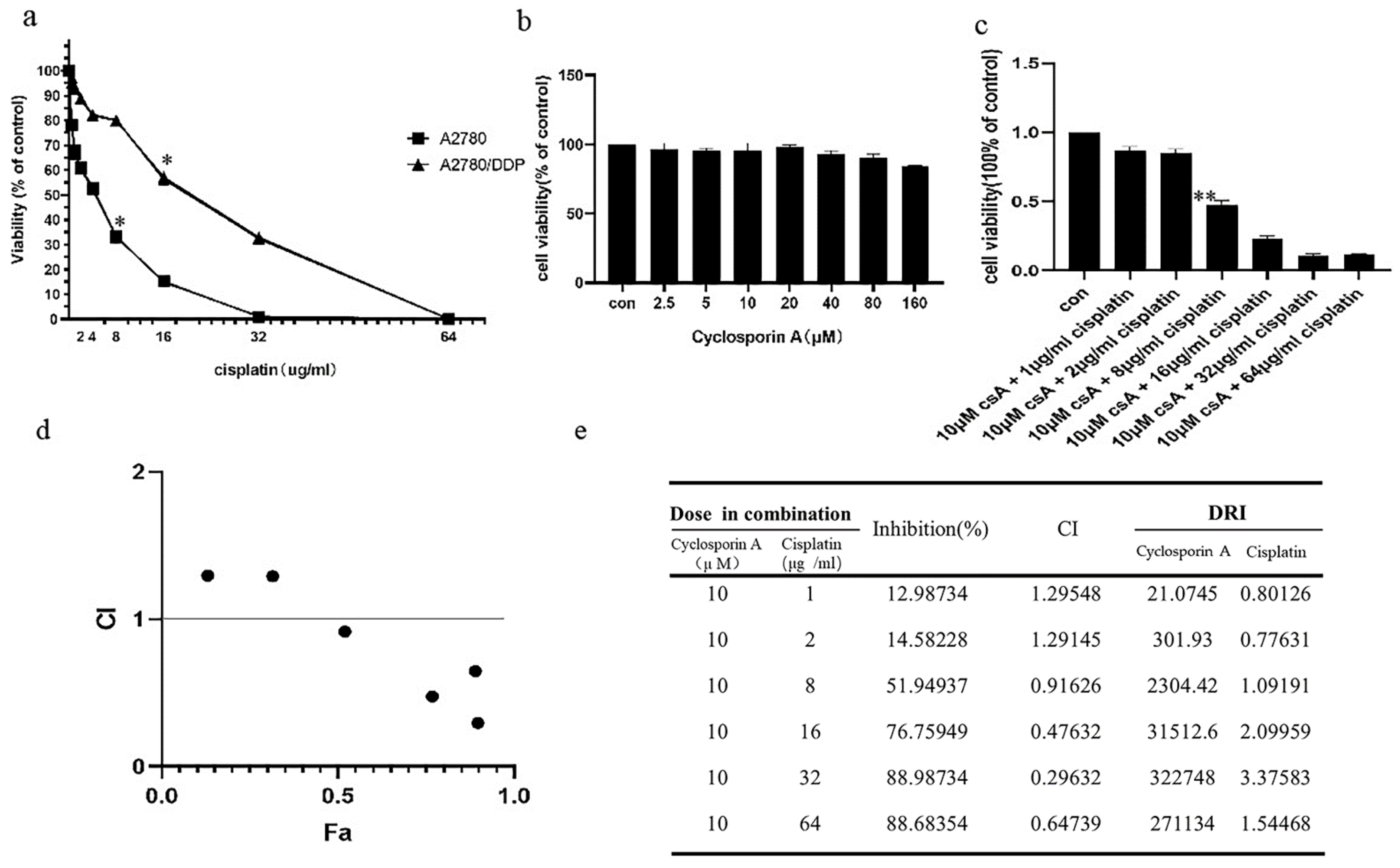
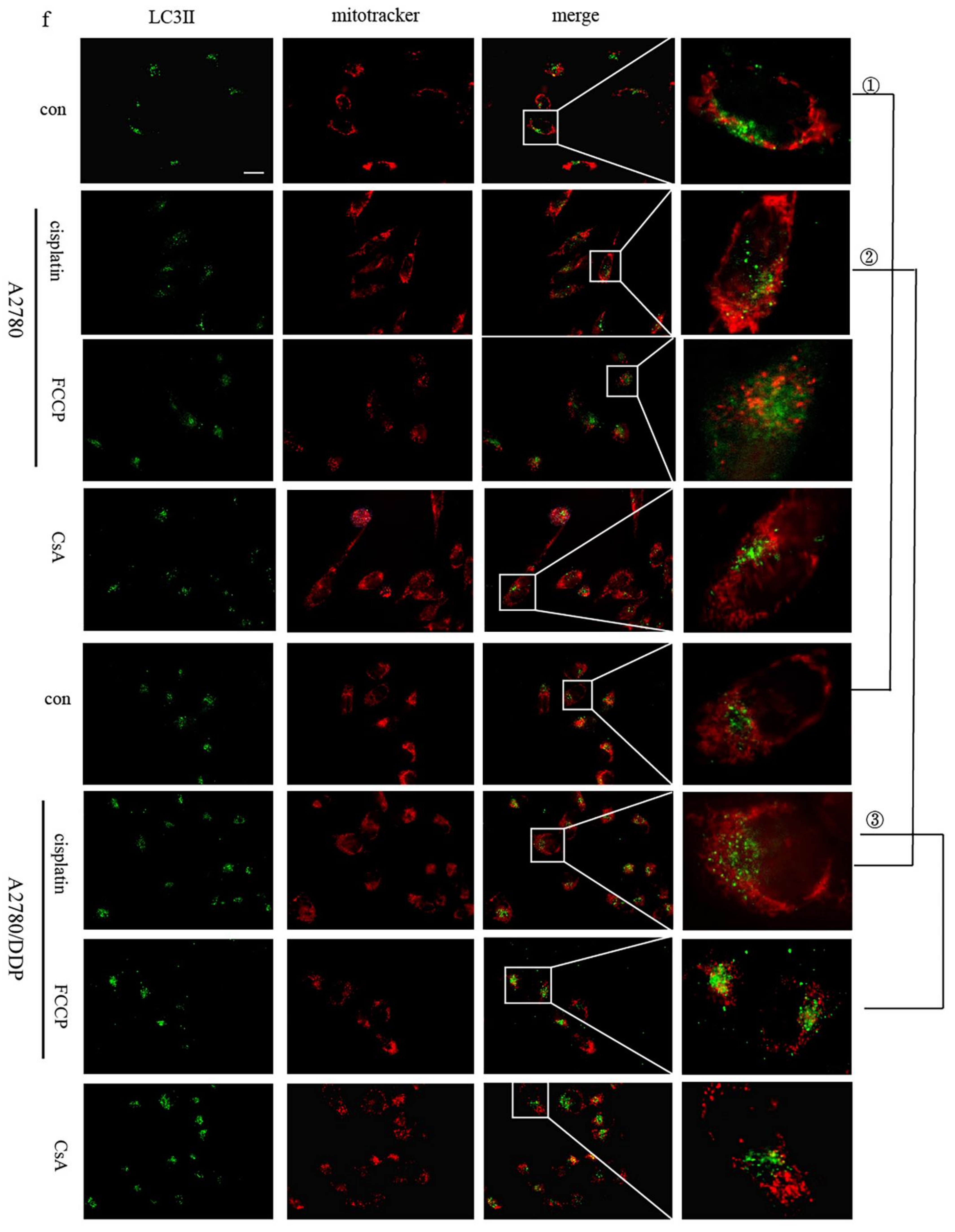
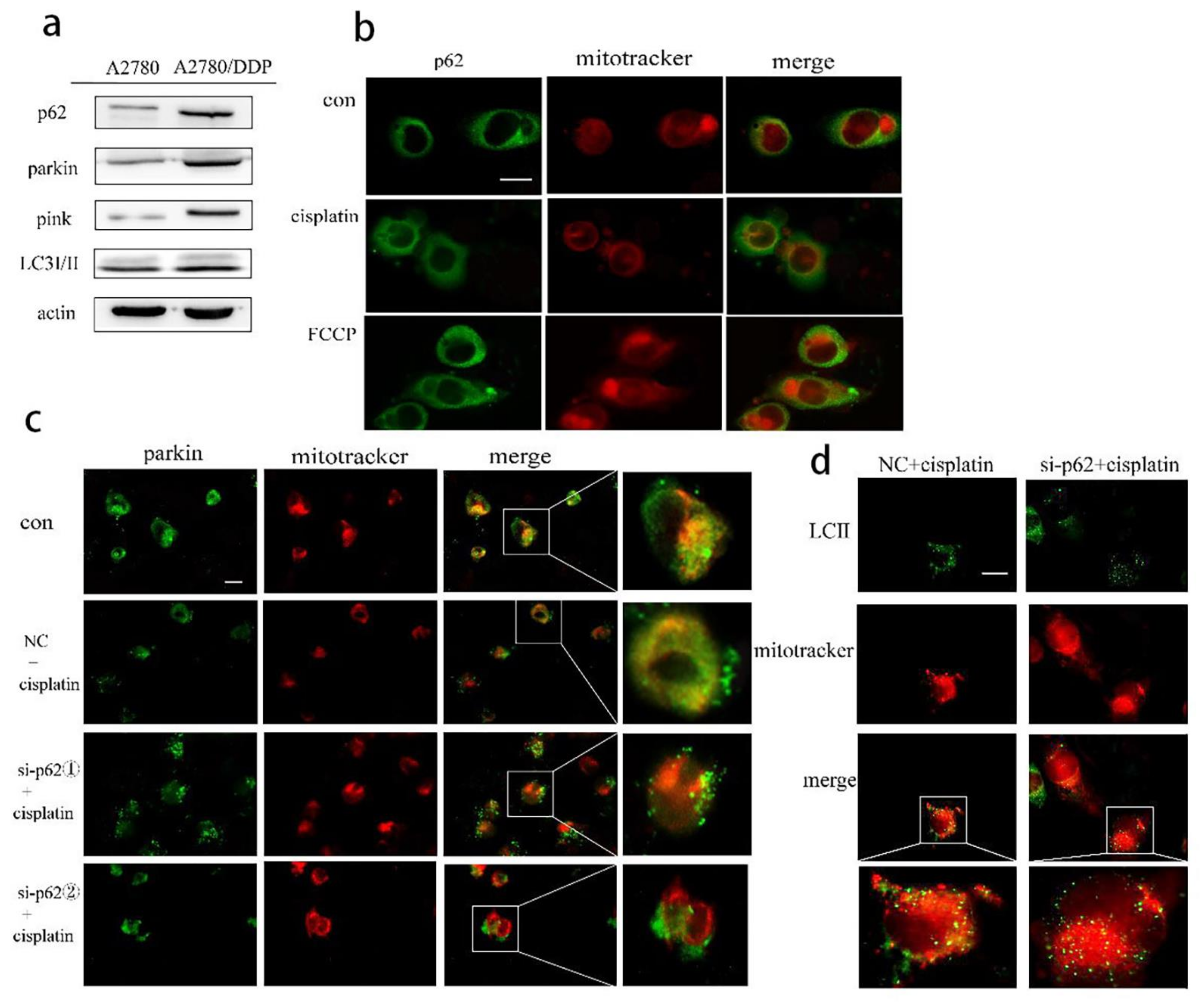
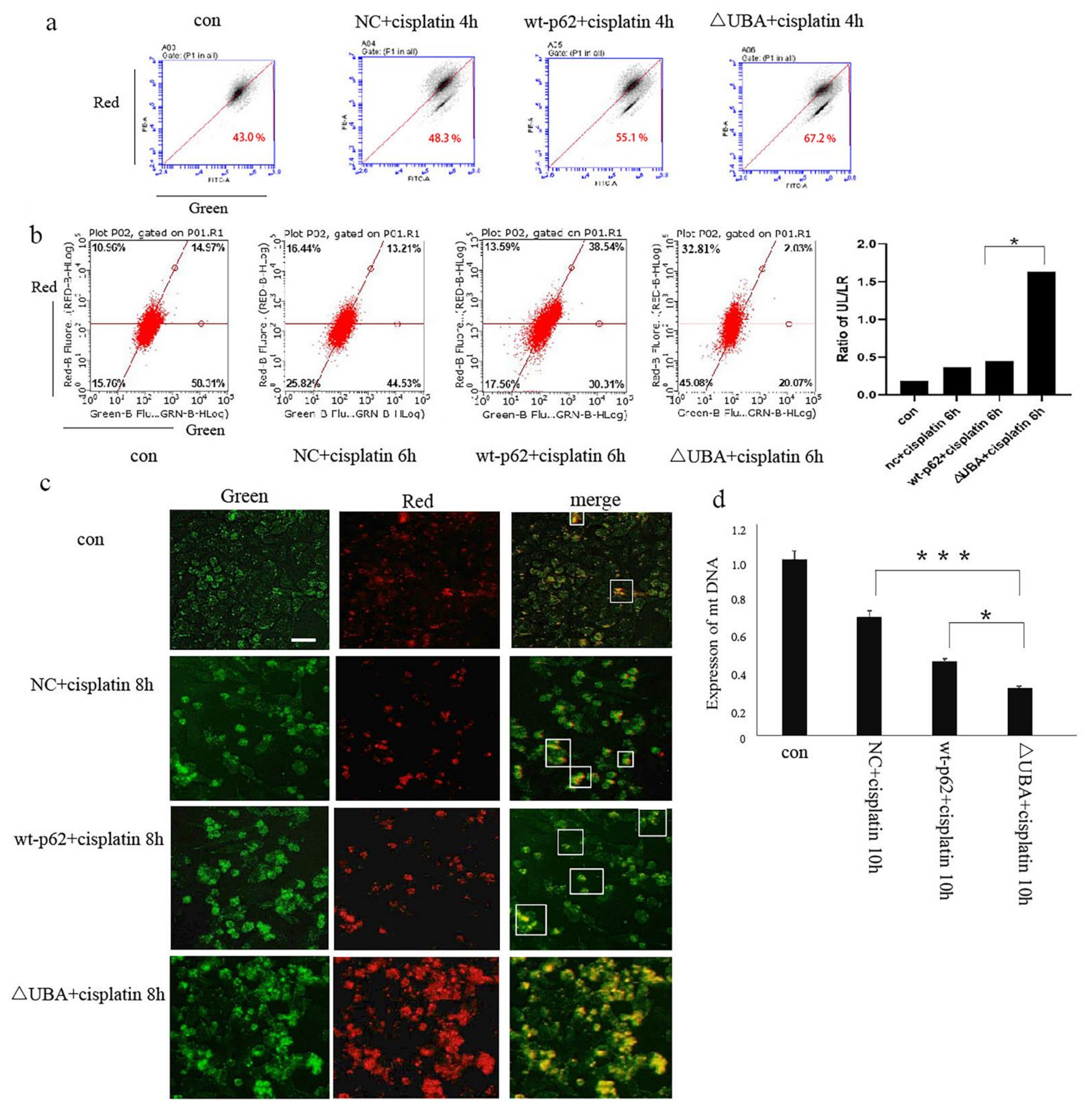
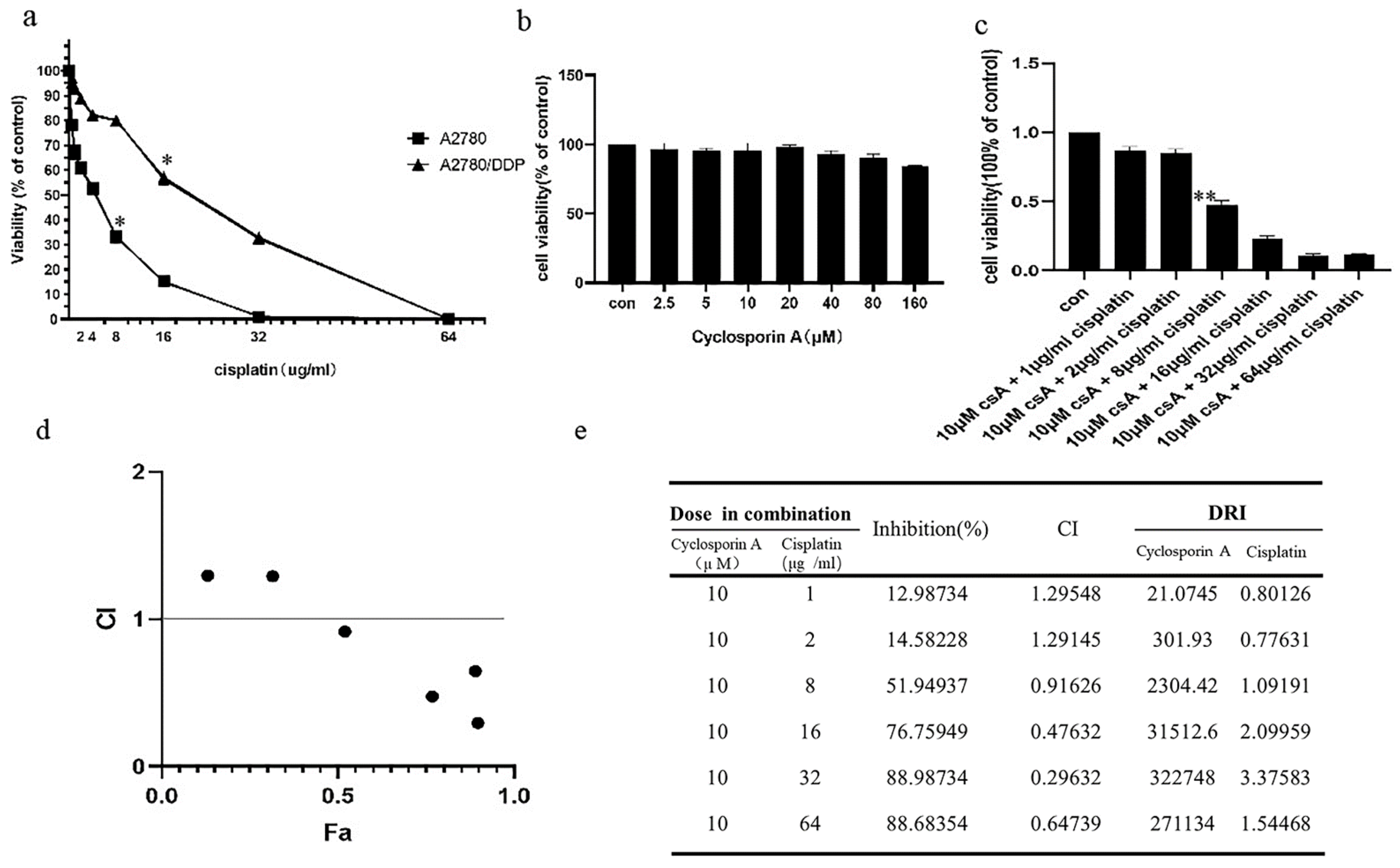
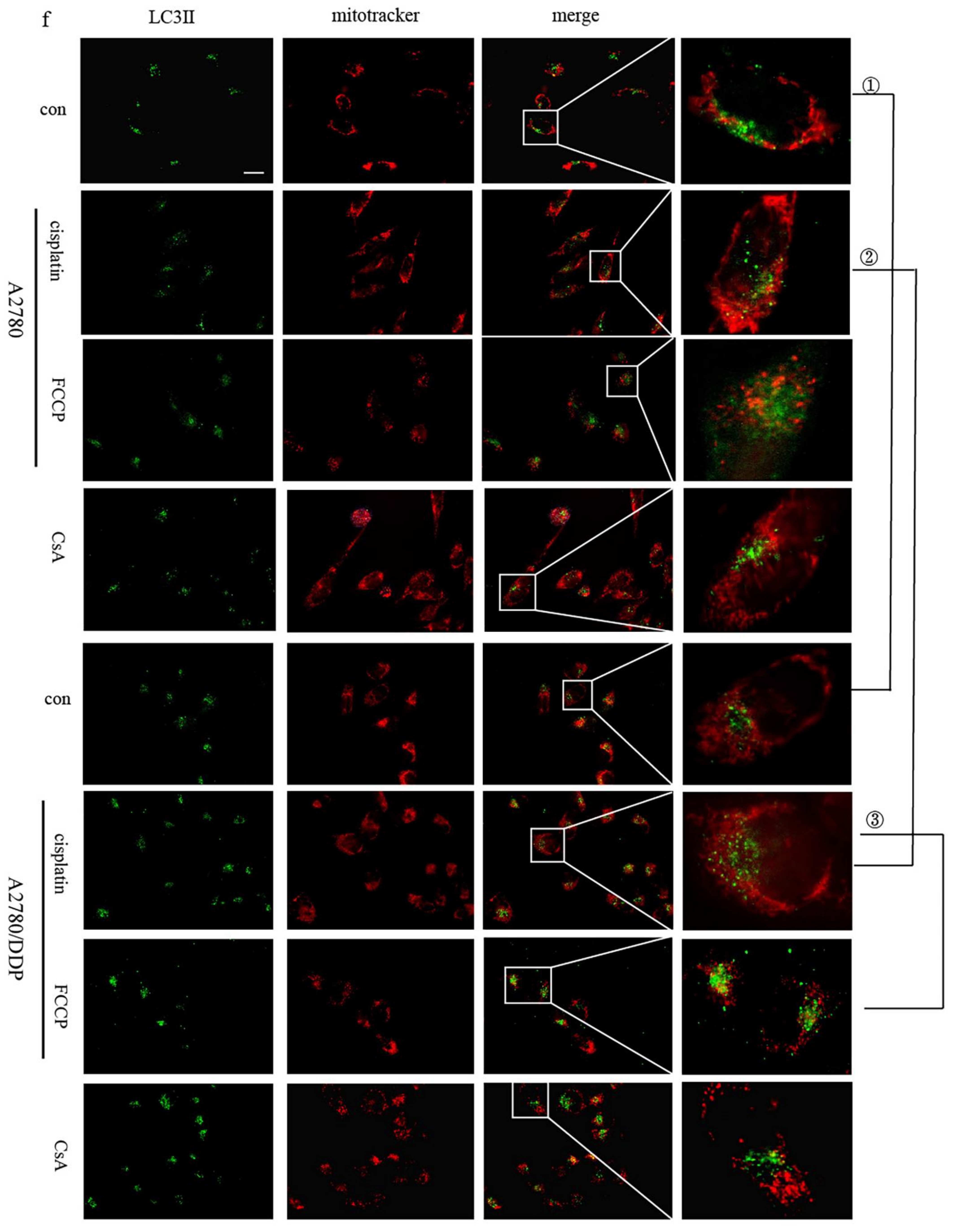
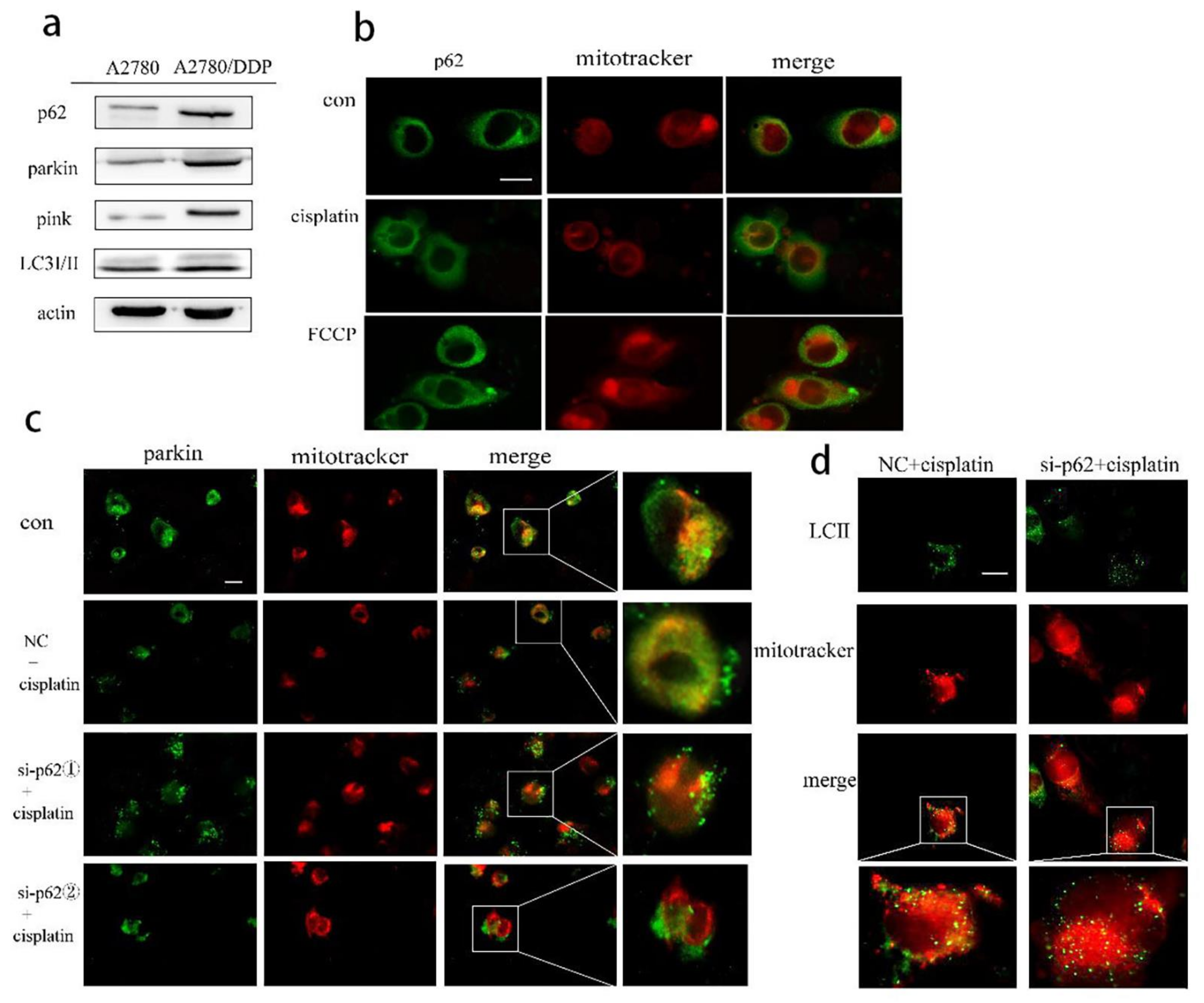
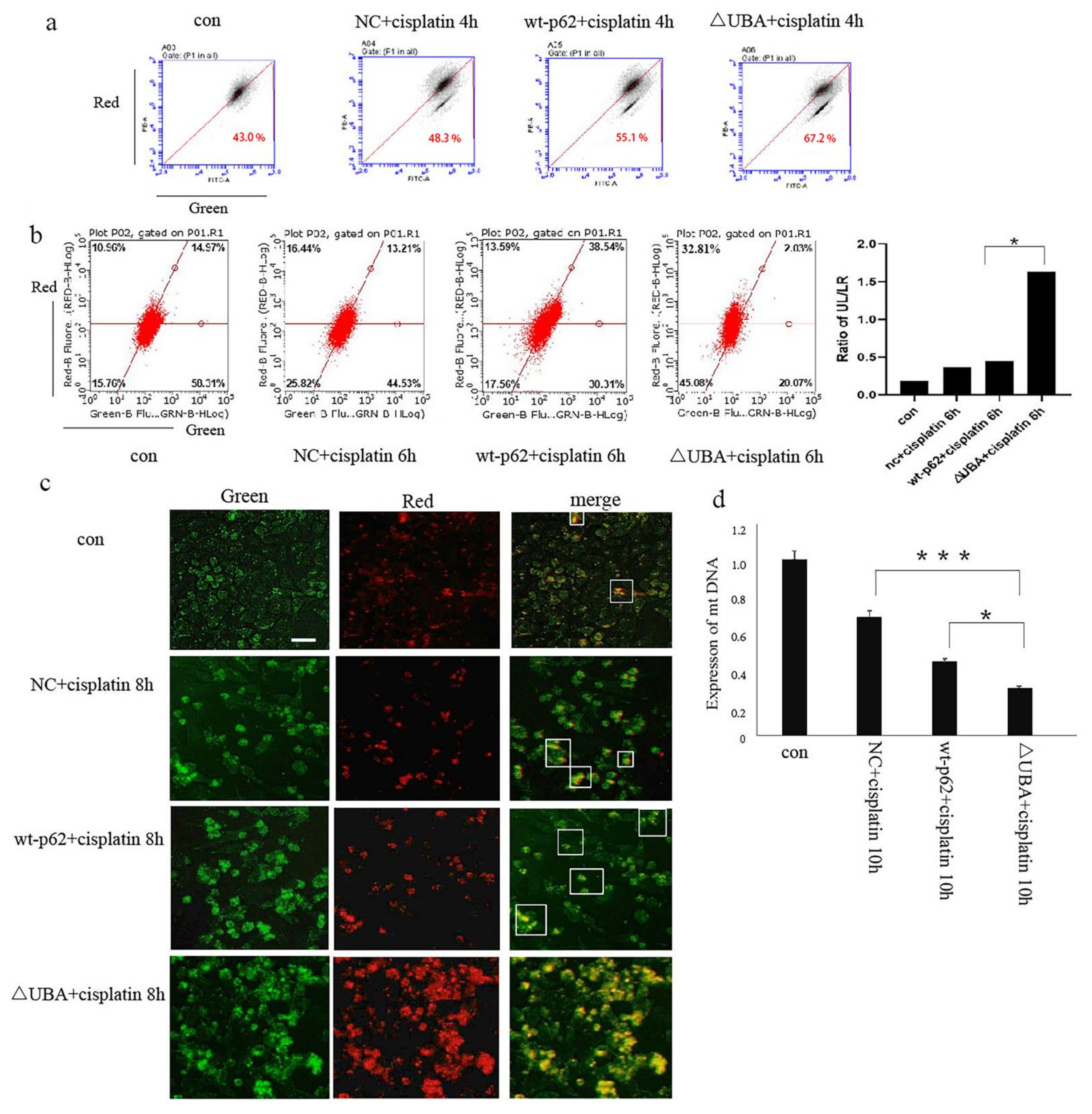
2.1. p62 Is Involved in the Mitochondrial Clearance of Ovarian Cancer Cells Stimulated by Cisplatin
2.2. Regulation of Mitophagy by p62 Does Not Depend on the Location of p62 within the Mitochondria
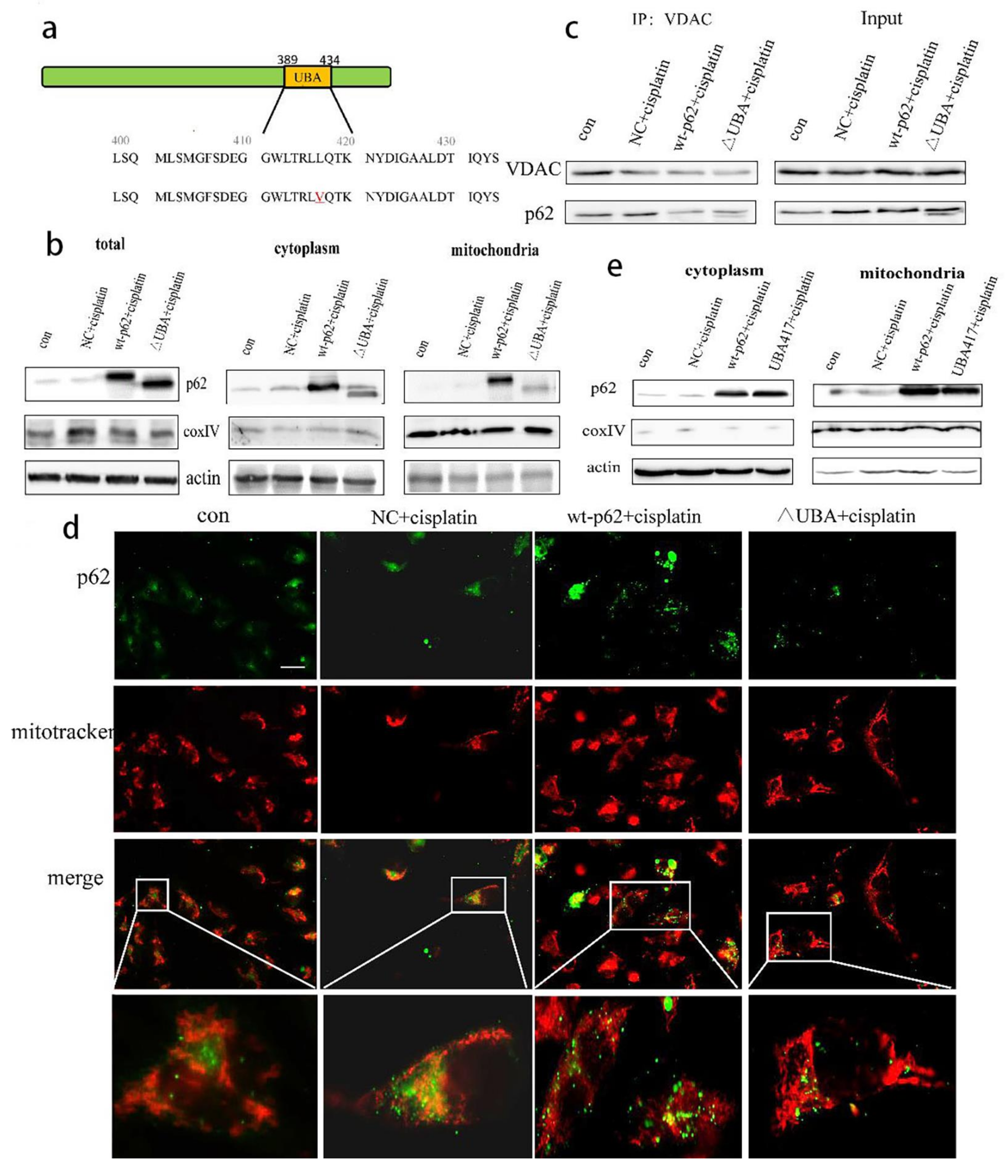
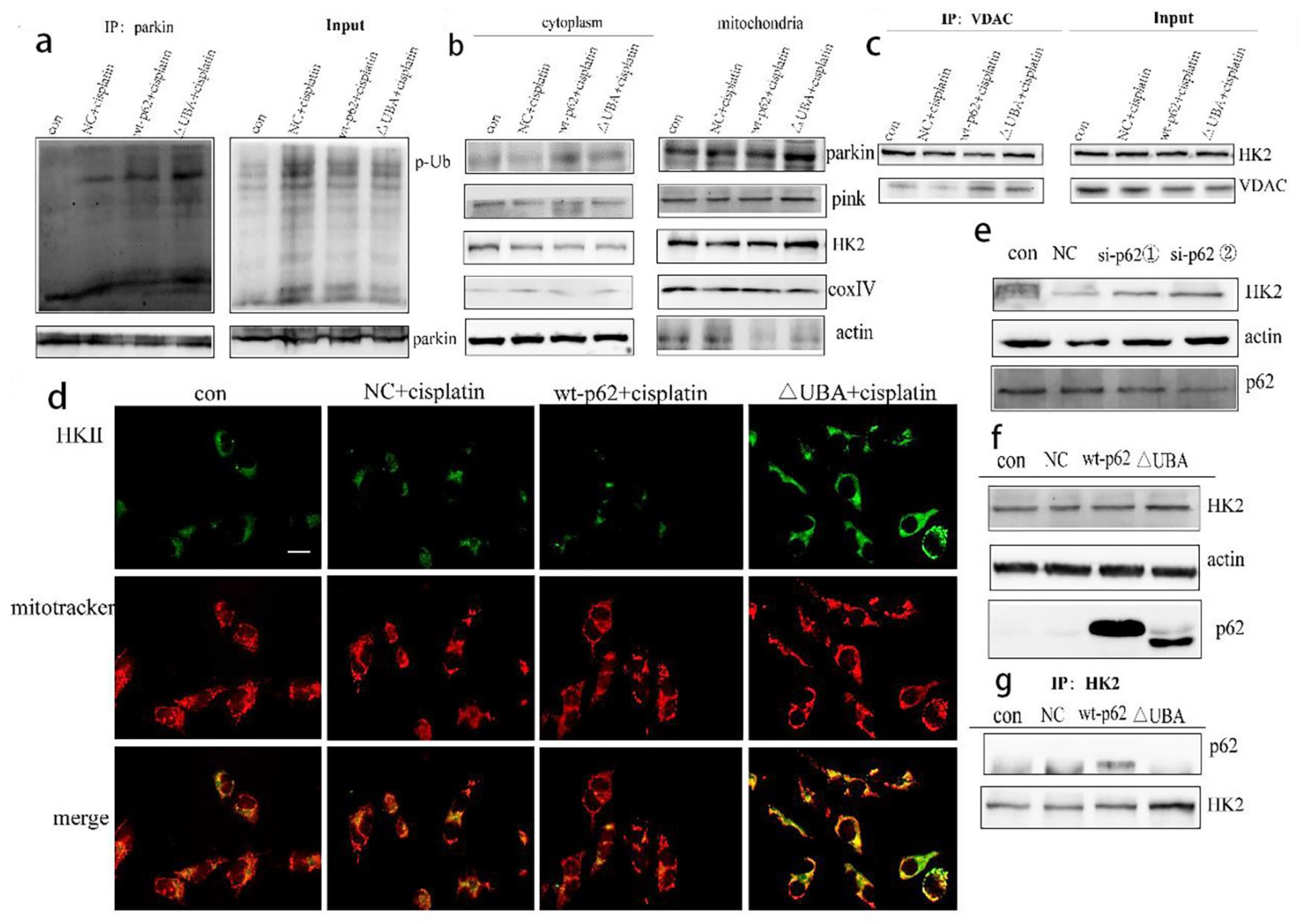
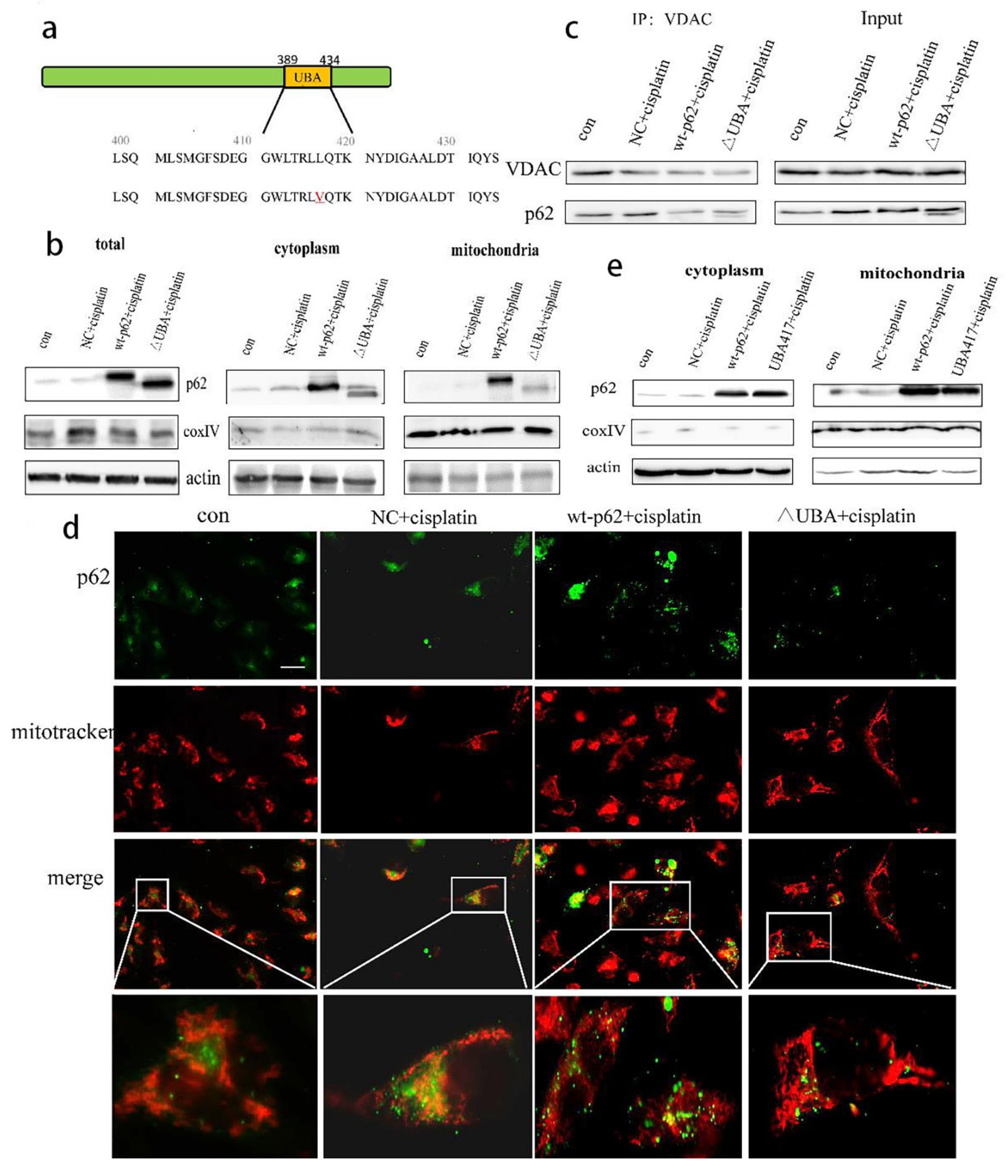
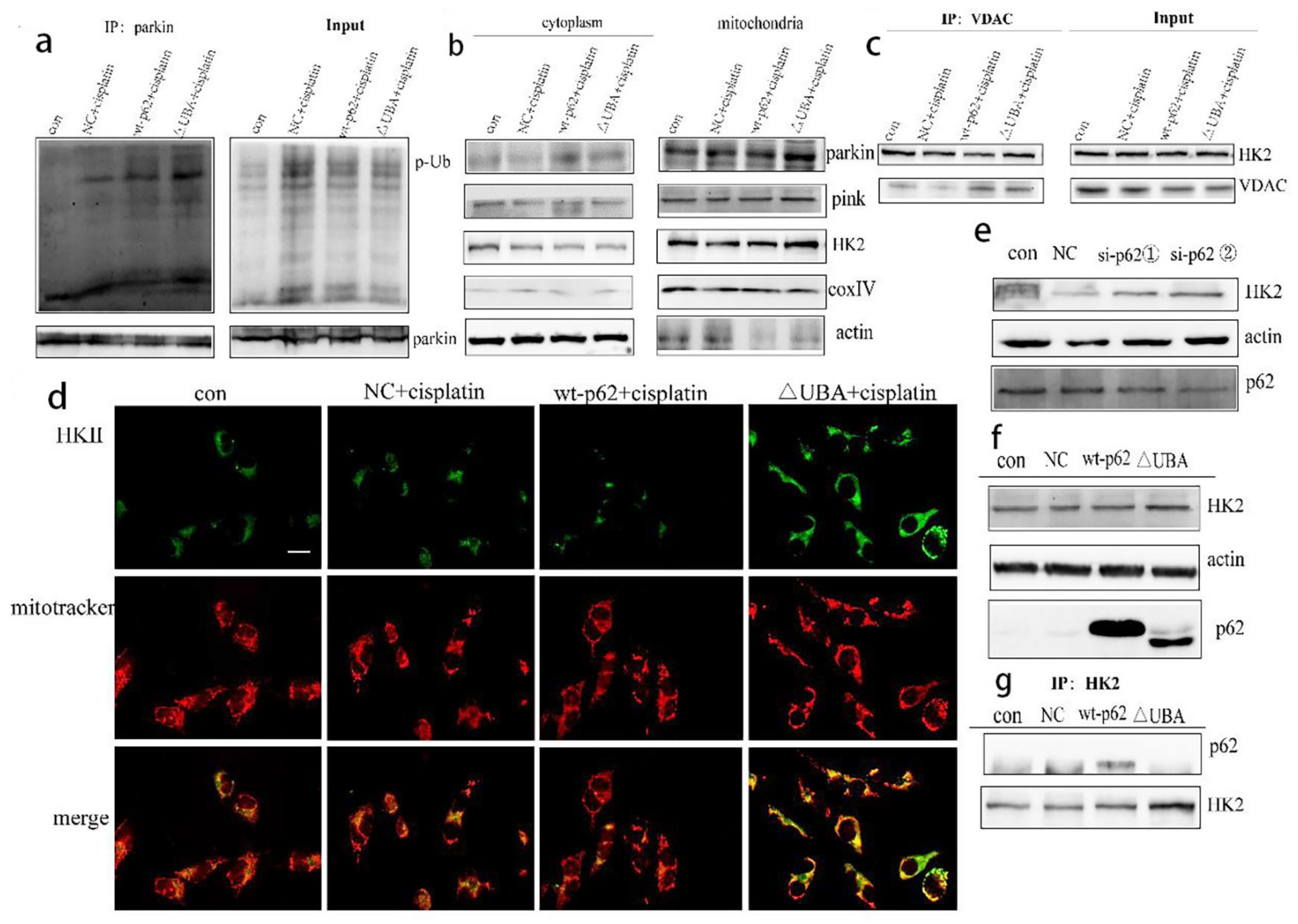
2.3. Recruitment of HK2 to the Mitochondria Is the Key to the UBA Domain-Mediated Mitophagy Regulation in Ovarian Cancer
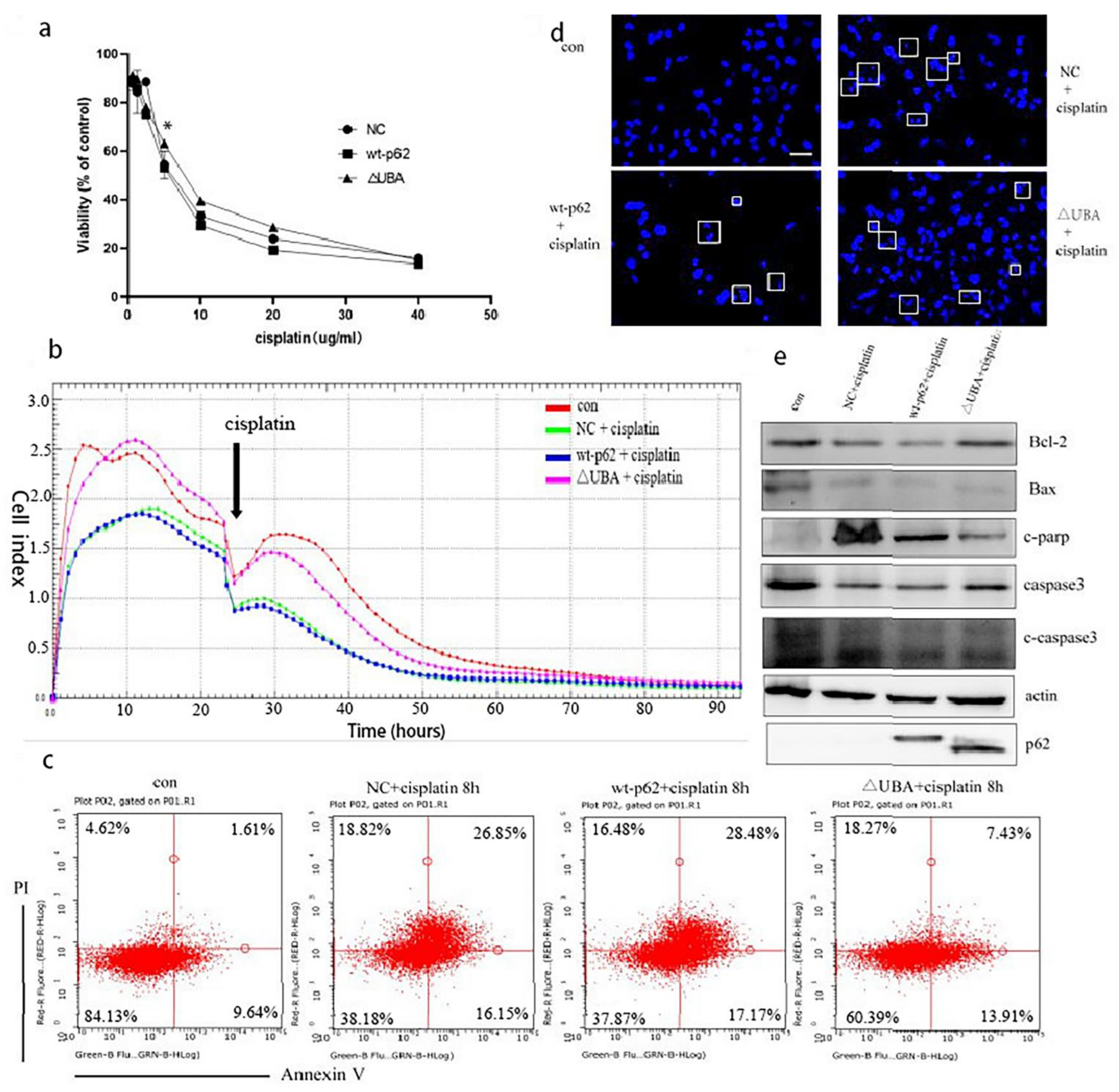
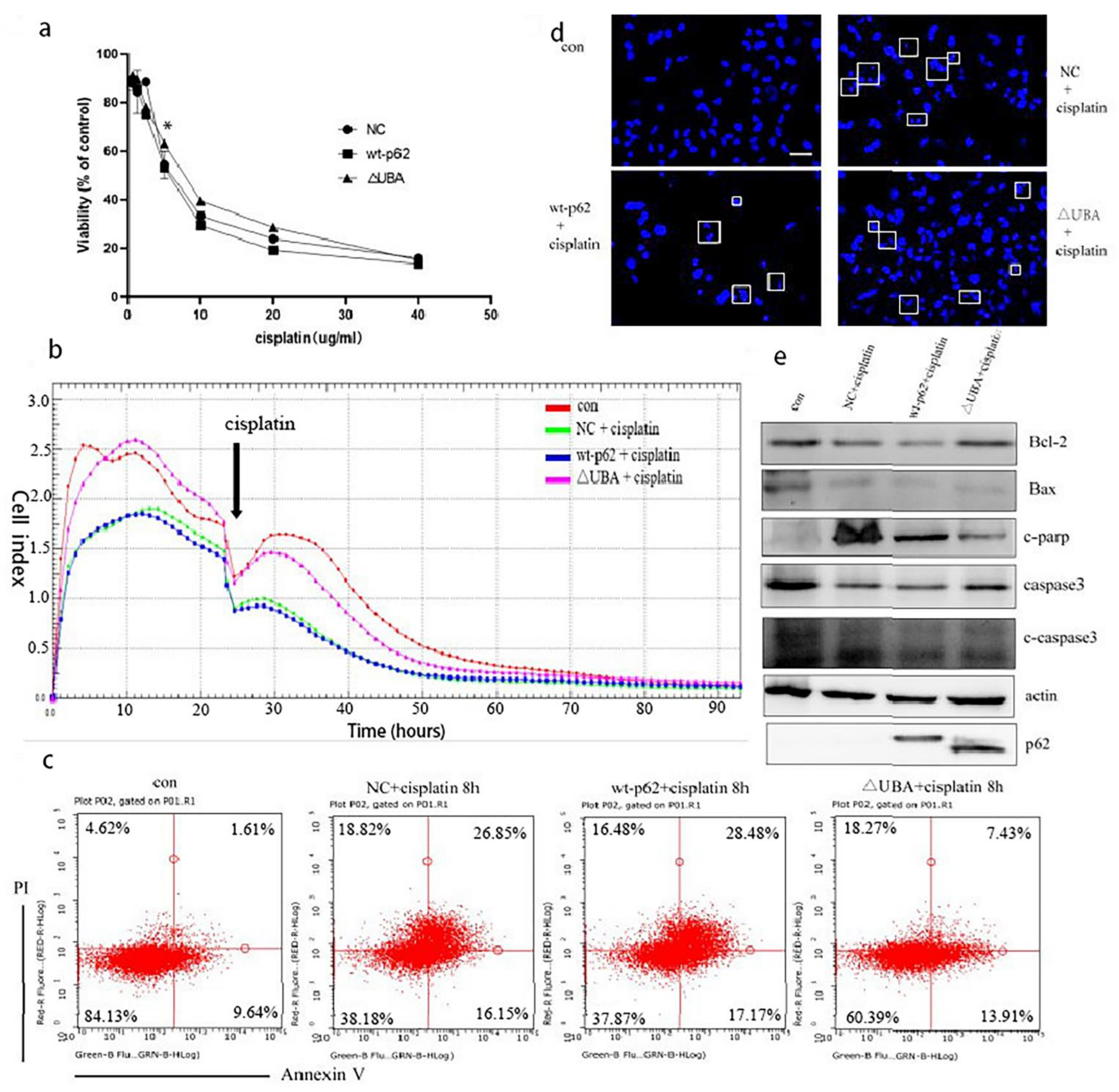
2.4. A Mutation in the UBA Domain of p62 Causes Balanced Regulation of Mitophagy by Altering the Cisplatin Sensitivity of Ovarian Cancer Cells
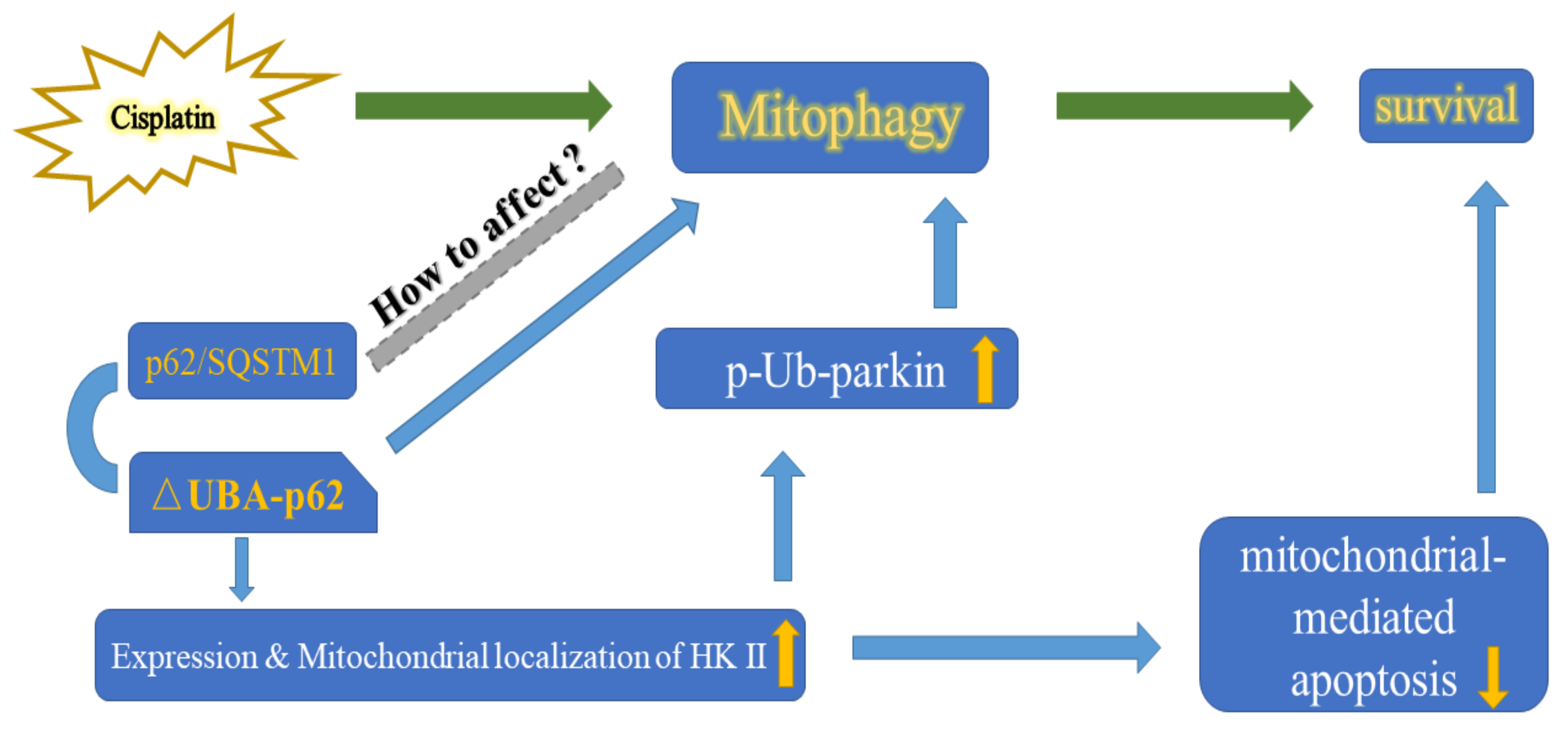
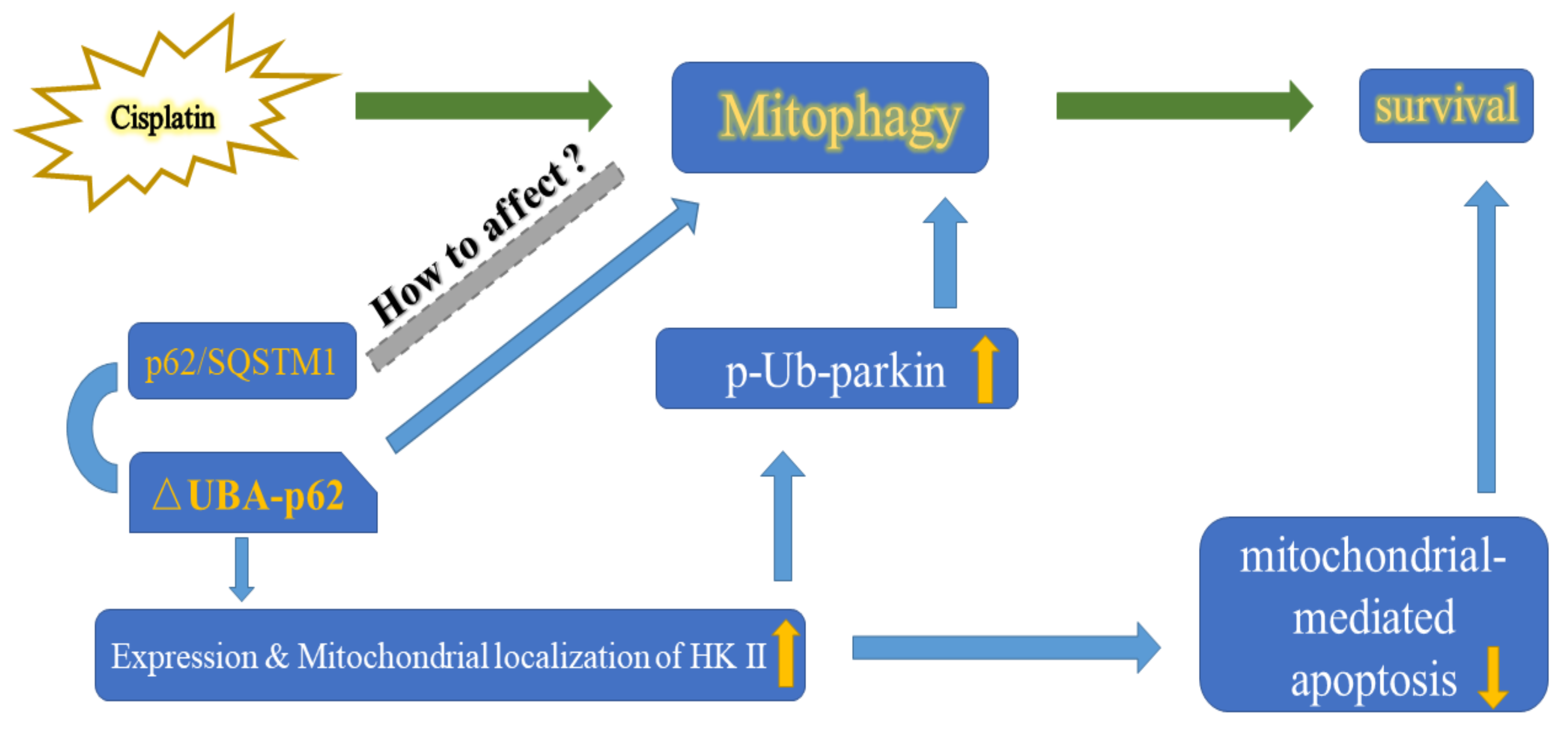
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Plasmid and Transfection
4.3. Cytotoxicity Assays
4.4. Western Blotting
4.5. Mitochondria Separation
