
Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment?




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Energetic Metabolism in Brain
3. Cytotoxic and Vascular Edema
4. Inflammation in Ischemic Stroke
5. Treatment of Ischemic Stroke
5.1. Present Treatment of Stroke
5.2. Inflammation and Classic Anti-Inflammatory Treatment
5.3. Potential Role of Aquaporins in Stroke Treatment
5.4. Penumbra as an Important Target of Treatment
5.5. Future Directions of Stroke Treatment
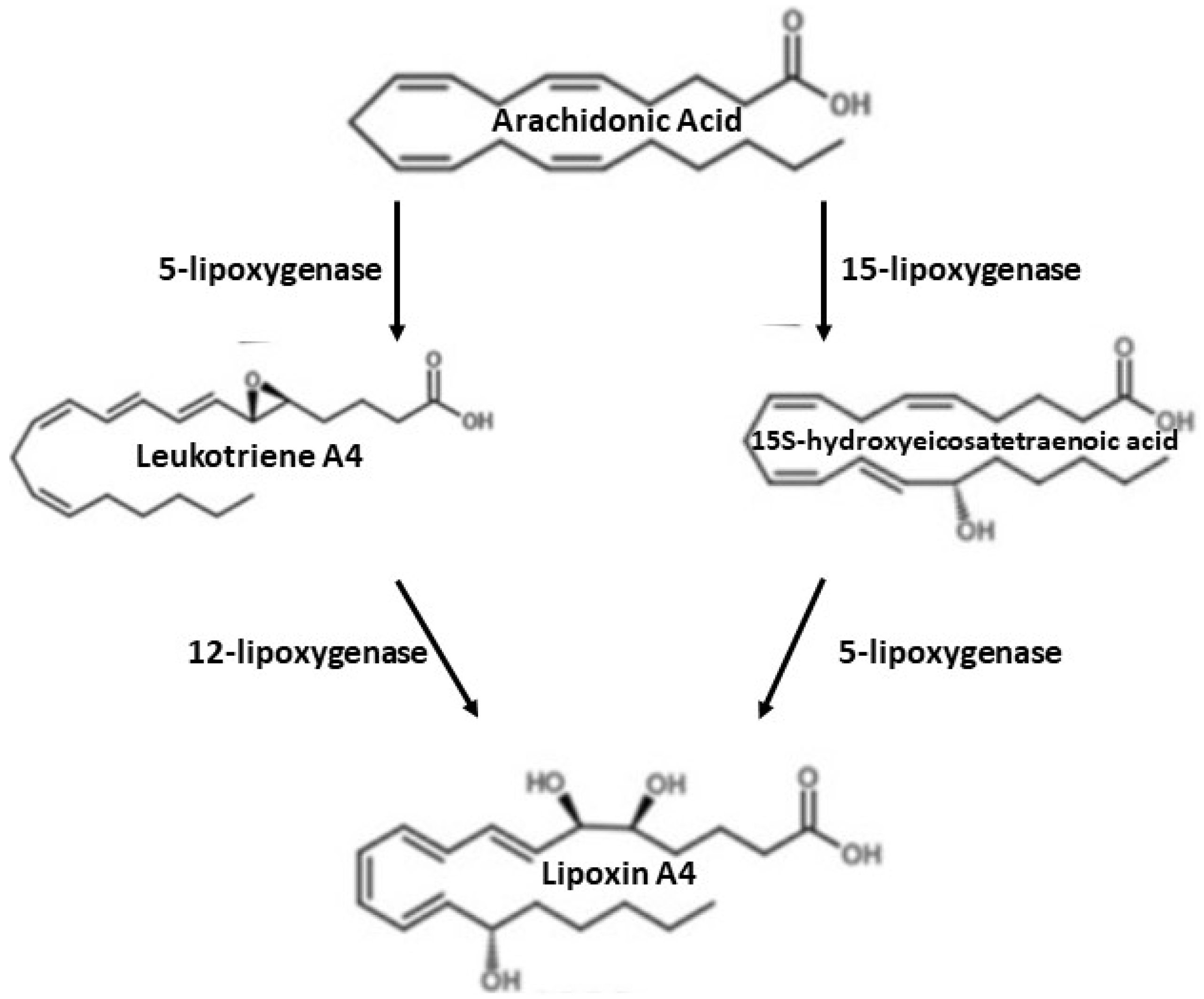
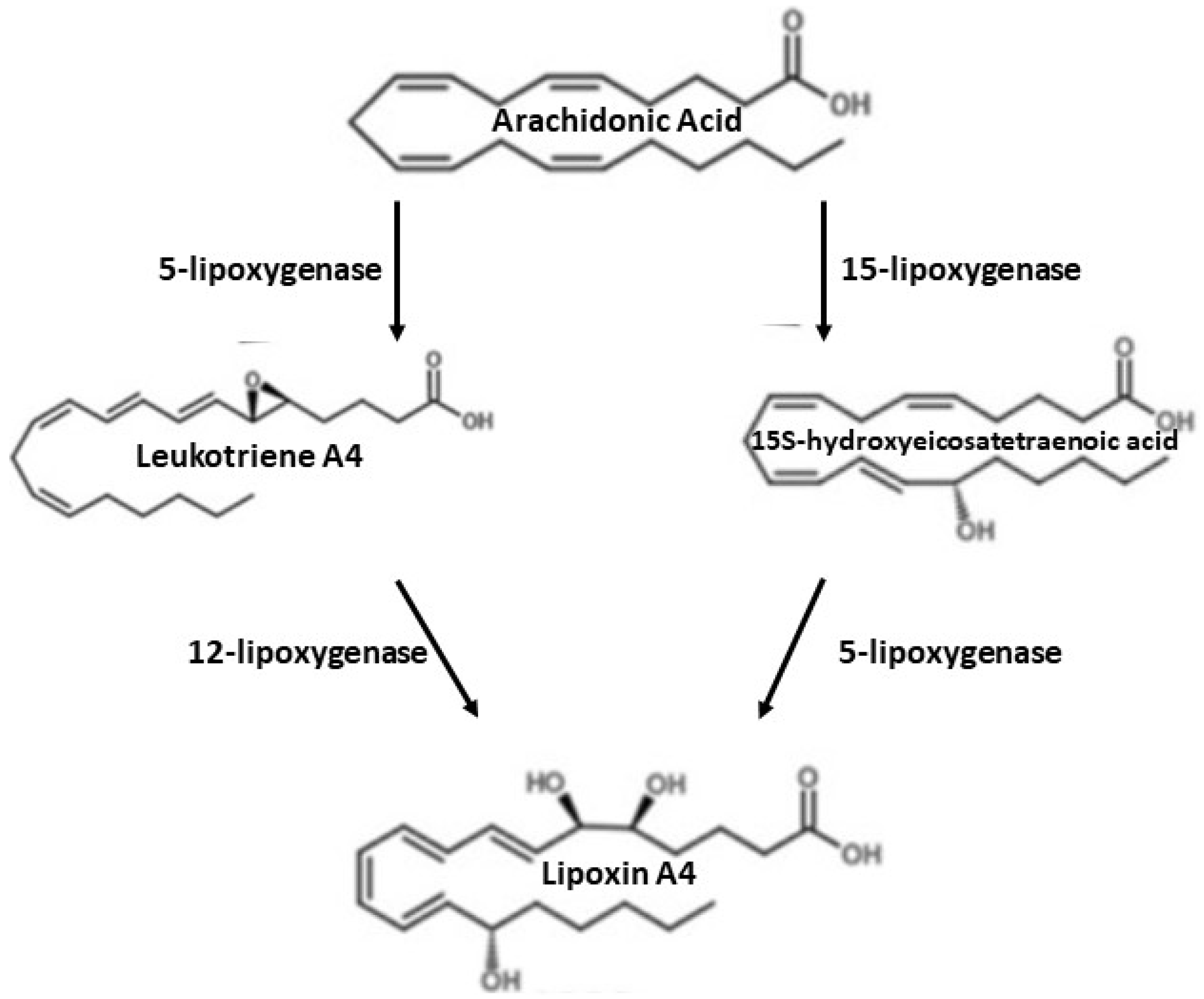
6. Arachidonic Acid Derivatives
7. Synthesis of Lipoxin A4 (LXA4)
8. Lipoxin A4
9. Receptors for Lipoxins
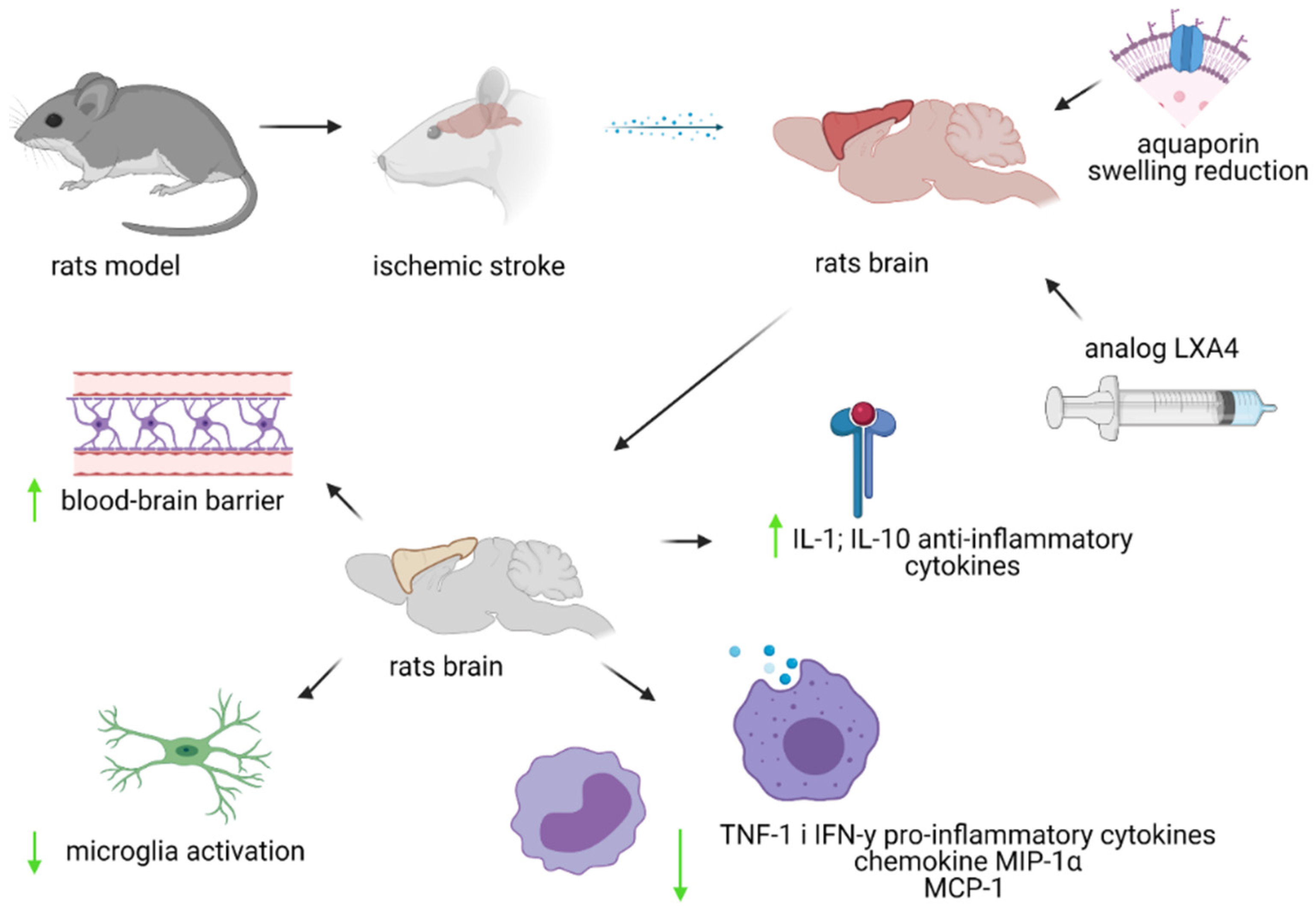
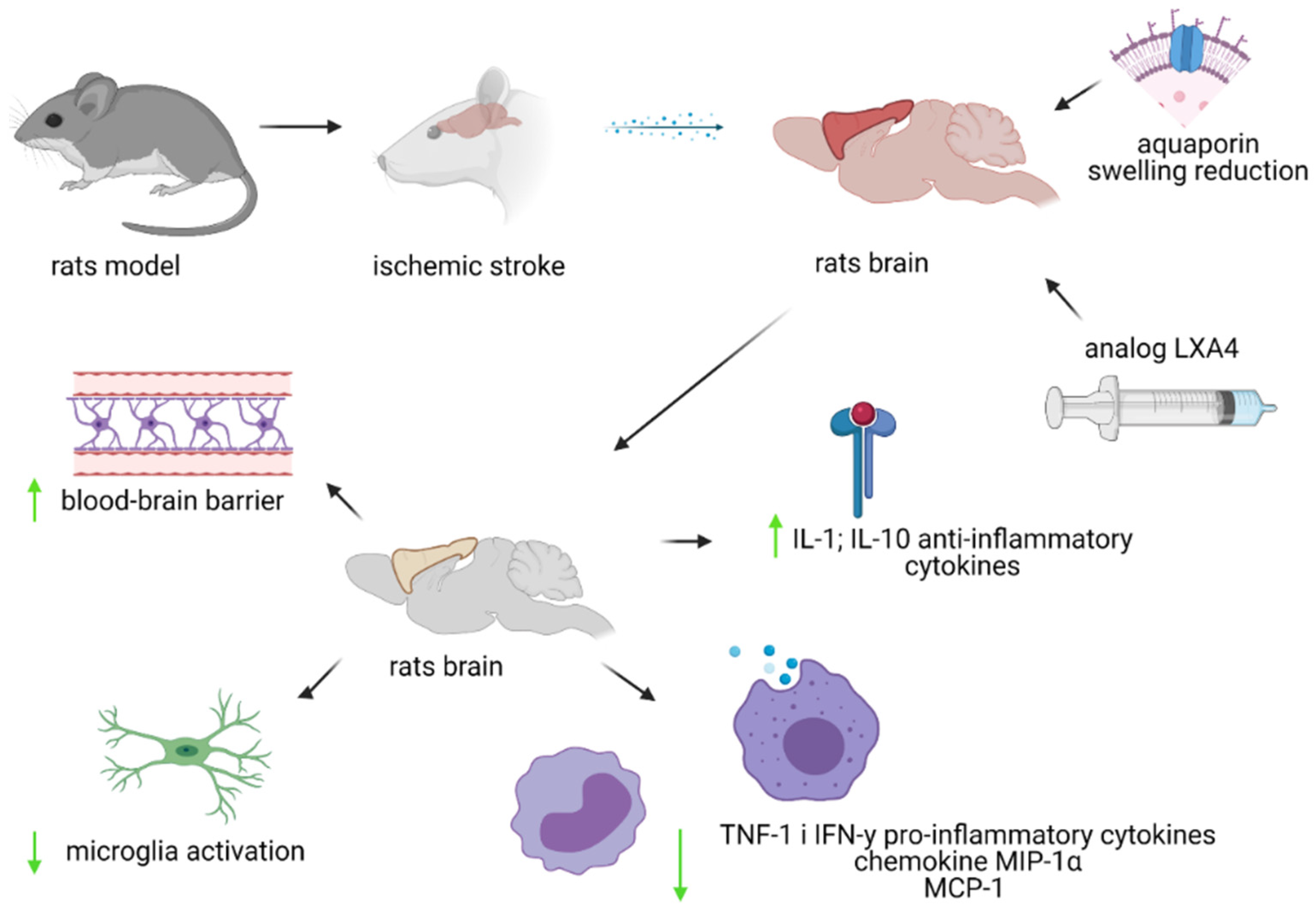
10. LXA4 and Ischemic Stroke
11. BML-111—Lipoxin A4 Analogue
12. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamilton, J.; Hasturk, H.; Kantarci, A.; Serhan, C.; Van Dyke, T. Atherosclerosis, Periodontal Disease, and Treatement with Resolvins. Curr. Atherloscler. Rep. 2017, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, H.; Sherchan, P.; Klebe, D.; Zhao, G.; Sun, X.; Zhang, J.; Tang, J.; Zhang, J.H. Controversises and evolving new mechanisms in subarachnoid hemorrhage. Prog. Neurobiol. 2014, 115, 64–91. [Google Scholar] [CrossRef] [Green Version]
- Tschoe, C.; Bushnell, C.D.; Duncan, P.W.; Alexander-Miller, M.A.; Wolfe, S.Q. Neuroinflammation after Intracerebral Hemorrhage and Potential Therapeutic Targets. J. Stroke 2020, 22, 29–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayman, L.A.; Pagani, J.J.; Kirkpatrick, J.B.; Hinck, V.C. Pathophysiology of Acute Intracerebral and Subarachnoid Hemorrhage: Applications to MR Imaging. AJNR 1989, 10, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Hu, Q.; Xu, L.; Guo, Z.; Ou, Y.; He, Y.; Yin, C.; Sun, X.; Tang, J.; Zhang, J.H. Lipoxin A4 Reduces Inflammation through Formyl Peptide Receptor 2/p38 MAPK Signaling Pathwat in Subarachnoid Hemorrhage Rats. Stroke 2016, 47, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ye, X.; Guo, P.; Xu, S.; Wang, J.; Yuan, S.; Yao, S.-L.; Shang, Y. Neuroprotective effect of lipoxin A4 methyl ester in a rat model of permanent focal cerebral ischemia. J. Mol. Neurosci. 2010, 42, 226–234. [Google Scholar] [CrossRef]
