
The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications



{kind=link}
Abstract
:1. Introduction
2. uPAR Structure, Interactors and Expression
2.1. uPAR Structure
2.2. uPAR Extracellular Ligands
2.3. uPAR Cellular Ligands
2.4. uPAR Expression
3. uPAR and Cancer Hallmarks
3.1. uPAR in Invasion and Metastasis
3.2. uPAR and Angiogenesis in Cancer
3.3. uPAR and Replicative Immortality
3.4. uPAR in Cell Proliferation and Survival
3.5. uPAR in Cancer Metabolism
4. The Clinical Value of uPAR: Emerging Opportunities and Current Challenges in Cancer
4.1. Diagnostic Potential of uPAR in Malignancy
4.2. Prognostic Potential of uPAR in Malignancy
4.3. uPAR as Target
4.3.1. Peptides- and Small Molecules-Derived Antagonists of uPAR
4.3.2. Antibody-Based uPAR Inhibitors
4.3.3. uPAR-Targeting in Nanotechnologies
4.3.4. uPAR as Gene-Therapy Target: From Antisense Methodologies to Novel Gene-Editing Technologies
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef] [Green Version]
- Fortelny, N.; Cox, J.H.; Kappelhoff, R.; Starr, A.E.; Lange, P.F.; Pavlidis, P.; Overall, C.M. Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol. 2014, 12, e1001869. [Google Scholar] [CrossRef]
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development. Methods Mol. Biol. 2009, 539, 1–32. [Google Scholar] [PubMed]
- Ellis, V.; Scully, M.F.; Kakkar, V.V. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J. Biol. Chem. 1989, 264, 2185–2188. [Google Scholar] [CrossRef]
- Hannocks, M.J.; Oliver, L.; Gabrilove, J.L.; Wilson, E.L. Regulation of proteolytic activity in human bone marrow stromal cells by basic fibroblast growth factor, interleukin-1, and transforming growth factor beta. Blood 1992, 79, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Del Rosso, M.; Anichini, E.; Pedersen, N.; Blasi, F.; Fibbi, G.; Pucci, M.; Ruggiero, M. Urokinase-urokinase receptor interaction: Non-mitogenic signal transduction in human epidermal cells. Biochem. Biophys. Res. Commun. 1993, 190, 347–352. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy-Morrogh, L.; Martin, P. The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. 2020, 13, eaay8690. [Google Scholar] [CrossRef] [PubMed]
- Loughner, C.L.; Bruford, E.A.; McAndrews, M.S.; Delp, E.E.; Swamynathan, S.; Swamynathan, S.K. Organization, evolution and functions of the human and mouse Ly6/uPAR family genes. Hum. Genom. 2016, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef]
- Mertens, H.D.; Kjaergaard, M.; Mysling, S.; Gårdsvoll, H.; Jørgensen, T.J.; Svergun, D.I.; Ploug, M. A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 2012, 287, 34304–34315. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Lin, L.; Høyer-Hansen, G.; Ploug, M.; Li, H.; Jiang, L.; Yuan, C.; Li, J.; Huang, M. Crystal structure of the unoccupied murine urokinase-type plasminogen activator receptor (uPAR) reveals a tightly packed DII-DIII unit. FEBS Lett. 2019, 593, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. 2009, 14, 2494–2503. [Google Scholar] [CrossRef] [Green Version]
- Ploug, M.; Rønne, E.; Behrendt, N.; Jensen, A.L.; Blasi, F.; Danø, K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J. Biol. Chem. 1991, 266, 1926–1933. [Google Scholar] [CrossRef]
- Greenlee, J.D.; Subramanian, T.; Liu, K.; King, M.R. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2021, 81, 5–17. [Google Scholar]
- Wilhelm, O.G.; Wilhelm, S.; Escott, G.M.; Lutz, V.; Magdolen, V.; Schmitt, M.; Rifkin, D.B.; Wilson, E.L.; Graeff, H.; Brunner, G. Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J. Cell. Physiol. 1999, 180, 225–235. [Google Scholar] [CrossRef]
- Van Veen, M.; Matas-Rico, E.; van de Wetering, K.; Leyton-Puig, D.; Kedziora, K.M.; De Lorenzi, V.; Stijf-Bultsma, Y.; van den Broek, B.; Jalink, K.; Sidenius, N.; et al. Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells. Elife 2017, 6, e23649. [Google Scholar] [CrossRef] [PubMed]
- Pyke, C.; Eriksen, J.; Solberg, H.; Nielsen, B.S.; Kristensen, P.; Lund, L.R.; Danø, K. An alternatively spliced variant of mRNA for the human receptor for urokinase plasminogen activator. FEBS Lett. 1993, 326, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Montuori, N.; Visconte, V.; Rossi, G.; Ragno, P. Soluble and cleaved forms of the urokinase-receptor: Degradation products or active molecules? Thromb. Haemost. 2005, 93, 192–198. [Google Scholar] [CrossRef]
- Høyer-Hansen, G.; Ploug, M.; Behrendt, N.; Rønne, E.; Danø, K. Cell-surface acceleration of urokinase-catalyzed receptor cleavage. Eur. J. Biochem. 1997, 243, 21–26. [Google Scholar] [CrossRef]
- Ragno, P. The urokinase receptor: A ligand or a receptor? Story of a sociable molecule. Cell. Mol. Life Sci. 2006, 63, 1028–1037. [Google Scholar] [CrossRef]
- Wei, Y.; Waltz, D.A.; Rao, N.; Drummond, R.J.; Rosenberg, S.; Chapman, H.A. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 1994, 269, 32380–32388. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Ploug, M. Mapping of the vitronectin-binding site on the urokinase receptor: Involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 2007, 282, 13561–13572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høyer-Hansen, G.; Behrendt, N.; Ploug, M.; Danø, K.; Preissner, K.T. The intact urokinase receptor is required for efficient vitronectin binding: Receptor cleavage prevents ligand interaction. FEBS Lett. 1997, 420, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Royle, G.; Wang, S.; Crain, K.; Loskutoff, D.J. Structural and functional analysis of the plasminogen activator inhibitor-1 binding motif in the somatomedin B domain of vitronectin. J. Biol. Chem. 1996, 271, 12716–12723. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, O.; Andolfo, A.; Santovito, M.L.; Iuzzolino, L.; Blasi, F.; Sidenius, N. Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J. 2003, 22, 5994–6003. [Google Scholar] [CrossRef] [Green Version]
