
smartPARE: An R Package for Efficient Identification of True mRNA Cleavage Sites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overview of Data Processing
2.2. A total of 7.3% of All Cleavage Windows Represented True Cleavages
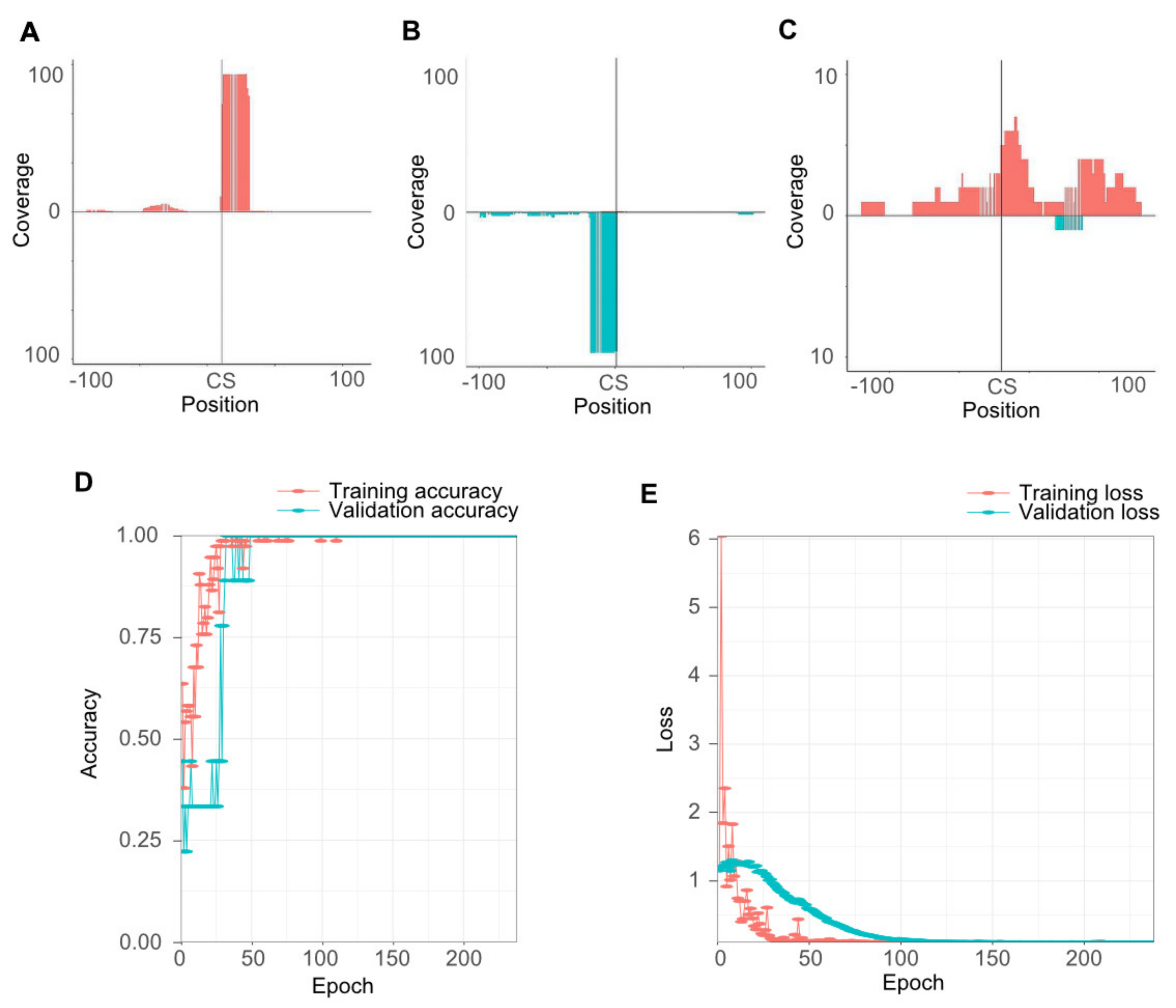
2.3. Evaluation of smartPARE and