- Hansson, G.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Kaźmierski, P.; Kacperska, M.; Figlus, M.; Tomasik, B. The formation of atherosclerotic plaque, its destabilisation and diagnostics. Aktualn Neurol. 2014, 14, 43–53. [Google Scholar] [CrossRef]
- Vilahur, G.; Padro, T.; Badimon, L. Atherlosclerosis and Thrombosis: Insights from Large Animal Models. J. Biomed. Biotechnol. 2011, 2011, 907575. [Google Scholar] [CrossRef] [Green Version]
- Tułowiecka, N.; Kotlęga, D.; Prowans, P.; Szczuko, M. The Role of Resolvins: EPA and DHA Derivatives Can Be Useful in the Prevention and Treatment of Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 7628. [Google Scholar] [CrossRef]
- Serhan, C. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Rosiers, M.D.; Patlak, C.S.; Pettigrew, K.; Sakurada, O.; Shinohara, M. The [14C] deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat 1. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolma, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Castro, V.; Skowronska, M.; Lombardi, J.; He, J.; Seth, N.; Velichkovska, M.; Toborek, M. Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J. Cereb. Blood Flow Metab. 2018, 38, 317–332. [Google Scholar] [CrossRef] [Green Version]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Freitas, A.E. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Taylor, L.H.J.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-induced Relocalization of the Aquaporin 4 Channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, J.; Kent, T.; Chen, M.; Tarasov, K.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Stokum, J.; Gerzanich, V.; Simard, J. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [Green Version]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Vecchié, A.; Casula, M.; Carbone, F.; Dallegri, F.; Montecucco, F. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int. J. Mol. Sci. 2016, 17, 1967. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [Green Version]
- Hladky, S.B.; Barrand, M.A. Elimination of substances from the brain parenchyma: Efflux via perivascular pathways and via the blood-brain barrier. Fluids Barriers CNS 2018, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Wiszniewska, M.; Kobayashi, A.; Członkowska, A. Stroke management Summary of the Guidelines of the Group of Experts of the Section of Vascular Diseases of the Polish Neurological Society of 2012. Pol. Przegląd Neurol. 2012, 8, 161–175. [Google Scholar]