- Huai, Q.; Zhou, A.; Lin, L.; Mazar, A.P.; Parry, G.C.; Callahan, J.; Shaw, D.E.; Furie, B.; Furie, B.C.; Huang, M. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 2008, 15, 422–423. [Google Scholar] [CrossRef] [Green Version]
- Gudewicz, P.W.; Gilboa, N. Human urokinase-type plasminogen activator stimulates chemotaxis of human neutrophils. Biochem. Biophys. Res. Commun. 1987, 147, 1176–1181. [Google Scholar] [CrossRef]
- Fibbi, G.; Ziche, M.; Morbidelli, L.; Magnelli, L.; Del Rosso, M. Interaction of urokinase with specific receptors stimulates mobilization of bovine adrenal capillary endothelial cells. Exp. Cell Res. 1988, 179, 385–395. [Google Scholar] [CrossRef]
- Bohuslav, J.; Horejsí, V.; Hansmann, C.; Stöckl, J.; Weidle, U.H.; Majdic, O.; Bartke, I.; Knapp, W.; Stockinger, H. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. J. Exp. Med. 1995, 181, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Baart, V.M.; Houvast, R.D.; de Geus-Oei, L.F.; Quax, P.H.A.; Kuppen, P.J.K.; Vahrmeijer, A.L.; Sier, C.F.M. Molecular imaging of the urokinase plasminogen activator receptor: Opportunities beyond cancer. EJNMMI Res. 2020, 10, 87. [Google Scholar] [CrossRef]
- Chaurasia, P.; Aguirre-Ghiso, J.A.; Liang, O.D.; Gardsvoll, H.; Ploug, M.; Ossowski, L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J. Biol. Chem. 2006, 281, 14852–14863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Kindzelskii, A.L.; Todd, R.F., 3rd; Petty, H.R. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J. Immunol. 1994, 152, 4630–4640. [Google Scholar] [PubMed]
- Wei, Y.; Lukashev, M.; Simon, D.I.; Bodary, S.C.; Rosenberg, S.; Doyle, M.V.; Chapman, H.A. Regulation of integrin function by the urokinase receptor. Science 1996, 273, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I.; Wei, Y.; Zhang, L.; Rao, N.K.; Xu, H.; Chen, Z.; Liu, Q.; Rosenberg, S.; Chapman, H.A. Identification of a urokinase receptor-integrin interaction site. Promiscuous regulator of integrin function. J. Biol. Chem. 2000, 275, 10228–10234. [Google Scholar] [CrossRef] [Green Version]
- Degryse, B.; Resnati, M.; Czekay, R.P.; Loskutoff, D.J.; Blasi, F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: Generation of a new integrin inhibitor. J. Biol. Chem. 2005, 280, 24792–24803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, G.; Archinti, M.; Arnaudova, R.; Andreotti, G.; Motta, A.; Furlan, F.; Citro, V.; Cubellis, M.V.; Degryse, B. D2A sequence of the urokinase receptor induces cell growth through alphavbeta3 integrin and EGFR. Cell. Mol. Life Sci. 2018, 75, 1889–1907. [Google Scholar] [CrossRef]
- Montuori, N.; Carriero, M.V.; Salzano, S.; Rossi, G.; Ragno, P. The cleavage of the urokinase receptor regulates its multiple functions. J. Biol. Chem. 2002, 277, 46932–46939. [Google Scholar] [CrossRef] [Green Version]
- Van der Pluijm, G.; Sijmons, B.; Vloedgraven, H.; van der Bent, C.; Drijfhout, J.W.; Verheijen, J.; Quax, P.; Karperien, M.; Papapoulos, S.; Löwik, C. Urokinase-receptor/integrin complexes are functionally involved in adhesion and progression of human breast cancer in vivo. Am. J. Pathol. 2001, 159, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Aguirre Ghiso, J.; Estrada, Y.; Ossowski, L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 2002, 1, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef]
- Liang, W.; Chen, K.; Gong, W.; Yoshimura, T.; Le, Y.; Wang, Y.; Wang, J.M. The Contribution of Chemoattractant GPCRs, Formylpeptide Receptors, to Inflammation and Cancer. Front. Endocrinol. 2020, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Gyetko, M.R.; Todd, R.F., 3rd; Wilkinson, C.C.; Sitrin, R.G. The urokinase receptor is required for human monocyte chemotaxis in vitro. J. Clin. Investig. 1994, 93, 1380–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degryse, B.; Resnati, M.; Rabbani, S.A.; Villa, A.; Fazioli, F.; Blasi, F. Src-dependence and pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. Blood 1999, 94, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 2004, 173, 5739–5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høyer-Hansen, G.; Pessara, U.; Holm, A.; Pass, J.; Weidle, U.; Danø, K.; Behrendt, N. Urokinase-catalysed cleavage of the urokinase receptor requires an intact glycolipid anchor. Biochem. J. 2001, 358, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Gorrasi, A.; Li Santi, A.; Amodio, G.; Alfano, D.; Remondelli, P.; Montuori, N.; Ragno, P. The urokinase receptor takes control of cell migration by recruiting integrins and FPR1 on the cell surface. PLoS ONE 2014, 9, e86352. [Google Scholar] [CrossRef] [Green Version]
- Nagamine, Y.; Medcalf, R.L.; Muñoz-Cánoves, P. Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb. Haemost. 2005, 93, 661–675. [Google Scholar]
