
From Stem Cells to Bone-Forming Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Defining Bone-Forming Cells
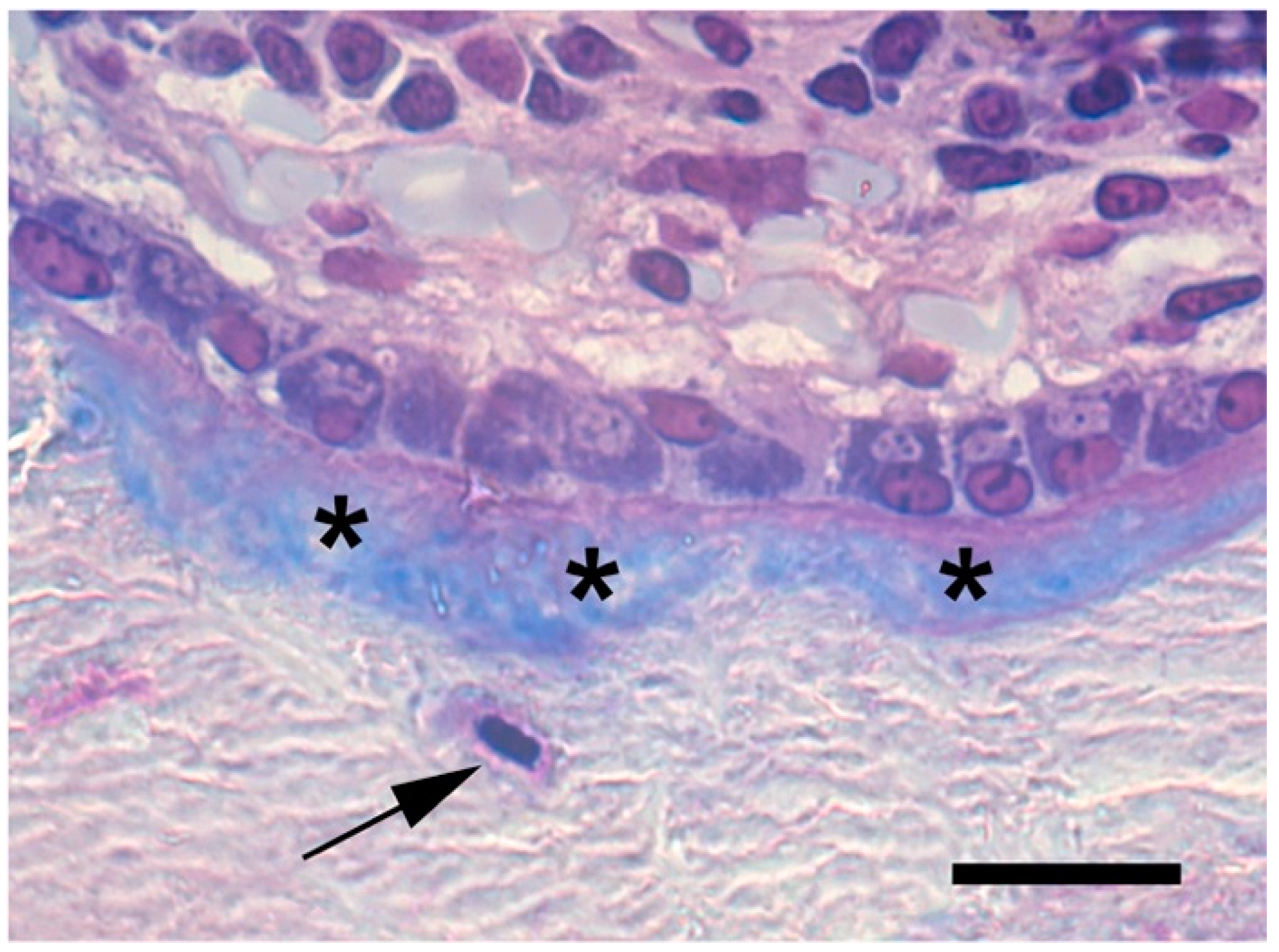
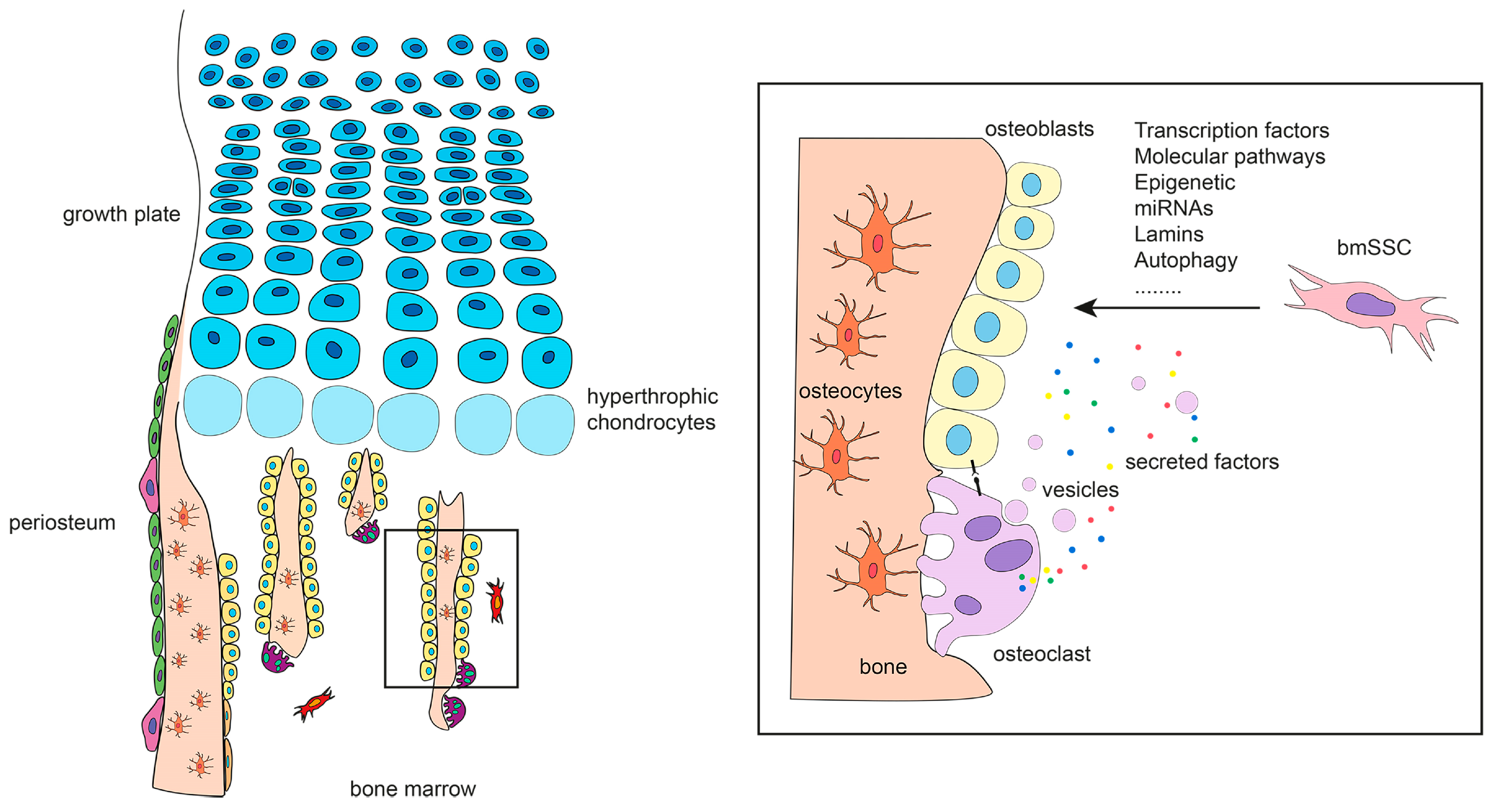
3. The Origin of Bone-Forming Cells in Developing Bones
4. The Origin of Bone-Forming Cells in Fetal and Adult Bones
4.1. Bone Marrow Skeletal Stem Cells
4.2. Other Sources of Bone-Forming Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | |||
|---|---|---|---|
| Source | Progenitor | Progeny (In Vivo) | Reference |
| Adult bone marrow | CD146+ | Osteoblasts, adipocytes and stromal cells | [21] |
| Adult bone marrow | CD271+ | Osteoblasts and stromal cells | [22] |
| Fetal bone marrow | Nestin+/CD45−/ Ter119−/CD31−/ PDGFRα+/CD51+ | Osteoblasts, chondrocytes and adipocytes | [23] |
| Fetal and adult bone | PDPN+/CD146−/ CD73+/CD164+ | Osteoblasts, chondrocytes and stromal cells | [40] |
| Periosteum | Lin−/CD90−/ CD200+/CD105− | Osteoblasts | [42] |
| Mouse | |||
| Source | Progenitor | Progeny (In Vivo) | Reference |
| Limb bud mesenchyme | Sox9-Cre | Osteoblasts and chondrocytes | [51] |
| Adult bone marrow | a-SMA+ | Osteoblasts | [52] |
| Adult bone marrow | PDGFRa+/Sca-1+/ CD45−/TER119− | Osteoblasts, adipocytes and stromal cells | [53] |
| Adult bone marrow | Nestin-GFP | Osteoblasts and chondrocytes | [54] |
| Adult bone marrow | CXCL12+ | Osteoblasts and adipocytes | [55] |
| Adult bone marrow | Osx-Cre | Osteoblasts, adipocytes and stromal cells | [56] |
| Adult bone marrow | LepR-Cre | Osteoblasts, chondrocytes and adipocytes | [57] |
| Fetal and adult bone marrow | Osx-Cre | Osteoblasts and stromal cells | [58] |
| Adult bone marrow (metaphysis) | Gremlin1-Cre | Osteoblasts, chondrocytes and stromal cells | [59] |
| Growth plate | CD45−/Ter119−/Tie2−/ Thy1−/6C3−/CD51+/ CD105−/CD200+ | Osteoblasts, chondrocytes and stromal cells | [41] |
| Adult bone marrow (methaphysis) | Gli1-CreERT2 | Osteoblasts, chondrocytes and adipocytes | [60] |
| Adult bone marrow | CD45−/CD31−/ Sca1+/CD24+ | Osteoblasts, chondrocytes and adipocytes | [61] |
| Periosteum | Catepsin K-Cre | Osteoblasts | [42] |
| Postnatal growth plate resting chondrocytes | Pthrp-mCherry | Osteoblasts, chondrocytes and stromal cells | [62] |
| Perichondrium, periosteum and bone marrow | Hoxa11-CreERT2 | Osteoblasts, chondrocytes and adipocytes | [63] |
| Fetal bone marrow | Kit-MerCreMer | Osteoblasts, chondrocytes and stromal cells | [64] |
5. Specification of Bone-Forming Cells
5.1. Transcription Factors
5.2. Molecular Pathways
5.3. Gsα-cAMP Signaling Pathway
5.4. Epigenetic Regulation and miRNAs
5.5. Lamins and Autophagy
5.6. Osteoclasts as Modulators of Osteoblastogenesis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [Green Version]
- Valenti, M.T.; Dalle Carbonare, L.; Mottes, M. Osteogenic Differentiation in Healthy and Pathological Conditions. Int. J. Mol. Sci. 2016, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Stenslokken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, M.; Yuen, T.; Sun, L.; Rosen, C.J. Regulation of skeletal homeostasis. Endocr. Rev. 2018, 39, 701–718. [Google Scholar] [CrossRef]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone—The functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; Parfitt, A.M. What old means to bone. Trends Endocrinol. Metab. 2010, 21, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.C.; De Saint-Georges, L.; Bowman, B.M.; Jee, W.S.S. Bone lining cells: Structure and function. Scanning Microsc. 1989, 3, 953–960. [Google Scholar]
- Kim, S.W.; Lu, Y.; Williams, E.A.; Lai, F.; Lee, J.Y.; Enishi, T.; Balani, D.H.; Ominsky, M.S.; Ke, H.Z.; Kronenberg, H.M.; et al. Sclerostin Antibody Administration Converts Bone Lining Cells Into Active Osteoblasts. J. Bone Miner. Res. 2017, 32, 892–901. [Google Scholar] [CrossRef]
- Miller, S.C.; Bowman, B.M. Medullary bone osteogenesis following estrogen administration to mature male Japanese quail. Dev. Biol. 1981, 87, 52–63. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000, 21, 115–137. [Google Scholar]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Currey, J.D. The many adaptations of bone. J. Biomech. 2003, 36, 1487–1495. [Google Scholar] [CrossRef]
- Hall, B.K.; Miyake, T. The membranous skeleton: The role of cell condensations in vertebrate skeletogenesis. Anat. Embryol. 1992, 186, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Iseki, S.; Wilkie, A.O.M.; Heath, J.K.; Ishimaru, T.; Eto, K.; Morriss-Kay, G.M. Fgfr2 and osteopontin domains in the developing skull vault are mutually exclusive and can be altered by locally applied FGF2. Development 1997, 124, 3375–3384. [Google Scholar]