- Bhaskar, S.B. Clinical trial registration: A practical perspective. Indian J Anaesth. 2018, 62, 10–15. [Google Scholar] [CrossRef]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Sprigg, N.; Renton, C.J.; Dineen, R.A.; Kwong, Y.; Bath, P.M.W. Tranexamic acid for spontaneous intracerebral hemorrhage: A randomized controlled pilot trial (ISRCTN50867461). J. Stroke Cerebrovasc. Dis. 2014, 23, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Schink, T.; Kollhorst, B.; Lorenzo, C.V.; Arfè, A.; Herings, R.; Lucchi, S.; Romio, S.; Schade, R.; Schuemie, M.J.; Straatman, H.; et al. Risk of ischemic stroke and the use of individual non-steroidal anti-inflammatory drugs: A multi-country European database study within the SOS Project. PLoS ONE 2018, 13, e0203362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ASCEND Study Collaborative Group; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [Green Version]
- Szczuko, M.; Palma, J.; Kikut, J.; Komorniak, N.; Ziętek, M. Changes of lipoxin levels during pregnancy and the monthly-cycle, condition the normal course of pregnancy or pathology. Inflamm. Res. 2020, 69, 869–881. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Hirsh, J.; Spencer, F.A.; Baglin, T.P.; Weitz, J.I. Antiplatelet drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e89S–e119S. [Google Scholar] [CrossRef] [Green Version]
- Mancia, G. Short- and long-term blood pressure variability: Present and future. Hypertension 2012, 60, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Day, E.; Salman, M.; Conner, M.; Bill, R.; Conner, A. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.; Pickel, S.; Jennings, J.; Tornroth-Horsefield, S.; Conner, M.; Bill, R.; Conner, A. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 20369. [Google Scholar] [CrossRef] [Green Version]