- Montuori, N.; Cosimato, V.; Rinaldi, L.; Rea, V.E.; Alfano, D.; Ragno, P. uPAR regulates pericellular proteolysis through a mechanism involving integrins and fMLF-receptors. Thromb. Haemost. 2013, 109, 309–318. [Google Scholar]
- Gorrasi, A.; Petrone, A.M.; Li Santi, A.; Alfieri, M.; Montuori, N.; Ragno, P. New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms. Cells 2020, 9, 2531. [Google Scholar] [CrossRef]
- Noh, H.; Hong, S.; Huang, S. Role of urokinase receptor in tumor progression and development. Theranostics 2013, 3, 487–495. [Google Scholar] [CrossRef]
- Alfano, D.; Gorrasi, A.; Li Santi, A.; Ricci, P.; Montuori, N.; Selleri, C.; Ragno, P. Urokinase receptor and CXCR4 are regulated by common microRNAs in leukaemia cells. J. Cell. Mol. Med. 2015, 19, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, F.; Rossi, F.W.; Pesapane, A.; Varricchio, S.; Ilardi, G.; Mascolo, M.; Staibano, S.; Lavecchia, A.; Ragno, P.; Selleri, C.; et al. N-Formyl Peptide Receptors Induce Radical Oxygen Production in Fibroblasts Derived From Systemic Sclerosis by Interacting With a Cleaved Form of Urokinase Receptor. Front. Immunol. 2018, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Illemann, M.; Laerum, O.D.; Hasselby, J.P.; Thurison, T.; Høyer-Hansen, G.; Nielsen, H.J. Danish Study Group on Early Detection of Colorectal Cancer, Christensen IJ. Urokinase-type plasminogen activator receptor (uPAR) on tumor-associated macrophages is a marker of poor prognosis in colorectal cancer. Cancer Med. 2014, 3, 855–864. [Google Scholar] [CrossRef]
- Liu, K.L.; Fan, J.H.; Jing, W. Prognostic Role of Circulating Soluble uPAR in Various Cancers: A Systematic Review and Meta-Analysis. Clin. Lab. 2017, 63, 871–880. [Google Scholar] [CrossRef]
- Carriero, M.V.; Stoppelli, M.P. The urokinase-type plasminogen activator and the generation of inhibitors of urokinase activity and signaling. Curr. Pharm. Des. 2011, 17, 1944–1961. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, K.; Votta, G.; Ingangi, V.; Di Carluccio, G.; Rea, D.; Losito, S.; Montuori, N.; Ragno, P.; Stoppelli, M.P.; Arra, C.; et al. Urokinase receptor promotes ovarian cancer cell dissemination through its 84-95 sequence. Oncotarget 2014, 5, 4154–4169. [Google Scholar] [CrossRef] [Green Version]
- Montuori, N.; Bifulco, K.; Carriero, M.V.; La Penna, C.; Visconte, V.; Alfano, D.; Pesapane, A.; Rossi, F.W.; Salzano, S.; Rossi, G.; et al. The cross-talk between the urokinase receptor and fMLP receptors regulates the activity of the CXCR4 chemokine receptor. Cell. Mol. Life Sci. 2011, 68, 2453–2467. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci. 2020, 249, 117534. [Google Scholar] [CrossRef]
- Margheri, F.; Luciani, C.; Taddei, M.L.; Giannoni, E.; Laurenzana, A.; Biagioni, A.; Chillà, A.; Chiarugi, P.; Fibbi, G.; Del Rosso, M. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget 2014, 5, 1538–1553. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Chetty, C.; Bhoopathi, P.; Lakka, S.; Mohanam, S.; Rao, J.S.; Dinh, D.E. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial-mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int. J. Oncol. 2011, 38, 733–744. [Google Scholar] [PubMed] [Green Version]
- Bao, Y.N.; Cao, X.; Luo, D.H.; Sun, R.; Peng, L.X.; Wang, L.; Yan, Y.P.; Zheng, L.S.; Xie, P.; Cao, Y.; et al. Urokinase-type plasminogen activator receptor signaling is critical in nasopharyngeal carcinoma cell growth and metastasis. Cell Cycle 2014, 13, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, M.; Eastman, B.M.; Webb, D.L.; Stoletov, K.; Klemke, R.; Gonias, S.L. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res. 2010, 70, 8948–8958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Laurenzana, A.; Biagioni, A.; Bianchini, F.; Peppicelli, S.; Chillà, A.; Margheri, F.; Luciani, C.; Pimpinelli, N.; Del Rosso, M.; Calorini, L.; et al. Inhibition of uPAR-TGFbeta crosstalk blocks MSC-dependent EMT in melanoma cells. J. Mol. Med. 2015, 93, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Santibanez, J.F.; Obradović, H.; Kukolj, T.; Krstić, J. Transforming growth factor-β, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Dyn 2018, 247, 382–395. [Google Scholar] [CrossRef] [Green Version]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Sharpe, E.E.; Maupin, A.B.; Teleron, A.A.; Pyle, A.L.; Carmeliet, P.; Young, P.P. VEGF and PlGF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascularization. FASEB J. 2006, 20, 1495–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selleri, C.; Montuori, N.; Ricci, P.; Visconte, V.; Carriero, M.V.; Sidenius, N.; Serio, B.; Blasi, F.; Rotoli, B.; Rossi, G.; et al. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 2005, 105, 2198–2205. [Google Scholar] [CrossRef] [Green Version]
- Montuori, N.; Ragno, P. Role of uPA/uPAR in the modulation of angiogenesis. Chem. Immunol. Allergy 2014, 99, 105–122. [Google Scholar]
- Larusch, G.A.; Merkulova, A.; Mahdi, F.; Shariat-Madar, Z.; Sitrin, R.G.; Cines, D.B.; Schmaier, A.H. Domain 2 of uPAR regulates single-chain urokinase-mediated angiogenesis through β1-integrin and VEGFR2. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H305–H320. [Google Scholar] [CrossRef] [Green Version]
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.Q.; et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117. [Google Scholar] [CrossRef] [PubMed]