4.6. Co-Immunoprecipitation
4.7. Mitochondrial Depolarisation Assessment
4.8. mt-Keima-COX8 A2780 Cell Line to Assess the Level of Mitochondrial Autophagy
4.8.1. Construction of mt-Keima-COX8 A2780 Cell Line
4.8.2. Flow Cytometry
4.8.3. Fluorescence Microscopy Evaluation
4.9. RT-PCR to Detect Mitochondrial Copy Number
4.10. Real-Time Cell Analysis (RTCA)
4.11. Flow Cytometry Analysis
4.12. Immunofluorescence and Microscopy
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian Cancer. Nat. Rev. Dis. Primers. 2016, 2, 16061. [Google Scholar] [CrossRef]
- Tian, M.; Chen, .X.-S.; Li, L.-Y.; Wu, H.-Z.; Zeng, D.; Wang, X.-L.; Zhang, Y.; Xiao, S.S.; Cheng, Y. Inhibition of AXL Enhances Chemosensitivity of Human Ovarian Cancer Cells to Cisplatin via Decreasing Glycolysis. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef]
- Ma, S.; Liu, D.; Tan, W.; Du, B.; Liu, W.; Li, W.; Jiao, Y. Interference with SMO Increases Chemotherapy Drug Sensitivity of A2780/DDP Cells by Inhibiting the Hh/Gli Signaling Pathway. J. Cell. Biochem. 2020, 121, 3256–3265. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [Green Version]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Abdrakhmanov, A.; Kulikov, A.V.; Luchkina, E.A.; Zhivotovsky, B.; Gogvadze, V. Involvement of mitophagy in cisplatin-induced cell death regulation. Biol. Chem. 2019, 400, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and Cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy. Autophagy 2021, 17, 1–382. [Google Scholar]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and Mitophagy in Cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, Z.; Xu, X.; An, X.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zhang, B.; Zhang, A.; et al. Pink1/Parkin-Mediated Mitophagy Play a Protective Role in Cisplatin Induced Renal Tubular Epithelial Cells Injury. Exp. Cell. Res. 2017, 350, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-Mediated Mitophagy is Activated in Cisplatin Nephrotoxicity to Protect Against Kidney Injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, Y.; Zhang, J.; He, H.; Ren, X. Circumvention of Cisplatin Resistance in Ovarian Cancer by Combination of Cyclosporin A and Low-Intensity Ultrasound. Eur. J. Pharm. Biopharm. 2015, 91, 103–110. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.; Chu, C.T. After the Banquet: Mitochondrial Biogenesis, Mitophagy, and Cell Survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The Mitochondrial Intramembrane Protease PARL Cleaves Human Pink1 to Regulate Pink1 Trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial Homeostasis: The Interplay between Mitophagy and Mitochondrial Biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.N.; Yu, B.B.; Li, J.L.; Guo, R.; Zhang, L.C.; Sun, L.K.; Liu, Y.N.; Li, Y. Zinc and p53 Disrupt Mitochondrial Binding of HK2 by Phosphorylating VDAC1. Exp. Cell Res. 2019, 374, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Przedborski, S. PINK1 Points Parkin to Mitochondria. Autophagy 2010, 6, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Ishikawa, H.; Furuoka, M.; Yokozeki, M.; Matsuda, N.; Tanimura, S.; Takeda, K. Cleaved PGAM5 is Released from Mitochondria Depending on Proteasome-Mediated Rupture of the Outer Mitochondrial Membrane during Mitophagy. J. Biochem. 2019, 165, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Sheng, M. Mechanisms of Mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Heo, J.-M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining Roles of PARKIN and Ubiquitin Phosphorylation by PINK1 in Mitochondrial Quality Control Using a Ubiquitin Replacement Strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II Positively Regulates Glucose Starvation-Induced Autophagy through TORC1 Inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and TORCing to Autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, J.G.; Hoek, J.B. Regulation of Hexokinase Binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of Glycolytic Metabolism by Autophagy in Liver Cancer Involves Selective Autophagic Degradation of HK2 (Hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular Localization of Hexokinases I and II Directs the Metabolic Fate of Glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Wang, R.; Zhang, Y.; Zhou, L.; Wang, W.; Liu, H.; Li, W. Skp2-Mediated Ubiquitination and Mitochondrial Localization of Akt Drive Tumor Growth and Chemoresistance to Cisplatin. Oncogene 2019, 38, 7457–7472. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Long, M.; Li, X.; Li, L.; Dodson, M.; Zhang, D.D.; Zheng, H. Multifunctional p62 Effects Underlie Diverse Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of Selective Autophagy: The p62/SQSTM1 Paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 Activates its Autophagy Receptor Function and Controls Selective Autophagy upon Ubiquitin Stress. Cell Res. 2017, 27, 657–674. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-Mediated Mitophagy is Dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Gallego, I.; Soria, M.E.; Gregori, J.; de Avila, A.I.; Garcia-Crespo, C.; Moreno, E.; Gadea, I.; Esteban, J.; Fernández-Roblas, R.; Esteban, J.I.; et al. Synergistic Lethal Mutagenesis of Hepatitis C Virus. Antimicrob. Agents Chemother. 2019, 63, e01653-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, D.; Liu, X.; Wang, Q.; Chen, X.; Hu, X.; Wang, Q.; Jin, C.; Wen, L.; Zhang, L. Targeting Laryngeal Cancer Cells with 5-Fluorouracil and Curcumin Using Mesoporous Silica Nanoparticles. Technol. Cancer Res. Treat. 2020, 19, 1533033820962114. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate Role of Mitochondrial Lipid in Mitophagy and Mitochondrial Apoptosis: Its Implication in Cancer Therapeutics. Cell Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring in Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 Functions as a Signaling Hub and An Autophagy Adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell Mol. Biol. Lett. 2016, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1-Steering the Cell through Health and Disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Yin, S.; Zhang, E.; Hu, H. Role of p62 in the Regulation of Cell Death Induction. Apoptosis 2018, 23, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.Y.; Zhang, Y.; Zhang, J.J.; Zhang, L.C.; Liu, Y.N.; Wu, Y.; Xue, Y.N.; Lu, S.Y.; Su, J.; Sun, L.K. p62/SQSTM1 as An Oncotarget Mediates Cisplatin Resistance through Activating RIP1-NF-kappaB Pathway in Human Ovarian Cancer Cells. Cancer Sci. 2017, 108, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Y.; Zhong, X.R.; Yu, S.H.; Zhang, L.C.; Liu, Y.N.; Zhang, Y.; Sun, L.K.; Su, J. p62 Aggregates Mediated Caspase 8 Activation is Responsible for Progression of Ovarian Cancer. J. Cell Mol. Med. 2019, 23, 4030–4042. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial Elongation-Mediated Glucose Metabolism Reprogramming is Essential for Tumour Cell Survival during Energy Stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Shi, S.P.; Qiu, J.D.; Sun, X.Y.; Huang, J.H.; Huang, S.Y.; Suo, S.B.; Liang, R.P.; Zhang, L. Identify Submitochondria and Subchloroplast Locations with Pseudo Amino Acid Composition: Approach from the Strategy of Discrete Wavelet Transform Feature Extraction. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, G.-L.; Li, Q.-Z. Predicting Protein Submitochondria Locations by Combining Different Descriptors into the General Form of Chou’s Pseudo Amino Acid Composition. Amino Acids 2012, 43, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yip, Y.M.; Li, L. In Silico Construction of HK2-VDAC1 Complex and Investigating the HK2 Binding-Induced Molecular Gating Mechanism of VDAC1. Mitochondrion 2016, 30, 222–228. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J. Am. Heart Assoc. 2017, 6, e005328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Yan, X.; Tian, R.; Xu, L.; Zhao, Y.; Sun, L.; Su, J. An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells. Int. J. Mol. Sci. 2021, 22, 3983. https://doi.org/10.3390/ijms22083983
Yu S, Yan X, Tian R, Xu L, Zhao Y, Sun L, Su J. An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells. International Journal of Molecular Sciences. 2021; 22(8):3983. https://doi.org/10.3390/ijms22083983
Chicago/Turabian StyleYu, Sihang, Xiaoyu Yan, Rui Tian, Long Xu, Yuanxin Zhao, Liankun Sun, and Jing Su. 2021. "An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells" International Journal of Molecular Sciences 22, no. 8: 3983. https://doi.org/10.3390/ijms22083983
APA StyleYu, S., Yan, X., Tian, R., Xu, L., Zhao, Y., Sun, L., & Su, J. (2021). An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells. International Journal of Molecular Sciences, 22(8), 3983. https://doi.org/10.3390/ijms22083983