Validation of miRNA Cleavage Sites
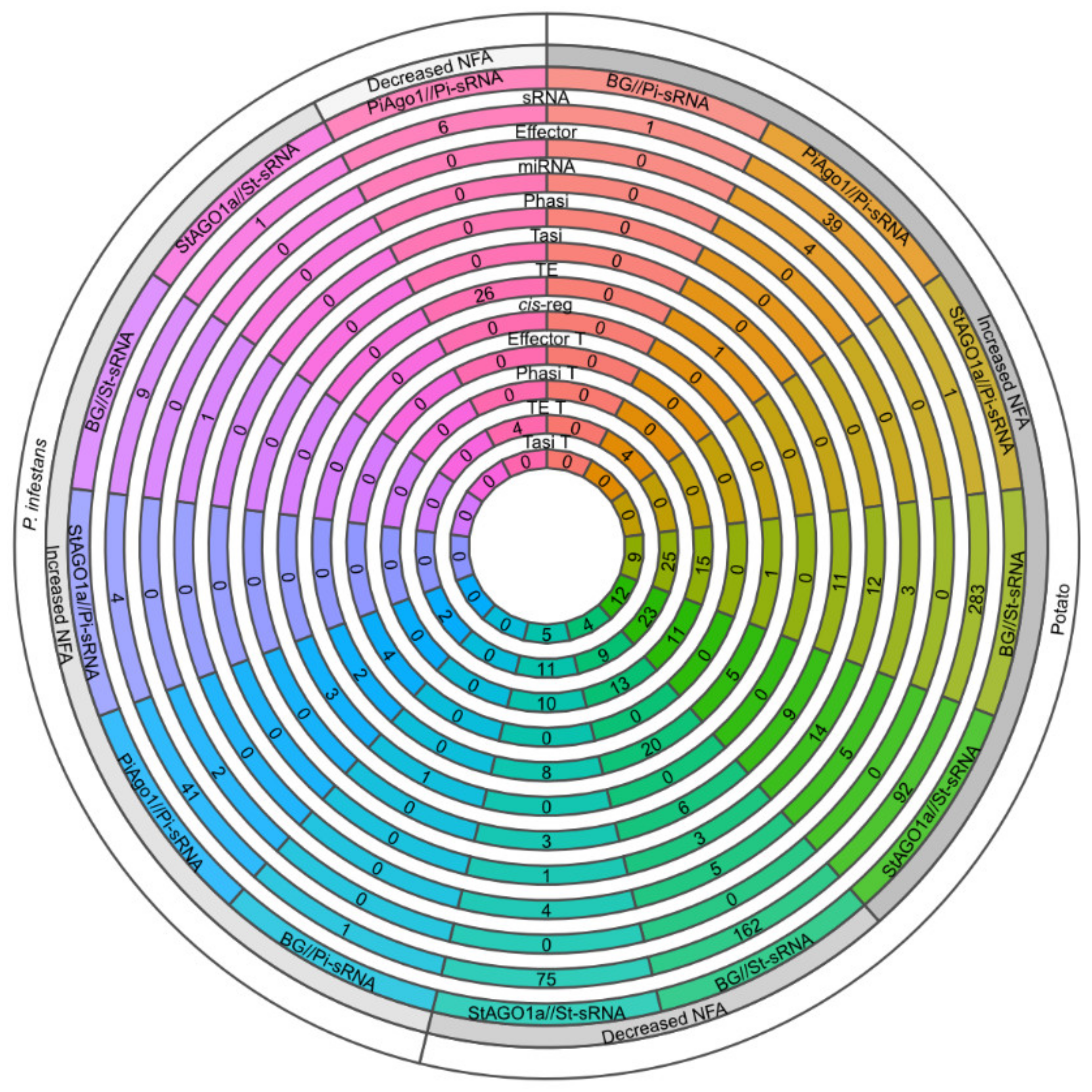
2.4. Crinkler Effectors Are Involved in sRNA Cascades during Infection