- Ting, M.C.; Wu, N.L.; Roybal, P.G.; Sun, J.; Liu, L.; Yen, Y.; Maxson, R.E. EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 2009, 136, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell. 2010, 19, 329–344. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Latsinik, N.V.; Panasyvk, A.F.; Keiliss-Borok, I.V. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues: Cloning in vitro and retransplantation in vivo. Transplantation 1974, 17, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.A.; Krebsbach, P.H.; Satomura, K.; Kerr, J.; Riminucci, M.; Benayahu, D.; Robey, P.G. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J. Bone Miner. Res. 1997, 12, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Gerasimov, U.V. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987, 20, 263–272. [Google Scholar] [CrossRef]
- Johnstone, B.; Hering, T.M.; Caplan, A.I.; Goldberg, V.M.; Yoo, J.U. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp. Cell Res. 1998, 238, 265–272. [Google Scholar] [CrossRef]
- Fitter, S.; Gronthos, S.; Ooi, S.S.; Zannettino, A.C. The Mesenchymal Precursor Cell Marker Antibody STRO-1 binds to cell surface Heat Schock Cognate 70. Stem Cells 2017, 35, 940–951. [Google Scholar] [CrossRef] [Green Version]
- Simmons, P.J.; Torok-Storb, B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood 1991, 78, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Tormin, A.; Li, O.; Brune, J.C.; Walsh, S.; Schutz, B.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011, 117, 5067–5077. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFRα and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 6, 897–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, T.H.; Longaker, M.T.; Chan, C.K.F. A Revised Perspective of Skeletal Stem Cell Biology. Front. Cell Dev. Biol. 2019, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Justesen, J.; Stenderup, K.; Ebbesen, E.N.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef]
- Bianco, P.; Bonucci, E. Endosteal surfaces in hyperparathyroidism: An enzyme cytochemical study on low-temperature-processed, glycol-methacrylate-embedded bone biopsies. Virchows Arch. A Pathol. Anat. Histopathol. 1991, 419, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Fisher, L.W.; Shenker, A.; Spiegel, A.M.; Bianco, P.; Robey, P.G. Fibrous dysplasia of bone in the McCune-Albright syndrome: Abnormalities in bone formation. Am. J. Pathol. 1997, 151, 1587–1600. [Google Scholar] [PubMed]
- Owen, M.; MacPerson, S. Cell population kinetics of an osteogenic tissue. II. J. Cell Biol. 1963, 19, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.; Friedenstein, A.J. Stromal stem cells: Marrow-derived osteogenic precursors. Ciba Found. Symp. 1988, 136, 42–60. [Google Scholar]
- Sworder, B.J.; Yoshizawa, S.; Mishra, P.J.; Cherman, N.; Kuznetsov, S.A.; Merlino, G.; Balakumaran, A.; Robey, P.G. Molecular profile of clonal strains of human skeletal stem/progenitor cells with different potencies. Stem Cell Res. 2015, 14, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932. [Google Scholar] [CrossRef]
- Roach, H.I.; Erenpreisa, J.; Aigner, T. Osteogenic differentiation of hypertophic chondrocytes involves asymmetric cell divisions and apoptosis. J. Cell Biol. 1995, 131, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Bradbeer, J.N.; Corsi, A.; Gentili, C.; Descalzi, F.; Cancedda, R.; Bianco, P. Vis-à-vis cells and the priming of bone formation. J. Bone Miner. Res. 1998, 13, 1852–1861. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the Human Skeletal Stem Cell. Cell 2018, 175, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.K.F.; Seo, E.Y.; Chen, J.Y.; Lo, D.; McArdle, A.; Sinha, R.; Tevlin, R.; Seita, J.; Vincent-Tompkins, J.; Wearda, T.; et al. Identification and specification of the mouse skeletal stem cell. Cell 2015, 160, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, S.; Yallowitz, A.R.; McCormick, J.; Lalani, S.; Zhang, T.; Xu, R.; Li, N.; Liu, Y.; Yang, Y.S.; Eiseman, M.; et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 2018, 562, 133–139. [Google Scholar] [CrossRef]
- Duchamp De Lageneste, O.; Julien, A.; Abou-Khalil, R.; Frangi, G.; Carvalho, C.; Cagnard, N.; Cordier, C.; Conway, S.J.; Colnot, C. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 2018, 9, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, S.; Sacchetti, B.; Laitinen, A.; Donsante, S.; Klöckers, R.; Laitinen, S.; Riminucci, M.; Kogler, G. Low oxygen tension reveals distinct HOX codes in human cord blood-derived stromal cells associated with specific endochondral ossification capacities in vitro and in vivo. J. Tissue Eng. Regen. Med. 2017, 11, 2725–2736. [Google Scholar] [CrossRef]