- Unseld, M.; Chilla, A.; Pausz, C.; Mawas, R.; Breuss, J.; Zielinski, C.; Schabbauer, G.; Prager, G.W. PTEN expression in endothelial cells is down-regulated by uPAR to promote angiogenesis. Thromb. Haemost. 2015, 114, 379–389. [Google Scholar] [CrossRef]
- Margheri, F.; Chillà, A.; Laurenzana, A.; Serratì, S.; Mazzanti, B.; Saccardi, R.; Santosuosso, M.; Danza, G.; Sturli, N.; Rosati, F.; et al. Endothelial progenitor cell-dependent angiogenesis requires localization of the full-length form of uPAR in caveolae. Blood 2011, 118, 3743–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margheri, F.; Papucci, L.; Schiavone, N.; D’Agostino, R.; Trigari, S.; Serratì, S.; Laurenzana, A.; Biagioni, A.; Luciani, C.; Chillà, A.; et al. Differential uPAR recruitment in caveolar-lipid rafts by GM1 and GM3 gangliosides regulates endothelial progenitor cells angiogenesis. J. Cell. Mol. Med. 2015, 19, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, K.; Longanesi-Cattani, I.; Gala, M.; DI Carluccio, G.; Masucci, M.T.; Pavone, V.; Lista, L.; Arra, C.; Stoppelli, M.P.; Carriero, M.V. The soluble form of urokinase receptor promotes angiogenesis through its Ser⁸⁸-Arg-Ser-Arg-Tyr⁹² chemotactic sequence. J. Thromb. Haemost. 2010, 8, 2789–2799. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serratì, S.; Mocali, A.; et al. uPAR-expressing melanoma exosomes promote angiogenesis by VE-Cadherin, EGFR and uPAR overexpression and rise of ERK1,2 signaling in endothelial cells. Cell. Mol. Life Sci. 2020, 78, 3057–3072. [Google Scholar] [CrossRef]
- Sun, Y.; Coppé, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Hodjat, M.; Haller, H.; Dumler, I.; Kiyan, Y. Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J. Vasc. Res. 2013, 50, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, P.B.; Hodjat, M.; Haller, H.; Dumler, I.; Kiyan, Y. Loss of urokinase receptor sensitizes cells to DNA damage and delays DNA repair. PLoS ONE 2014, 9, e101529. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Zhao, H.; Darzynkiewicz, Z.; Moscatello, A.; Shin, E.; Schantz, S.; Tiwari, R.K.; Geliebter, J. Downregulation of uPAR inhibits migration, invasion, proliferation, FAK/PI3K/Akt signaling and induces senescence in papillary thyroid carcinoma cells. Cell Cycle 2011, 10, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Kirchheimer, J.C.; Wojta, J.; Christ, G.; Binder, B.R. Proliferation of a human epidermal tumor cell line stimulated by urokinase. FASEB J. 1987, 1, 125–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L.; Tamagnone, L.; Vigna, E.; Sachs, M.; Hartmann, G.; Birchmeir, W.; Daikuhara, Y.; Tsubouchi, H.; Blasi, F.; Comoglio, P.M. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992, 11, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Gerasi, L.; Blasi, F. uPAR-deficient mouse keratinocytes fail to produce EGFR-dependent laminin-5, affecting migration in vivo and in vitro. J. Cell Sci. 2008, 121 Pt 23, 3922–3932. [Google Scholar]
- Mazzieri, R.; Furlan, F.; D’Alessio, S.; Zonari, E.; Talotta, F.; Verde, P.; Blasi, F. A direct link between expression of urokinase plasminogen activator receptor, growth rate and oncogenic transformation in mouse embryonic fibroblasts. Oncogene 2007, 26, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Kiyan, J.; Kiyan, R.; Haller, H.; Dumler, I. Urokinase-induced signaling in human vascular smooth muscle cells is mediated by PDGFR-beta. EMBO J. 2005, 24, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Webb, D.J.; Jo, M.; Gonias, S.L. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J. Cell Sci. 2001, 114 Pt 18, 3387–3396. [Google Scholar]
- Alfano, D.; Iaccarino, I.; Stoppelli, M.P. Urokinase signaling through its receptor protects against anoikis by increasing BCL-xL expression levels. J. Biol. Chem. 2006, 281, 17758–17767. [Google Scholar] [CrossRef] [Green Version]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.; Pizzo, D.; VandenBerg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 2015, 75, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rysenkova, K.D.; Klimovich, P.S.; Shmakova, A.A.; Karagyaur, M.N.; Ivanova, K.A.; Aleksandrushkina, N.A.; Tkachuk, V.A.; Rubina, K.A.; Semina, E.V. Urokinase receptor deficiency results in EGFR-mediated failure to transmit signals for cell survival and neurite formation in mouse neuroblastoma cells. Cell Signal. 2020, 75, 109741. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, B.; Chen, L.; Ju, Y.; Li, C.; Meng, S. Urokinase-type plasminogen activator receptor inhibits apoptosis in triple-negative breast cancer through miR-17/20a suppression of death receptors 4 and 5. Oncotarget 2017, 8, 88645–88657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, V.; Iommelli, F.; Monti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of Warburg Effect and Reactivation of Oxidative Phosphorylation by Differential Inhibition of EGFR Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnala, S.; Chetty, C.; Veeravalli, K.K.; Dinh, D.H.; Klopfenstein, J.D.; Rao, J.S. Metabolic remodeling precedes mitochondrial outer membrane permeabilization in human glioma xenograft cells. Int. J. Oncol. 2012, 40, 509–518. [Google Scholar]