- Zador, Z.; Stiver, S.; Wang, V.; Manley, G. Role of aquaporin-4 in cerebral edema and stroke. Handb. Exp. Pharmacol. 2009, 190, 159–170. [Google Scholar]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Huishu Hou, H.; Vedashree Meher, V.; Herlo, R.; Lissa, P.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Vella, J.; Zammit, C.; Giovanni, G.; Muscat, R.; Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell. Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciappelloni, S.; Bouchet, D.; Dubourdieu, N.; Boué-Grabot, E.; Kellermayer, B.; Manso, C.; Marignier, R.; Oliet, S.H.R.; Tourdias, T.; Groc, L. Aquaporin-4 Surface Trafficking Regulates Astrocytic Process Motility and Synaptic Activity in Health and Autoimmune Disease. Cell Rep. 2019, 27, 3860–3872.e4. [Google Scholar] [CrossRef] [Green Version]
- Gauberti, M.; De Lizarrondo, S.M.; Vivien, D. The “inflammatory penumbra” in ischemic stroke: From clinical data to experimental evidence. Eur. Stroke J. 2016, 1, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Ladecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. International Natalizumab Multiple Sclerosis Trial Group. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Elkind, M.S.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Kasliwal, R.; Elkins, J. Natalizumab in acute ischemic stroke (ACTION II) A randomized, placebo-controlled trial. Neurology 2020, 95, e1091–e1104. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, N.; Ren, L.; Yan, Y.; Sun, N.; Li, Y.J.; Han, W.; Xue, R.; Liu, Q.; Hao, J.; et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 18315–18320. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Fu, Y.; Tian, D.; Sun, N.; Han, W.; Chang, G.; Dong, Y.; Xu, X.; Liu, Q.; Huang, D.; et al. Combination of the Immune Modulator Fingolimod With Alteplase in Acute Ischemic Stroke: A Pilot Trial. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, M.M.; Marsh, G.; Kusters, I.; Matthieu Delincé, M.; Caprio, G.D.; Upadhyayula, S.; de Nola, G.; Ronan Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innes, J.; Calder, P. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachier, I.; Chanez, P.; Bonnans, C.; Godard, P.; Bousquet, J.; Chavis, C. Mediators from Arachidonate in Human Neutrophils. Biochem. Biophys. Res. Commun. 2002, 290, 219–224. [Google Scholar] [CrossRef]
- Szczuko, M.; Kotlęga, D.; Palma, J.; Zembroń-Łacny, A.; Tylutka, A.; Gołąb-Janowska, M.; Drozd, A. Lipoxins, RevD1 and 9, 13 HODE as the most important derivatives after an early incident of ischemic stroke. Sci. Rep. 2020, 10, 12849. [Google Scholar] [CrossRef] [PubMed]