- Galotto, M.; Campanile, G.; Robino, G.; Cancedda, F.D.; Bianco, P.; Cancedda, R. Hypertrophic chondrocytes undergo further differentiation to osteoblast-like cells and participate in the initial bone formation in developing chick embryo. J. Bone Miner. Res. 1994, 9, 1239–1249. [Google Scholar] [CrossRef]
- Holmbeck, K.; Bianco, P.; Chrysovergis, K.; Yamada, S.; Birkedal-Hansen, H. MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: A critical process in skeletal growth. J. Cell Biol. 2003, 163, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, N.; Ono, W.; Nagasawa, T.; Kronenberg, H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014, 16, 1157–1167. [Google Scholar] [CrossRef]
- Yang, G.; Zhu, L.; Hou, N.; Lan, Y.; Wu, X.M.; Zhou, B.; Teng, Y.; Yang, X. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014, 24, 1266–1269. [Google Scholar] [CrossRef] [Green Version]
- Muraglia, A.; Corsi, A.; Riminucci, M.; Mastrogiacomo, M.; Cancedda, R.; Bianco, P.; Quarto, R. Formation of a chondro-osseous rudiment in micromass cultures of human bone-marrow stromal cells. J. Cell Sci. 2003, 116, 2949–2955. [Google Scholar] [CrossRef] [Green Version]
- Serafini, M.; Sacchetti, B.; Pievani, A.; Redaelli, D.; Remoli, C.; Biondi, A.; Riminucci, M.; Bianco, P. Establishment of bone marrow and hematopoietic niches in vivo by reversion of chondrocyte differentiation of human bone marrow stromal cells. Stem Cell Res. 2014, 12, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Kim, J.E.; Nakashima, K.; Balmes, G.; Iwai, N.; Deng, J.M.; Zhang, Z.P.; Martin, J.F.; Behringer, R.R.; Nakamura, T.; et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 2005, 102, 14665–14670. [Google Scholar] [CrossRef] [Green Version]
- Kalajzic, Z.; Li, H.; Wang, L.P.; Jiang, X.; Lamothe, K.; Adams, D.J.; Aguila, H.L.; Rowe, D.W.; Kalajzic, I. Use of an alpha-smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone 2008, 43, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, S.; Mabuchi, Y.; Kubota, Y.; Nagai, Y.; Niibe, K.; Hiratsu, E.; Suzuki, S.; Miyauchi-Hara, C.; Nagoshi, N.; Sunabori, T.; et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 2009, 206, 2483–2496. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The Essential Functions of Adipo-osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.L.; Strecker, S.; Wang, L.P.; Kronenberg, M.S.; Wang, W.; Rowe, D.W.; Maye, P. Osterix-Cre Labeled Progenitor Cells Contribute to the Formation and Maintenance of the Bone Marrow Stroma. PLoS ONE 2013, 8, e71318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.O.; Yue, R.; Murphy, M.M.; Peyer, J.G.; Morrison, S.J. Leptin-Receptor-Expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell 2014, 15, 154–168. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, T.; Pinho, S.; Ahmed, J.; Kunisaki, Y.; Hanoun, M.; Mendelson, A.; Ono, N.; Kronenberg, H.M.; Frenette, P.S. Osterix Marks Distinct Waves of Primitive and Definitive Stromal Progenitors during Bone Marrow Development. Dev. Cell. 2014, 29, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.L.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 Identifies a Skeletal Stem Cell with Bone, Cartilage, and Reticular Stromal Potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; He, G.X.; Lee, W.C.; McKenzie, J.A.; Silva, M.J.; Long, F.X. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017, 8, 2043. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schurmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Mizuhashi, K.; Ono, W.; Matsushita, Y.; Sakagami, N.; Takahashi, A.; Saunders, T.L.; Nagasawa, T.; Kronenberg, H.M.; Ono, N. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 2018, 563, 254–258. [Google Scholar] [CrossRef]
- Pineault, K.M.; Song, J.Y.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun. 2019, 10, 3168. [Google Scholar] [CrossRef] [Green Version]
- He, D.D.; Tang, X.T.; Dong, W.J.; Cui, G.Z.; Peng, G.D.; Yin, X.J.; Chen, Y.J.; Jing, N.H.; Zhou, B.O. C-KIT Expression Distinguishes Fetal from Postnatal Skeletal Progenitors. Stem Cell Rep. 2020, 14, 614–630. [Google Scholar] [CrossRef]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; De Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Graves, R.A.; Budavari, A.I.; Erdjument-bromage, H.; Lui, M.; Hu, E.; Tempst, P.; Spiegelman, B.M. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPAR7 and RXRa. Nucleic Acids Res. 1994, 22, 5628–5634. [Google Scholar] [CrossRef] [Green Version]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.M.