- Laurenzana, A.; Chillà, A.; Luciani, C.; Peppicelli, S.; Biagioni, A.; Bianchini, F.; Tenedini, E.; Torre, E.; Mocali, A.; Calorini, L.; et al. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int. J. Cancer 2017, 141, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Biagioni, A.; Laurenzana, A.; Chillà, A.; Del Rosso, M.; Andreucci, E.; Poteti, M.; Bani, D.; Guasti, D.; Fibbi, G.; Margheri, F. uPAR Knockout Results in a Deep Glycolytic and OXPHOS Reprogramming in Melanoma and Colon Carcinoma Cell Lines. Cells 2020, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [PubMed]
- Li, D.; Liu, S.; Shan, H.; Conti, P.; Li, Z. Urokinase plasminogen activator receptor (uPAR) targeted nuclear imaging and radionuclide therapy. Theranostics 2013, 3, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, S.A.; Gladu, J. Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res. 2002, 62, 2390–2397. [Google Scholar] [PubMed]
- Dullin, C.; Zientkowska, M.; Napp, J.; Missbach-Guentner, J.; Krell, H.W.; Müller, F.; Grabbe, E.; Tietze, L.F.; Alves, F. Semiautomatic landmark-based two-dimensional-three-dimensional image fusion in living mice: Correlation of near-infrared fluorescence imaging of Cy5.5-labeled antibodies with flat-panel volume computed tomography. Mol. Imaging 2009, 8, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M.; Østergaard, S.; Gårdsvoll, H.; Kovalski, K.; Holst-Hansen, C.; Holm, A.; Ossowski, L.; Danø, K. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 2001, 40, 12157–12168. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.B.; Niu, G.; Wang, H.; He, L.; Yang, L.; Ploug, M.; Chen, X. Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist by microPET. Clin. Cancer Res. 2008, 14, 4758–4766. [Google Scholar] [CrossRef] [Green Version]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The molecular crystal ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Chounta, A.; Ellinas, C.; Tzanetakou, V.; Pliarhopoulou, F.; Mplani, V.; Oikonomou, A.; Leventogiannis, K.; Giamarellos-Bourboulis, E.J. Serum soluble urokinase plasminogen activator receptor as a screening test for the early diagnosis of hepatocellular carcinoma. Liver Int. 2015, 35, 601–607. [Google Scholar] [CrossRef]
- Kaya, S.; Köksal, I.; Menteşe, A.; Sönmez, M.; Sümer, A.; Yıldırım, S.S.; Yılmaz, G. The significance of serum urokinase plasminogen activation receptor (suPAR) in the diagnosis and follow-up of febrile neutropenic patients with hematologic malignancies. Int. J. Infect. Dis. 2013, 17, e1056–e1059. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, L.J.H.; Schultz, M.; Gaardsting, A.; Ladelund, S.; Garred, P.; Iversen, K.; Eugen-Olsen, J.; Helms, M.; David, K.P.; Kjaer, A.; et al. Inflammatory biomarkers and cancer: CRP and suPAR as markers of incident cancer in patients with serious nonspecific symptoms and signs of cancer. Int. J. Cancer 2017, 141, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Aronen, A.; Aittoniemi, J.; Huttunen, R.; Nikkola, A.; Rinta-Kiikka, I.; Nikkola, J.; Limnell, O.; Nordback, I.; Sand, J.; Laukkarinen, J. Plasma suPAR may help to distinguish between chronic pancreatitis and pancreatic cancer. Scand. J. Gastroenterol. 2021, 56, 81–85. [Google Scholar] [CrossRef] [PubMed]
- De Petro, G.; Tavian, D.; Copeta, A.; Portolani, N.; Giulini, S.M.; Barlati, S. Expression of urokinase-type plasminogen activator (u-PA), u-PA receptor, and tissue-type PA messenger RNAs in human hepatocellular carcinoma. Cancer Res. 1998, 58, 2234–2239. [Google Scholar]
- Dubuisson, L.; Monvoisin, A.; Nielsen, B.S.; Le Bail, B.; Bioulac-Sage, P.; Rosenbaum, J. Expression and cellular localization of the urokinase-type plasminogen activator and its receptor in human hepatocellular carcinoma. J. Pathol. 2000, 190, 190–195. [Google Scholar] [CrossRef]
- Riisbro, R.; Christensen, I.J.; Nielsen, H.J.; Brunner, N.; Nilbert, M.; Fernebro, E. Preoperative plasma soluble urokinase plasminogen activator receptor as a prognostic marker in rectal cancer patients. An EORTC-Receptor and Biomarker Group collaboration. Int. J. Biol. Markers 2005, 20, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Loosen, S.H.; Tacke, F.; Binnebosel, M.; Leyh, C.; Vucur, M.; Heitkamp, F.; Schoening, W.; Ulmer, T.F.; Alizai, P.H.; Trautwein, C.; et al. Serum levels of soluble urokinase plasminogen activator receptor (suPAR) predict outcome after resection of colorectal liver metastases. Oncotarget 2018, 9, 27027–27038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sier, C.F.; Stephens, R.; Bizik, J.; Mariani, A.; Bassan, M.; Pedersen, N.; Frigerio, L.; Ferrari, A.; Danø, K.; Brünner, N.; et al. The level of urokinase-type plasminogen activator receptor is increased in serum of ovarian cancer patients. Cancer Res. 1998, 58, 1843–1849. [Google Scholar]
- Sier, C.F.; Nicoletti, I.; Santovito, M.L.; Frandsen, T.; Aletti, G.; Ferrari, A.; Lissoni, A.; Giavazzi, R.; Blasi, F.; Sidenius, N. Metabolism of tumour-derived urokinase receptor and receptor fragments in cancer patients and xenografted mice. Thromb. Haemost. 2004, 91, 403–411. [Google Scholar]
- Ljuca, D.; Fatusić, Z.; Iljazović, E.; Ahmetović, B. Monitoring of chemotherapy successfulness of platina/taxol chemotherapy protocol by using determination of serum urokinase plasminogen activator (uPA) and soluble urokinase plasminogen activator receptor (suPAR) in patients with ovarian carcinoma FIGO II and III stage. Bosn. J. Basic Med. Sci. 2007, 7, 111–116. [Google Scholar]