- Kotlęga, D.; Zembron-Lacny, A.; Morawin, B.; Golab-Janowska, M.; Nowacki, P.; Szczuko, M. Free Fatty Acids and Their Inflammatory Derivatives Affect BDNF in Stroke Patients. Mediat. Inflamm. 2020, 2020, 6676247. [Google Scholar] [CrossRef]
- Capra, V.; Bäck, M.; Barbieri, S.; Camera, M.; Tremoli, E.; Rovati, G. Eicosanoids and Their Drugs in Cardiovascular Diseases: Focus on Atherlosclerosis and Stroke. Med. Res. Rev. 2012, 33, 363–438. [Google Scholar] [CrossRef]
- Romano, M.; Serhan, C. Lipoxin generation by permeabilized human platelets. Biochemistry 1992, 31, 8269–8277. [Google Scholar] [CrossRef]
- Serhan, C.; Romano, M. Lipoxin biosynthesis and actions: Role of the human platelet LX-synthase. J. Lipid Mediat. Cell Signal 1995, 12, 293–306. [Google Scholar] [CrossRef]
- Claria, J.; Serhan, C. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.; Chiang, N.; van Dyke, T. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Bäck, M.; Dahlen, S.; Drazen, J.; Evans, J.; Sergan, C.; Shimizu, T.; Yokomizo, T.; Rovati, G.E. International Union of Basic and Clinical Pharmacology. Leucotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol. Rev. 2011, 63, 539–584. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Fierro, I.; Gronert, K.; Serhan, C. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 2000, 191, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russel, R.; Gori, I.; Pellegrini, C.; Kumar, R.; Achtari, C.; Canny, G. Lipoxin A4 is a novel estrogen receptor modulator. FASEB J. 2011, 25, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Koczulla, R.; von Degenfeld, G.; Kupatt, C.; Krotz, F.; Zahler, S.; Gloe, T.; Issbrücker, K.; Unterberger, P.; Zaiou, M.; Lebherz, C.; et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J. Clin. Investig. 2003, 111, 1665–1672. [Google Scholar] [CrossRef]
- Svensson, C.; Zattoni, M.; Serhan, C. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J. Exp. Med. 2007, 204, 245–252. [Google Scholar] [CrossRef]
- Wada, K.; Arita, M.; Nakajima, A.; Katayama, K.; Kudo, C.; Kamisaki, Y.; Serhan, C.N. Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J. 2006, 20, 1785–1792. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.; Dahlen, S.; Drazen, J.; Hay, D.; Rovati, G.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef]