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Tosa, I.; Yamada, D.; Yasumatsu, M.; Hinoi, E.; Ono, M.; Oohashi, T.; Kuboki, T.; Takarada, T. Postnatal Runx2 deletion leads to low bone mass and adipocyte accumulation in mice bone tissues. Biochem. Biophys. Res. Commun. 2019, 516, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Starbuck, M.; Priemel, M.; Shen, J.; Pinero, G.; Geoffroy, V.; Amling, M.; Karsenty, G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999, 13, 1025–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Toyosawa, S.; Furuichi, T.; Kanatani, N.; Yoshida, C.; Liu, Y.; Himeno, M.; Narai, S.; Yamaguchi, A.; Komori, T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J. Cell Biol. 2001, 155, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, Z.; Yoshida, C.A.; Furuichi, T.; Amizuka, N.; Ito, M.; Fukuyama, R.; Miyazaki, T.; Kitaura, H.; Nakamura, K.; Fujita, T.; et al. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dev. Dyn. 2007, 236, 1876–1890. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Hinoi, E.; Nakazato, R.; Ochi, H.; Xu, C.; Tsuchikane, A.; Takeda, S.; Karsenty, G.; Abe, T.; Kiyonari, H.; et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J. Bone Miner. Res. 2013, 28, 2064–2069. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; De Crombrugghe, B. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, T. Runx2 regulates cranial suture closure by inducing hedgehog, Fgf, Wnt and Pthlh signaling pathway gene expressions in suture mesenchymal cells. Hum. Mol. Genet. 2019, 28, 896–911. [Google Scholar] [CrossRef] [Green Version]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.d.S.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, Z.; Feng, J.Q.; Dusevich, V.M.; Sinha, K.; Zhang, H.; Darnay, B.G.; De Crombrugghe, B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12919–12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapunzina, P.; Aglan, M.; Temtamy, S.; Caparros-Martin, J.A.; Valencia, M.; Leton, R.; Martinez-Glez, V.; Elhossini, R.; Amr, K.; Vilaboa, N.; et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 87, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, C.A.; Komori, H.; Maruyama, Z.; Miyazaki, T.; Kawasaki, K.; Furuichi, T.; Fukuyama, R.; Mori, M.; Yamana, K.; Nakamura, K.; et al. Sp7 inhibits osteoblast differentiation at a late stage in mice. PLoS ONE 2012, 7, e32364. [Google Scholar] [CrossRef] [Green Version]
- Satokata, I.; Ma, L.; Ohshima, H.; Bei, M.; Ian, W.; Nishizawa, K.; Maeda, T.; Takano, Y.; Uchiyama, M.; Heaney, S.; et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 2000, 24, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Warman, M.L.; Mulliken, J.B.; Hayward, P.G.; Muller, U. Newly recognized autosomal dominant disorder with craniosynostosis. Am. J. Med. Genet. 1993, 46, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Minaur, S.; Mavrogiannis, L.A.; Rannan-Eliya, S.V.; Hendry, M.A.; Liston, W.A.; Porteous, M.E.M.; Wilkie, A.O.M. Parietal foramina with cleidocranial dysplasia is caused by mutation in MSX2. Eur. J. Hum. Genet. 2003, 11, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialek, P.; Kern, B.; Yang, X.; Schrock, M.; Sosic, D.; Hong, N.; Wu, H.; Yu, K.; Ornitz, D.M.; Olson, E.N.; et al. A twist code determines the onset of osteoblast differentiation. Dev. Cell. 2004, 6, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Howard, T.D.; Paznekas, W.A.; Green, E.D.; Chiang, L.C.; Ma, N.; Ortiz De Luna, R.I.; Delgado, C.G.; Gonzalez-Ramos, M.; Kline, A.D.; Jabs, E.W. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 1997, 15, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kveiborg, M.; Sabatakos, G.; Chiusaroli, R.; Wu, M.; Philbrick, W.M.; Horne, W.C.; Baron, R. DeltaFosB Induces Osteosclerosis and Decreases Adipogenesis by Two Independent Cell-Autonomous Mechanisms. Mol. Cell Biol. 2004, 24, 2820–2830. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Nakashima, T.; Takeda, S.; Isogai, M.; Hamada, M.; Kimura, A.; Kodama, T.; Yamaguchi, A.; Owen, M.J.; Takahashi, S.; et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Investig. 2010, 120, 3455–3465. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, P.; Xu, S.; Li, Y.; Dekker, J.D.; Li, B.; Fan, Y.; Zhang, Z.; Hong, Y.; Yang, G.; et al. FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J. Clin. Investig. 2017, 127, 1241–1253. [Google Scholar] [CrossRef]