- Grøndahl-Hansen, J.; Peters, H.A.; van Putten, W.L.; Look, M.P.; Pappot, H.; Rønne, E.; Dano, K.; Klijn, J.G.; Brünner, N.; Foekens, J.A. Prognostic significance of the receptor for urokinase plasminogen activator in breast cancer. Clin. Cancer Res. 1995, 1, 1079–1087. [Google Scholar]
- Riisbro, R.; Christensen, I.J.; Piironen, T.; Greenall, M.; Larsen, B.; Stephens, R.W.; Han, C.; Høyer-Hansen, G.; Smith, K.; Brünner, N.; et al. Prognostic significance of soluble urokinase plasminogen activator receptor in serum and cytosol of tumor tissue from patients with primary breast cancer. Clin. Cancer Res. 2002, 8, 1132–1141. [Google Scholar]
- Pierga, J.Y.; Bonneton, C.; Magdelénat, H.; Vincent-Salomon, A.; Nos, C.; Boudou, E.; Pouillart, P.; Thiery, J.P.; de Cremoux, P. Real-time quantitative PCR determination of urokinase-type plasminogen activator receptor (uPAR) expression of isolated micrometastatic cells from bone marrow of breast cancer patients. Int. J. Cancer 2005, 114, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Roehrborn, C.G.; McConnell, J.D.; Park, S.; Alam, N.; Wheeler, T.M.; Slawin, K.M. Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J. Clin. Oncol. 2007, 25, 349–355. [Google Scholar] [CrossRef]
- Gupta, A.; Lotan, Y.; Ashfaq, R.; Roehrborn, C.G.; Raj, G.V.; Aragaki, C.C.; Montorsi, F.; Shariat, S.F. Predictive value of the differential expression of the urokinase plasminogen activation axis in radical prostatectomy patients. Eur. Urol. 2009, 55, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Brünner, N.; Francis, D.; Osterlind, K.; Rønne, E.; Hansen, H.H.; Danø, K.; Grøndahl-Hansen, J. Prognostic impact of urokinase, urokinase receptor, and type 1 plasminogen activator inhibitor in squamous and large cell lung cancer tissue. Cancer Res. 1994, 54, 4671–4675. [Google Scholar] [PubMed]
- Almasi, C.E.; Drivsholm, L.; Pappot, H.; Høyer-Hansen, G.; Christensen, I.J. The liberated domain I of urokinase plasminogen activator receptor--a new tumour marker in small cell lung cancer. APMIS 2013, 121, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Werle, B.; Kotzsch, M.; Lah, T.T.; Kos, J.; Gabrijelcic-Geiger, D.; Spiess, E.; Schirren, J.; Ebert, W.; Fiehn, W.; Luther, T.; et al. Cathepsin B, plasminogenactivator-inhibitor (PAI-1) and plasminogenactivator-receptor (uPAR) are prognostic factors for patients with non-small cell lung cancer. Anticancer Res. 2004, 24, 4147–4161. [Google Scholar] [PubMed]
- Ostheimer, C.; Evers, C.; Bache, M.; Reese, T.; Vordermark, D. Prognostic implications of the co-detection of the urokinase plasminogen activator system and osteopontin in patients with non-small-cell lung cancer undergoing radiotherapy and correlation with gross tumor volume. Strahlenther. Onkol. 2018, 194, 539–551. [Google Scholar] [CrossRef]
- Di Mauro, C.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef] [PubMed]
- Mustjoki, S.; Sidenius, N.; Sier, C.F.; Blasi, F.; Elonen, E.; Alitalo, R.; Vaheri, A. Soluble urokinase receptor levels correlate with number of circulating tumor cells in acute myeloid leukemia and decrease rapidly during chemotherapy. Cancer Res. 2000, 60, 7126–7132. [Google Scholar] [PubMed]
- Erkut, N.; Menteşe, A.; Özbaş, H.M.; Ermantaş, N.; Sümer, A.; Örem, A.; Sönmez, M. The Prognostic Significance of Soluble Urokinase Plasminogen Activator Receptor in Acute Myeloid Leukemia. Turk. J. Haematol. 2016, 33, 135–140. [Google Scholar] [CrossRef]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. UniSa 2016, 15, 15–21. [Google Scholar]
- Tarighi, P.; Montazeri, H.; Khorramizadeh, M.R.; Sobhani, A.M.; Ostad, S.N.; Ghahremani, M.H. uPAR peptide antagonist alters regulation of MAP kinases and Bcl-2 family members in favor of apoptosis in MDA-MB-231 cell line. Res. Pharm. Sci. 2015, 10, 200–205. [Google Scholar] [PubMed]
- Bifulco, K.; Longanesi-Cattani, I.; Gargiulo, L.; Maglio, O.; Cataldi, M.; De Rosa, M.; Stoppelli, M.P.; Pavone, V.; Carriero, M.V. An urokinase receptor antagonist that inhibits cell migration by blocking the formyl peptide receptor. FEBS Lett. 2008, 582, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriero, M.V.; Longanesi-Cattani, I.; Bifulco, K.; Maglio, O.; Lista, L.; Barbieri, A.; Votta, G.; Masucci, M.T.; Arra, C.; Franco, R.; et al. Structure-based design of an urokinase-type plasminogen activator receptor-derived peptide inhibiting cell migration and lung metastasis. Mol. Cancer Ther. 2009, 8, 2708–2717. [Google Scholar] [CrossRef] [Green Version]
- Bifulco, K.; Longanesi-Cattani, I.; Liguori, E.; Arra, C.; Rea, D.; Masucci, M.T.; De Rosa, M.; Pavone, V.; Stoppelli, M.P.; Carriero, M.V. A urokinase receptor-derived peptide inhibiting VEGF-dependent directional migration and vascular sprouting. Mol. Cancer Ther. 2013, 12, 1981–1993. [Google Scholar] [CrossRef] [Green Version]
- Carriero, M.V.; Bifulco, K.; Minopoli, M.; Lista, L.; Maglio, O.; Mele, L.; Di Carluccio, G.; De Rosa, M.; Pavone, V. UPARANT: A urokinase receptor-derived peptide inhibitor of VEGF-driven angiogenesis with enhanced stability and in vitro and in vivo potency. Mol. Cancer Ther. 2014, 13, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Ingangi, V.; Bifulco, K.; Yousif, A.M.; Ragone, C.; Motti, M.L.; Rea, D.; Minopoli, M.; Botti, G.; Scognamiglio, G.; Fazioli, F.; et al. The urokinase receptor-derived cyclic peptide [SRSRY] suppresses neovascularization and intravasation of osteosarcoma and chondrosarcoma cells. Oncotarget 2016, 7, 54474–54487. [Google Scholar] [CrossRef] [Green Version]