- El Kebir, D.; Jozsef, L.; Khreiss, T.; Pan, W.; Petasis, N.; Serhan, C.; Filep, J.G. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: A novel mechanism for resolution of inflammation. J. Immunol. 2007, 179, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.; Hogg, N.; Brady, H. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Gronert, K.; Gewirtz, A.; Madara, J.; Serhan, C. Identification of a Human Enterocyte Lipoxin A4 Receptor That Is Regulated by Interleukin (IL)-13 and Interferon y and Inhibits Tumor Necrosis Factor α- induced Il-8 Release. J. Exp. Med. 1998, 187, 1285–1294. [Google Scholar] [CrossRef]
- Fierro, I.; Colgan, S.; Bernasconi, G.; Petasis, N.; Clish, C.; Arita, M.; Serhan, C.N. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: Comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J. Immunol. 2003, 170, 2688–2694. [Google Scholar] [CrossRef] [Green Version]
- El Kebir, D.; József, L.; Filep, J. Opposing regulation of neutrophil apoptosis through the formyl peptide receptor-like 1/lipoxin A4 receptor: Implications for resolution of inflammation. Soc. Leukoc. Biol. 2007, 84, 600–606. [Google Scholar] [CrossRef]
- Nascimento-Silva, V.; Arruda, M.; Barja-Fidalgo, C.; Fierro, I. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: A novel antioxidative mechanism. Thromb. Haemost. 2007, 97, 88–98. [Google Scholar] [PubMed]
- Zhou, X.; Wu, P.; Zhang, L.; Xiong, W.; Li, Y.; Feng, Y.; Ye, D.-Y. Effects of lipoxin A4 on lipopolysaccharide induced proliferation and reactive oxygen speciec production in RAW264,7 macrophages through modulation of G-CSF secretion. Inflamm. Res. 2007, 56, 324–333. [Google Scholar] [CrossRef]
- Pirault, J.; Back, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.; Marullo, S.; Getting, S.; Solito, E.; Serhan, C.N. Endogenous lipi- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Zhang, Q.; Bonnans, C.; Primo, V.; Reilly, J.; Perkins, D.; Liang, Y.; Arnaout, M.A.; Nikolic, B.; Serhan, C.N. The endogenous pro-resolving mediators lipoxin A4 and resolvin E1 preserve organ function in allograft rejection. Prostaglandins Leukot. Essent. Fat. Acids 2011, 84, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, K.; DeMars, K.; Singh, J.; Yang, C.; Cho, H.; Frankowski, J.; Doré, S.; Candelario-Jalil, E. Neurovascular protection by post-ischemic intravenous injections of the lipoxin A4 receptor agonist, BML-111, in a rat model of ischemic stroke. J. Neurochem. 