- Canalis, E. Growth Factor Control of Bone Mass. J. Cell Biochem. 2009, 108, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell. 2005, 8, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Rodda, S.J.; McMahon, A.P. Distinct roles for Hedgehog and caronical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 2006, 133, 3231–3244. [Google Scholar] [CrossRef] [Green Version]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell. 2005, 8, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Liu, M.; Ono, N.; Bringhurst, F.R.; Kronenberg, H.M.; Guo, J. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 2012, 27, 2344–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmen, S.L.; Zylstra, C.R.; Mukherjee, A.; Sigler, R.E.; Faugere, M.C.; Bouxsein, M.L.; Deng, L.; Clemens, T.L.; Williams, B.O. Essential role of β-catenin in postnatal bone acquisition. J. Biol. Chem. 2005, 280, 21162–21168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Zylstra-Diegel, C.R.; Schumacher, C.A.; Baker, J.J.; Carpenter, A.C.; Rao, S.; Yao, W.; Guan, M.; Helms, J.A.; Lane, N.E.; et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc. Natl. Acad. Sci. USA 2012, 109, E2197–E2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.A.; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell. 2005, 8, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Van Wesenbeeck, L.; Cleiren, E.; Gram, J.; Beals, R.K.; Benichou, O.; Scopelliti, D.; Key, L.; Renton, T.; Bartels, C.; Gong, Y.; et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 2003, 72, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.-H.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Moester, M.J.C.; Papapoulos, S.E.; Löwik, C.W.G.M.; Van Bezooijen, R.L. Sclerostin: Current knowledge and future perspectives. Calcif. Tissue Int. 2010, 87, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Joeng, K.S.; Long, F. Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev. Biol. 2012, 362, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Kitaura, Y.; Hojo, H.; Komiyama, Y.; Takato, T.; Chung, U.I.; Ohba, S. Gli1 haploinsufficiency leads to decreased bone mass with an uncoupling of bone metabolism in adult mice. PLoS ONE 2014, 9, e109597. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Guo, J.; She, C.; Shu, A.; Yang, M.; Tan, Z.; Yang, X.; Guo, S.; Feng, G.; He, L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 2001, 28, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF–β3 indicates defects of epithelial–mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-Β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, A.; Saito, T.; Tomita, H.; Makita, Y.; Yoshida, K.; Ghadami, M.; Yamada, K.; Kondo, S.; Ikegawa, S.; Nishimura, G.; et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat. Genet. 2000, 26, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Tsuji, K.; Cox, K.; Harfe, B.D.; Rosen, V.; Tabin, C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006, 2, e216. [Google Scholar] [CrossRef] [Green Version]
- Salazar, V.S.; Zarkadis, N.; Huang, L.; Norris, J.; Grimston, S.K.; Mbalaviele, G.; Civitelli, R. Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical wnt signaling and causes an osteogenesis-imperfectalike phenotype. J. Cell Sci. 2013, 126, 4974–4984. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Weng, T.; Zhang, J.; Wang, J.; Li, W.; Wan, H.; Lan, Y.; Cheng, X.; Hou, N.; Liu, H.; et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 2007, 120, 2162–2170. [Google Scholar] [CrossRef] [Green Version]
- Locklin, R.M.; Riggs, B.L.; Hicok, K.C.; Horton, H.F.; Byrne, M.C.; Khosla, S. Assessment of gene regulation by bone morphogenetic protein 2 in human marrow stromal cells using gene array technology. J. Bone Miner. Res. 2001, 16, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 regulates osterix through Msx2 and Runx2 during osteoblast differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, I.H.; Jeong, B.C.; Hur, S.W.; Choi, H.; Choi, S.H.; Ryu, J.H.; Hwang, Y.C.; Koh, J.T. MicroRNA-302a stimulates osteoblastic differentiation by repressing COUP-TFII expression. J. Cell Physiol. 2015, 230, 911–921. [Google Scholar] [CrossRef]
- Jun, J.H.; Yoon, W.J.; Seo, S.B.; Woo, K.M.; Kim, G.S.; Ryoo, H.M.; Baek, J.H. BMP2-activated Erk/MAP kinase stabilizes runx2 by increasing p300 levels and histone acetyltransferase activity. J. Biol. Chem. 2010, 285, 36410–36419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, T.; Yamaguchi, A.; Komaki, M.; Abe, E.; Takahashi, N.; Ikeda, T.; Rosen, V.; Wozney, J.M.; Fujisawa-Sehara, A.; Suda, T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 1994, 127, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Gejman, P.V.; Friedman, E.; Kadowaki, T.; Collins, R.M.; Gershon, E.S.; Spiegel, A.M. Mutations of the Gs alpha-subunit gene in Albright hereditary osteodystrophy detected by denaturing gradient gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 8287–8290. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, L.S.; Yu, S. The role of genomic imprinting of GS-alpha in the pathogenesis of Albright hereditary osteodystrophy. Trends Endocr. Metab. 1999, 10, 81–85. [Google Scholar] [CrossRef]