- Khanna, M.; Wang, F.; Jo, I.; Knabe, W.E.; Wilson, S.M.; Li, L.; Bum-Erdene, K.; Li, J.; Sledge, G.W.; Khanna, R.; et al. Targeting multiple conformations leads to small molecule inhibitors of the uPAR·uPA protein-protein interaction that block cancer cell invasion. ACS Chem. Biol. 2011, 6, 1232–1243. [Google Scholar] [CrossRef]
- Mani, T.; Wang, F.; Knabe, W.E.; Sinn, A.L.; Khanna, M.; Jo, I.; Sandusky, G.E.; Sledge, G.W.; Jones, D.R.; Khanna, R.; et al. Small-molecule inhibition of the uPAR·uPA interaction: Synthesis, biochemical, cellular, in vivo pharmacokinetics and efficacy studies in breast cancer metastasis. Bioorg. Med. Chem. 2013, 21, 2145–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bum-Erdene, K.; Liu, D.; Xu, D.; Ghozayel, M.K.; Meroueh, S.O. Design and Synthesis of Fragment Derivatives with a Unique Inhibition Mechanism of the uPAR·uPA Interaction. ACS Med. Chem. Lett. 2020, 12, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Bum-Erdene, K.; Leth, J.M.; Ghozayel, M.K.; Ploug, M.; Meroueh, S.O. Small-Molecule Inhibition of the uPAR ⋅ uPA Interaction by Conformational Selection. ChemMedChem 2021, 16, 377–387. [Google Scholar] [CrossRef]
- Rea, V.E.; Lavecchia, A.; Di Giovanni, C.; Rossi, F.W.; Gorrasi, A.; Pesapane, A.; de Paulis, A.; Ragno, P.; Montuori, N. Discovery of new small molecules targeting the vitronectin-binding site of the urokinase receptor that block cancer cell invasion. Mol. Cancer Ther. 2013, 12, 1402–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, S.A.; Ateeq, B.; Arakelian, A.; Valentino, M.L.; Shaw, D.E.; Dauffenbach, L.M.; Kerfoot, C.A.; Mazar, A.P. An anti-urokinase plasminogen activator receptor antibody (ATN-658) blocks prostate cancer invasion, migration, growth, and experimental skeletal metastasis in vitro and in vivo. Neoplasia 2010, 12, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Löwik, D.W.P.M.; Miller, A.D.; Thanou, M. Targeting the urokinase plasminogen activator receptor with synthetic self-assembly nanoparticles. Bioconjugate Chem. 2009, 20, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.O.; Karna, P.; Sajja, H.K.; Mao, H.; Yates, C.; Turner, T.; Aneja, R. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J. Control. Release 2011, 149, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Peng, X.H.; Wang, Y.A.; Wang, X.; Cao, Z.; Ni, C.; Karna, P.; Zhang, X.; Wood, W.C.; Xiaohu, G.; et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin. Cancer Res. 2009, 15, 4722–4732. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.; Larsen, E.K.U.; Nielsen, E.H.; Iversen, F.; Liu, Z.; Thomsen, K.; Pedersen, M.; Skrydstrup, T.; Nielsen, N.C.; Ploug, M.; et al. Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells. Nanoscale 2013, 5, 8192–8201. [Google Scholar] [CrossRef]
- Go, Y.; Chintala, S.K.; Mohanam, S.; Gokaslan, Z.; Venkaiah, B.; Bjerkvig, R.; Oka, K.; Nicolson, G.L.; Sawaya, R.; Rao, J.S. Inhibition of in vivo tumorigenicity and invasiveness of a human glioblastoma cell line transfected with antisense uPAR vectors. Clin. Exp. Metastasis 1997, 15, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Lakka, S.S.; Yanamandra, N.; Siddique, K.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene 2003, 22, 5967–5975. [Google Scholar] [CrossRef] [Green Version]
- Lakka, S.S.; Rajagopal, R.; Rajan, M.K.; Mohan, P.M.; Adachi, Y.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Ali-Osman, F.; Roth, J.A.; et al. Adenovirus-mediated antisense urokinase-type plasminogen activator receptor gene transfer reduces tumor cell invasion and metastasis in non-small cell lung cancer cell lines. Clin. Cancer Res. 2001, 7, 1087–1093. [Google Scholar] [PubMed]
- Kondraganti, S.; Gondi, C.S.; McCutcheon, I.; Dinh, D.H.; Gujrati, M.; Rao, J.S.; Olivero, W.C. RNAi-mediated downregulation of urokinase plasminogen activator and its receptor in human meningioma cells inhibits tumor invasion and growth. Int. J. Oncol. 2006, 28, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondi, C.S.; Lakka, S.S.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Intraperitoneal injection of a hairpin RNA-expressing plasmid targeting urokinase-type plasminogen activator (uPA) receptor and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin. Cancer Res. 2007, 13, 4051–4060. [Google Scholar] [CrossRef] [Green Version]
- Wach, S.; Brandl, M.; Borchardt, H.; Weigelt, K.; Lukat, S.; Nolte, E.; Al-Janabi, O.; Hart, M.; Grässer, F.; Giedl, J.; et al. Exploring the MIR143-UPAR Axis for the Inhibition of Human Prostate Cancer Cells In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2019, 16, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Xing, Z.H.; Jiang, Q.W.; Yang, Y.; Huang, J.R.; Yuan, M.L.; Wei, M.N.; Li, Y.; Wang, S.T.; Liu, K.; et al. Targeting uPAR by CRISPR/Cas9 System Attenuates Cancer Malignancy and Multidrug Resistance. Front. Oncol. 2019, 27, 9–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, R.; Zhao, L.; Zhang, X.; Xu, T.; Cui, M. Basing on uPAR-binding fragment to design chimeric antigen receptors triggers antitumor efficacy against uPAR expressing ovarian cancer cells. Biomed. Pharmacother. 2019, 117, 109173. [Google Scholar] [CrossRef]
 : inhibition.