2014, 129, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechiche, H.; Candenas, L.; Pinto, F.; Nazeyrollas, P.; Clement, C.; Devillier, P. Characterization of cysteinyl leukotriene receptors on human saphenous veins: Antagonist activity of montelukast and its metabolites. J. Cardiovasc. Pharmacol. 2004, 43, 113–120. [Google Scholar] [CrossRef]
- Gronert, K.; Martinsson-Niskanen, T.; Ravasi, S.; Chiang, N.; Serhan, C. Selectivity of recombinant human leukotriene D4, leukotriene B4, and lipoxin A4 receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am. J. Pathol. 2001, 158, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Wu, Y.; Guo, P.; Wang, J.; Yuan, S.; Shang, Y.; Yao, S.-L. Lipoxin A4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res. 2010, 1323, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Z.; Miao, S.; Zou, L.; Cai, L.; Wu, P.; Ye, D.Y.; Wu, Q.; Li, H.H. Lipoxin A4 ameliorates cerebral ischaemia/reperfusion injury through upregulation of nuclear factor erythroid 2-related factor 2. Neurol. Res. 2013, 35, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Miao, S.; Zou, L.; Wu, P.; Hao, H.; Tang, K.; Zeng, P.; Xiong, J.; Li, H.-H.; Wu, Q.; et al. Lipoxin A4 Inhibits 5-lipoxygenase Translocation and Leukotrienes Biosynthesis to Exert a Neuroprotective Effect in Cerebral Ischemia/Reperfusion Injury. J. Mol. Neurosci. 2012, 48, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Guo, P.; Ye, X.; Wang, J.; Yuan, S.; Yao, S.-L.; Shang, Y. A lipoxin A4 analog ameliorates blood-brain barier dysfunction and reduces MMP-9 expression in a rat model of focal cerebral ischemia-reperfusion injury. J. Mol. Neurosci. 2012, 46, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Yiqin, Y.; Meilin, X.; Jie, X.; Keping, Z. Aspirin inhibits MMP-2 and MMP-9 expression and activity through PPARalpha/gamma and TIMP-1-mediated mechanisms in cultured mouse celiac macrophages. Inflammation 2009, 32, 233–241. [Google Scholar] [CrossRef]