- Shore, E.M.; Ahn, J.; Jan de Beur, S.; Li, M.; Xu, M.; Gardiner, R.J.M.; Zasloff, M.A.; Whyte, M.P.; Levine, M.A.; Kaplan, F.S. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N. Engl. J. Med. 2002, 346, 99–106. [Google Scholar] [CrossRef]
- Riminucci, M.; Saggio, I.; Robey, P.G.; Bianco, P. Fibrous dysplasia as a stem cell disease. J. Bone Miner. Res. 2006, 21 (Suppl. 2), 125–131. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating Mutations of the Stimulatory G Protein in the McCune-Albright Syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Regard, J.B.; Malhotra, D.; Gvozdenovic-Jeremic, J.; Josey, M.; Chen, M.; Weinstein, L.S.; Lu, J.; Shore, E.M.; Kaplan, F.S.; Yang, Y. Activation of hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013, 19, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Khan, S.K.; Zhou, T.; Gao, B.; Zhou, Y.; Zhou, X.; Yang, Y. Gαs signaling controls intramembranous ossification during cranial bone development by regulating both Hedgehog and Wnt/β-catenin signaling. Bone Res. 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Aarnisalo, P.; Bastepe, M.; Sinha, P.; Fulzele, K.; Selig, M.K.; Chen, M.; Poulton, I.J.; Purton, L.E.; Sims, N.A.; et al. Gsα enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J. Clin. Investig. 2011, 121, 3492–3504. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Aarnisalo, P.; Chubb, R.; Ono, N.; Fulzele, K.; Selig, M.; Saeed, H.; Chen, M.; Weinstein, L.S.; Pajevic, P.D.; et al. Loss of Gsα early in the osteoblast lineage favors adipogenic differentiation of mesenchymal progenitors and committed osteoblast precursors. J. Bone Miner. Res. 2014, 29, 2414–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lietman, S.A.; Ding, C.; Cooke, D.W.; Levine, M.A. Reduction in Gsα induces osteogenic differentiation in human mesenchymal stem cells. Clin. Orthop. Relat. Res. 2005, 434, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Chen, M.; Nakamura, T.; Xie, T.; Karsenty, G.; Weinstein, L.S. Deficiency of the G-protein α-subunit Gsα in osteoblasts leads to differential effects on trabecular and cortical bone. J. Biol. Chem. 2005, 280, 21369–21375. [Google Scholar] [CrossRef] [Green Version]
- Fulzele, K.; Dedic, C.; Lai, F.; Bouxsein, M.; Lotinun, S.; Baron, R.; Divieti Pajevic, P. Loss of Gsα in osteocytes leads to osteopenia due to sclerostin induced suppression of osteoblast activity. Bone 2018, 117, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Remoli, C.; Michienzi, S.; Sacchetti, B.; Consiglio, A.D.; Cersosimo, S.; Spica, E.; Robey, P.G.; Holmbeck, K.; Cumano, A.; Boyde, A.; et al. Osteoblast-specific expression of the fibrous dysplasia (FD)-causing mutation GsαR201C produces a high bone mass phenotype but does not reproduce FD in the mouse. J. Bone Miner. Res. 2015, 30, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sugamori, K.S.; Claridge, C.; dela Cruz, A.; Grynpas, M.D.; Mitchell, J. Overexpression of GαS in Murine Osteoblasts In Vivo Leads to Increased Bone Mass and Decreased Bone. J. Bone Miner. Res. 2017, 32, 2171–2181. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Sims, N.A.; Hunzelman, J.L.; Knight, M.C.; Giovannetti, A.; Saxton, J.M.; Kronenberg, H.M.; Baron, R.; Schipani, E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Investig. 2001, 107, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Saggio, I.; Remoli, C.; Spica, E.; Sacchetti, B.; Robey, P.G.; Holmbeck, K.; Cumano, A.; Boyde, A.; Bianco, P.; Riminucci, M. Constitutive expression of gsar201c in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J. Bone Miner. Res. 2014, 29, 2357–2368. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.K.; Yadav, P.S.; Elliott, G.; Hu, D.Z.; Xu, R.; Yang, Y. Induced GnasR201H expression from the endogenous Gnas locus causes fibrous dysplasia by up-regulating Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E418–E427. [Google Scholar] [CrossRef] [Green Version]
- Candeliere, G.A.; Glorieux, F.H.; Prud’homme, J.; St.-Arnaud, R. Increased Expression of the c- fos Proto-Oncogene in Bone from Patients with Fibrous Dysplasia. N. Engl. J. Med. 1995, 332, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; De Pollak, C.; Chanson, P.; Lomri, A. Increased proliferation of osteoblastic cells expressing the activating Gsα mutation in monostotic and polyostotic fibrous dysplasia. Am. J. Pathol. 1997, 150, 1059–1069. [Google Scholar] [PubMed]