: inhibition.
: inhibition.
: inhibition.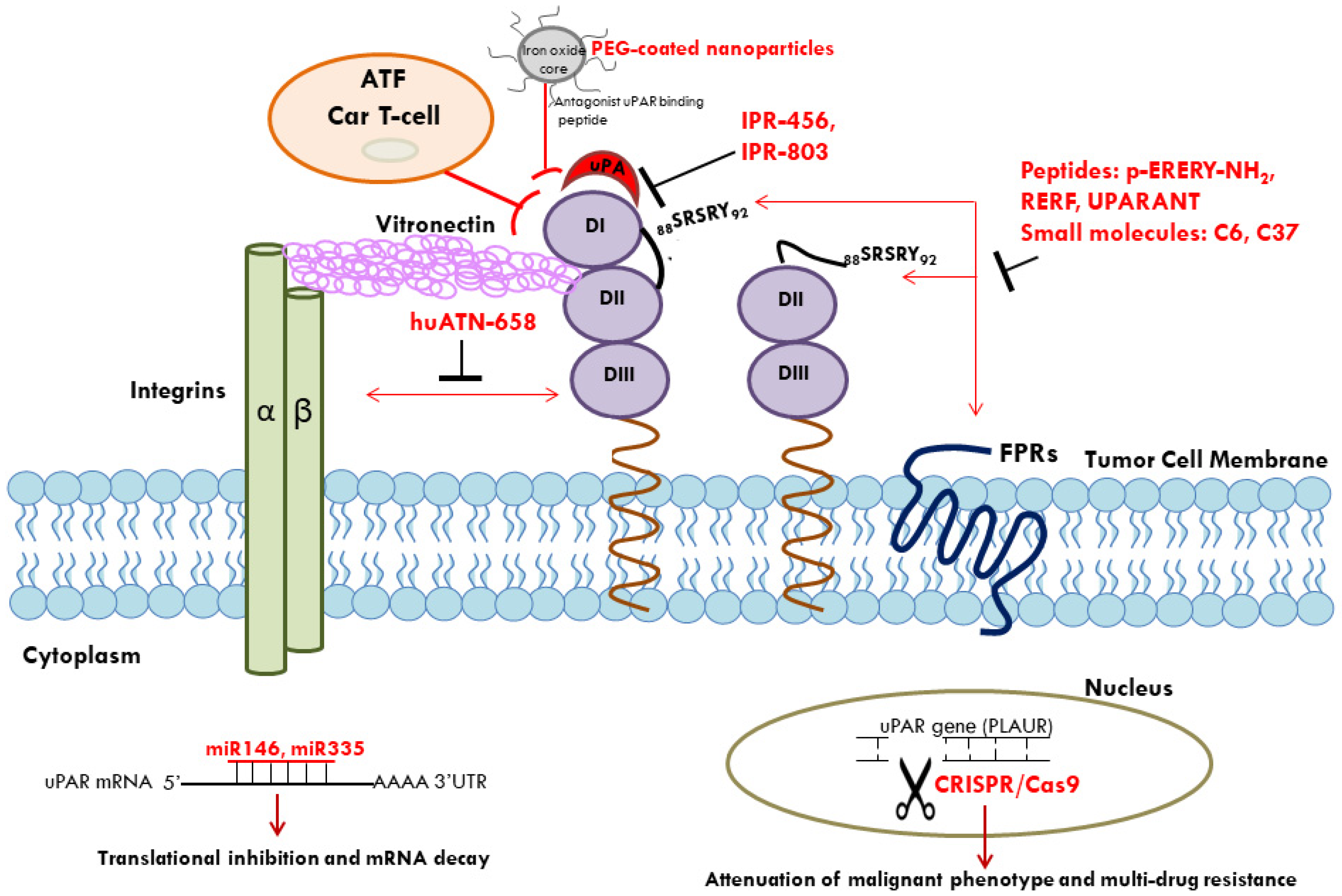
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 4111. https://doi.org/10.3390/ijms22084111
Li Santi A, Napolitano F, Montuori N, Ragno P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. International Journal of Molecular Sciences. 2021; 22(8):4111. https://doi.org/10.3390/ijms22084111
Chicago/Turabian StyleLi Santi, Anna, Filomena Napolitano, Nunzia Montuori, and Pia Ragno. 2021. "The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications" International Journal of Molecular Sciences 22, no. 8: 4111. https://doi.org/10.3390/ijms22084111
APA StyleLi Santi, A., Napolitano, F., Montuori, N., & Ragno, P. (2021). The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. International Journal of Molecular Sciences, 22(8), 4111. https://doi.org/10.3390/ijms22084111