- Huang, J.; Upadhyay, U.; Tamargo, R. Inflammation in stroke and focal cerebral ischemia. Surg. Neurol. 2006, 66, 232–245. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, J.; Li, H.; Wu, P.; Jin, S.; Zhou, X.; Yuan, P.; Xiong, W.; Li, Y.; Ye, D. Protective effects of BML-111, a lipoxin A4 receptor agonist, on carbon tetrachloride-induced liver injury in mice. Hepatol. Res. 2007, 37, 948–956. [Google Scholar] [CrossRef]
- Conte, F.; Menezes de Lima, O.; Verri, A., Jr.; Cunha, F.; Penido, C.; Henriques, M. Lipoxin A4 attenuates zymosan-induced artritis by modulating endothelin-1 and its effects. Br. J. Pharmacol. 2010, 161, 911–924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, X.; Wu, P.; Li, H.; Zhou, X.; Li, Y.; Ye, D.; Chen, B.; Wan, J. BML-111, a lipoxin receptor agonist, modulates the immune response and reduces the severity of collagen-induced arthritis. Inflamm. Res. 2008, 57, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Guo, S.; Li, H.; Yuan, S.; Shang, Y.; Yao, S. BML-111, a lipoxin receptor agonist, protects haemorrhagic shock-induced acute lung injury in rats. Resuscitation 2012, 83, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.; DeMars, K.; Alexander, J.; Leon, L.; Pacheco, S.; Graves, C.; Yang, C.; McCrea, A.O.; Frankowski, J.C.; Garrett, T.J.; et al. Targeting resolution of neuroinflammation after ischemic stroke with a lipoxin A4 analog: Protective mechanisms and long-term effects on neurological recovery. Brain Behav. 2017, 7, e00688. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tułowiecka, N.; Kotlęga, D.; Bohatyrewicz, A.; Szczuko, M. Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment? Int. J. Mol. Sci. 2021, 22, 4207. https://doi.org/10.3390/ijms22084207
Tułowiecka N, Kotlęga D, Bohatyrewicz A, Szczuko M. Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment? International Journal of Molecular Sciences. 2021; 22(8):4207. https://doi.org/10.3390/ijms22084207
Chicago/Turabian StyleTułowiecka, Nikola, Dariusz Kotlęga, Andrzej Bohatyrewicz, and Małgorzata Szczuko. 2021. "Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment?" International Journal of Molecular Sciences 22, no. 8: 4207. https://doi.org/10.3390/ijms22084207
APA StyleTułowiecka, N., Kotlęga, D., Bohatyrewicz, A., & Szczuko, M. (2021). Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment? International Journal of Molecular Sciences, 22(8), 4207. https://doi.org/10.3390/ijms22084207