- Corsi, A.; Collins, M.T.; Riminucci, M.; Howell, P.G.T.; Boyde, A.; Gehron Robey, P.; Bianco, P. Osteomalacic and Hyperparathyroid Changes in Fibrous Dysplasia of Bone: Core Biopsy Studies and Clinical Correlations. J. Bone Miner. Res. 2003, 18, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Ippolito, E.; Robey, P.G.; Riminucci, M.; Boyde, A. Bisphosphonate-induced zebra lines in fibrous dysplasia of bone: Histo-radiographic correlation in a case of McCune–Albright syndrome. Skeletal Radiol. 2017, 46, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Liu, B.; Corsi, A.; Shenker, A.; Spiegel, A.M.; Robey, P.G.; Bianco, P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gsa gene: Site-specific patterns and recurrent histological hallmarks. J. Pathol. 1999, 187, 249–258. [Google Scholar] [CrossRef]
- Hemming, S.; Cakouros, D.; Codrington, J.; Vandyke, K.; Arthur, A.; Zannettino, A.; Gronthos, S. EZH2 deletion in early mesenchyme compromises postnatal bone microarchitecture and structural integrity and accelerates remodeling. FASEB J. 2017, 31, 1011–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakouros, D.; Gronthos, S. Epigenetic Regulators of Mesenchymal Stem/Stromal Cell Lineage Determination. Curr. Osteoporos Rep. 2020, 18, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Pasumarthy, K.K.; Doni Jayavelu, N.; Kilpinen, L.; Andrus, C.; Battle, S.L.; Korhonen, M.; Lehenkari, P.; Lund, R.; Laitinen, S.; Hawkins, R.D. Methylome Analysis of Human Bone Marrow MSCs Reveals Extensive Age- and Culture-Induced Changes at Distal Regulatory Elements. Stem Cell Rep. 2017, 9, 999–1015. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, P.; El-Jawhari, J.J.; Giannoudis, P.V.; Burska, A.N.; Ponchel, F.; Jones, E.A. Age-related Changes in Bone Marrow Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development. Cell Transplant. 2017, 26, 1520–1529. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, T.; Neshati, Z. Role of microRNAs in osteogenesis of stem cells. J. Cell Biochem. 2019, 120, 14136–14155. [Google Scholar] [CrossRef]
- Li, Z.; Hassan, M.Q.; Volinia, S.; Van Wijnen, A.J.; Stein, J.L.; Croce, C.M.; Lian, J.B.; Stein, G.S. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA 2008, 105, 13906–13911. [Google Scholar] [CrossRef] [Green Version]
- Inose, H.; Ochi, H.; Kimura, A.; Fujita, K.; Xu, R.; Sato, S.; Iwasaki, M.; Sunamura, S.; Takeuchi, Y.; Fukumoto, S.; et al. A microRNA regulatory mechanism of osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 20794–20799. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Luo, G.; Bo, Z.; Liang, X.; Huang, J.; Li, D. Impaired osteogenic differentiation associated with connexin43/microRNA-206 in steroid-induced avascular necrosis of the femoral head. Exp. Mol. Pathol. 2016, 101, 89–99. [Google Scholar] [CrossRef]
- Eskildsen, T.; Taipaleenmäki, H.; Stenvang, J.; Abdallah, B.M.; Ditzel, N.; Nossent, A.Y.; Bak, M.; Kauppinen, S.; Kassem, M. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 6139–6144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candini, O.; Spano, C.; Murgia, A.; Grisendi, G.; Veronesi, E.; Piccinno, M.S.; Ferracin, M.; Negrini, M.; Giacobbi, F.; Bambi, F.; et al. Mesenchymal progenitors aging highlights a mir-196 switch targeting HOXB7 as master regulator of proliferation and osteogenesis. Stem Cells 2015, 33, 939–950. [Google Scholar] [CrossRef]
- Li, C.J.; Cheng, P.; Liang, M.K.; Chen, Y.S.; Lu, Q.; Wang, J.Y.; Xia, Z.Y.; Zhou, H.D.; Cao, X.; Xie, H.; et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 2015, 125, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Gargiuli, C.; Schena, E.; Mattioli, E.; Columbaro, M.; D’Apice, M.R.; Novelli, G.; Greggi, T.; Lattanzi, G. Lamins and bone disorders: Current understanding and perspectives. Oncotarget 2018, 9, 22817–22831. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Berendsen, A.D.; Jia, S.; Lotinun, S.; Baron, R.; Ferrara, N.; Olsen, B.R. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J. Clin. Investig. 2012, 122, 3101–3113. [Google Scholar] [CrossRef] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Akter, R.; Rivas, D.; Geneau, G.; Drissi, H.; Duque, G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J. Bone Miner. Res. 2009, 24, 283–293. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lai, J.K.; Xiong, Z.M.; Ren, M.; Moorer, M.C.; Stains, J.P.; Cao, K. Diminished Canonical β-Catenin Signaling During Osteoblast Differentiation Contributes to Osteopenia in Progeria. J. Bone Miner. Res. 2018, 33, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Infante, A.; Gago, A.; de Eguino, G.R.; Calvo-Fernández, T.; Gómez-Vallejo, V.; Llop, J.; Schlangen, K.; Fullaondo, A.; Aransay, A.M.; Martín, A.; et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging 2014, 6, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrefite-Carle, V.; Santucci-Darmanin, S.; Breuil, V.; Camuzard, O.; Carle, G.F. Autophagy in bone: Self-eating to stay in balance. Ageing Res. Rev. 2015, 24, 206–217. [Google Scholar] [CrossRef]
- Song, C.; Song, C.; Tong, F. Autophagy induction is a survival response against oxidative stress in bone marrow-derived mesenchymal stromal cells. Cytotherapy 2014, 16, 1361–1370. [Google Scholar] [CrossRef]
- Whitehouse, C.A.; Waters, S.; Marchbank, K.; Horner, A.; McGowan, N.W.A.; Jovanovic, J.V.; Xavier, G.M.; Kashima, T.G.; Cobourne, M.T.; Richards, G.O.; et al. Neighbor of Brca1 gene (Nbr1) functions as a negative regulator of postnatal osteoblastic bone formation and p38 MAPK activity. Proc. Natl. Acad. Sci. USA 2010, 107, 12913–12918. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Fang, F.; Yuan, H.; Yang, D.; Chen, Y.; Williams, L.; Goldstein, S.A.; Krebsbach, P.H.; Guan, J.L. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J. Bone Miner. Res. 2013, 28, 2414–2430. [Google Scholar] [CrossRef] [Green Version]
- Piemontese, M.; Onal, M.; Xiong, J.; Han, L.; Thostenson, J.D.; Almeida, M.; O’Brien, C.A. Low bone mass and changes in the osteocyte network in mice lacking autophagy in the osteoblast lineage. Sci. Rep. 2016, 6, 24262. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Ma, Z.; Qian, Z.; Kang, X.; Jin, X.; Li, F.; Wang, X.; Chen, Q.; Sun, H.; et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy 2018, 14, 1726–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onal, M.; Piemontese, M.; Xiong, J.; Wang, Y.; Han, L.; Ye, S.; Komatsu, M.; Selig, M.; Weinstein, R.S.; Zhao, H.; et al. Suppression of autophagy in osteocytes mimics skeletal aging. J. Biol. Chem. 2013, 288, 17432–17440. [Google Scholar] [CrossRef] [Green Version]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1977. [Google Scholar] [CrossRef]
- Guder, C.; Gravius, S.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. Osteoimmunology: A Current Update of the Interplay between Bone and the Immune System. Front. Immunol. 2020, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.J.; Sims, N.A. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol. Med. 2005, 11, 76–81. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef] [PubMed]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef]
- Li, D.; Liu, J.; Guo, B.; Liang, C.; Dang, L.; Lu, C.; He, X.; Cheung, H.Y.S.; Xu, L.; Lu, C.; et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat. Commun. 2016, 7, 10872. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Kim, H.J.; Chang, E.J.; Huang, H.; Banno, Y.; Kim, H.H. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006, 25, 5840–5851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Khoo, W.H.; Ng, P.Y.; Xiao, Y.; Zamerli, J.; Thatcher, P.; Kyaw, W.; Pathmanandavel, K.; Grootveld, A.K.; Moran, I.; et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021, 184, 1330–1347. [Google Scholar] [CrossRef]
- Palmisano, B.; Spica, E.; Remoli, C.; Labella, R.; Di Filippo, A.; Donsante, S.; Bini, F.; Raimondo, D.; Marinozzi, F.; Boyde, A.; et al. RANKL Inhibition in Fibrous Dysplasia of Bone: A Preclinical Study in a Mouse Model of the Human Disease. J. Bone Miner. Res. 2019, 34, 2171–2182. [Google Scholar] [CrossRef]
- Corsi, A.; Palmisano, B.; Spica, E.; Di Filippo, A.; Coletta, I.; Dello Spedale Venti, M.; Labella, R.; Fabretti, F.; Donsante, S.; Remoli, C.; et al. Zoledronic Acid in a Mouse Model of Human Fibrous Dysplasia: Ineffectiveness on Tissue Pathology, Formation of “Giant Osteoclasts” and Pathogenetic Implications. Calcif. Tissue Int. 2020, 107, 603–610. [Google Scholar] [CrossRef]
- Boyce, A.M.; Collins, M.T. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of G alpha(s) Activation. Endocr. Rev. 2020, 41, 345–370. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donsante, S.; Palmisano, B.; Serafini, M.; Robey, P.G.; Corsi, A.; Riminucci, M. From Stem Cells to Bone-Forming Cells. Int. J. Mol. Sci. 2021, 22, 3989. https://doi.org/10.3390/ijms22083989
Donsante S, Palmisano B, Serafini M, Robey PG, Corsi A, Riminucci M. From Stem Cells to Bone-Forming Cells. International Journal of Molecular Sciences. 2021; 22(8):3989. https://doi.org/10.3390/ijms22083989
Chicago/Turabian StyleDonsante, Samantha, Biagio Palmisano, Marta Serafini, Pamela G. Robey, Alessandro Corsi, and Mara Riminucci. 2021. "From Stem Cells to Bone-Forming Cells" International Journal of Molecular Sciences 22, no. 8: 3989. https://doi.org/10.3390/ijms22083989