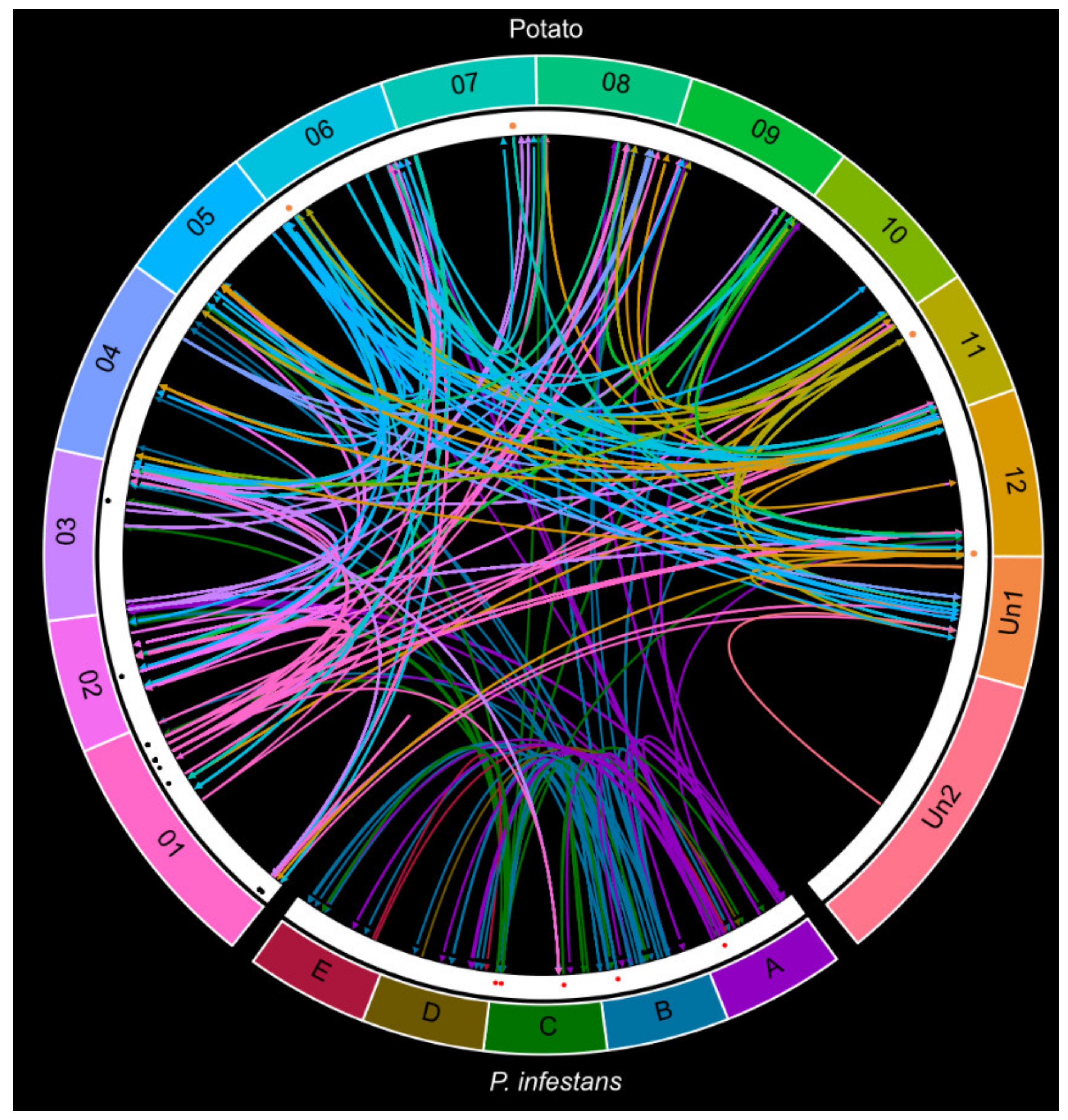
2.5. The Complex Small RNA Landscape during Potato–P. infestans Interaction
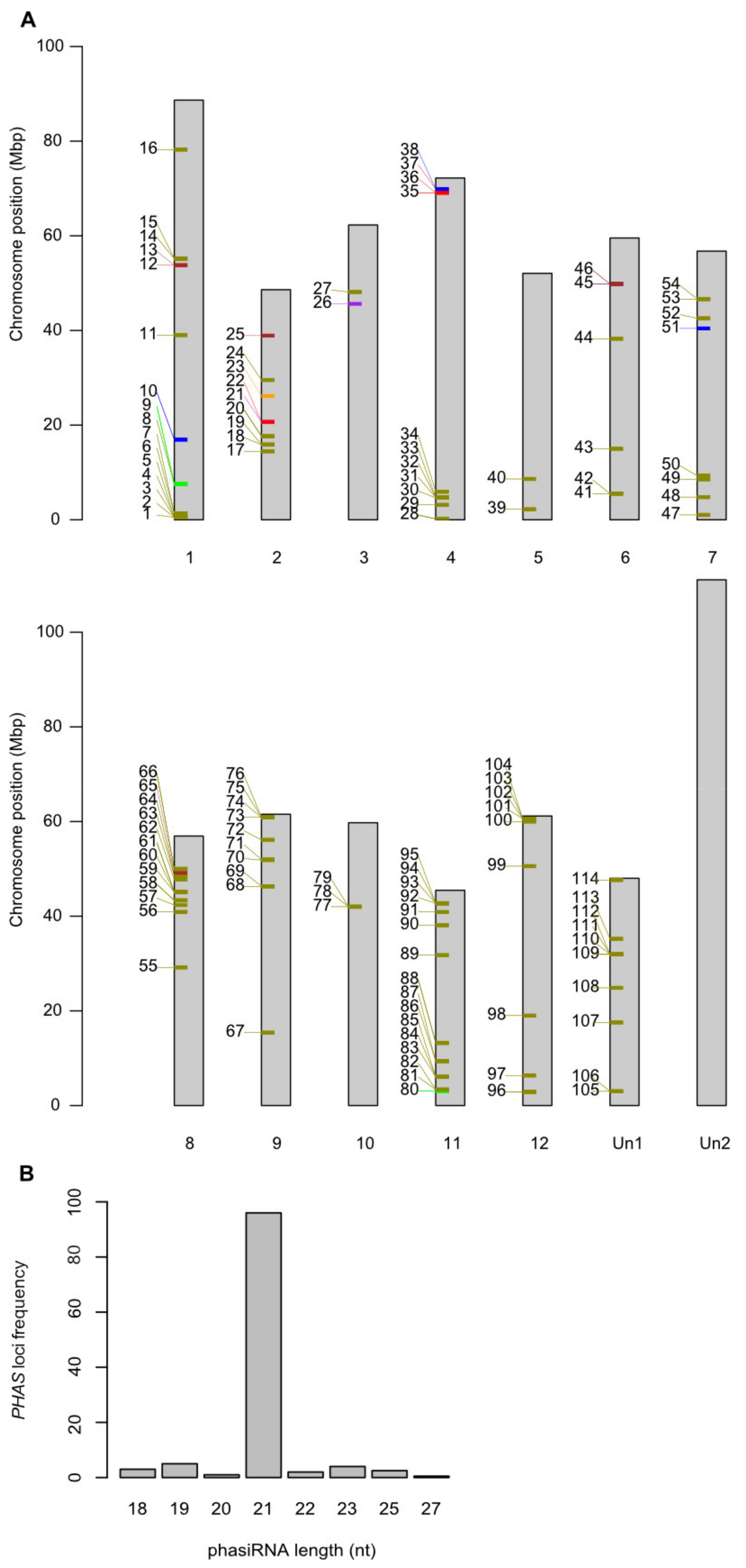
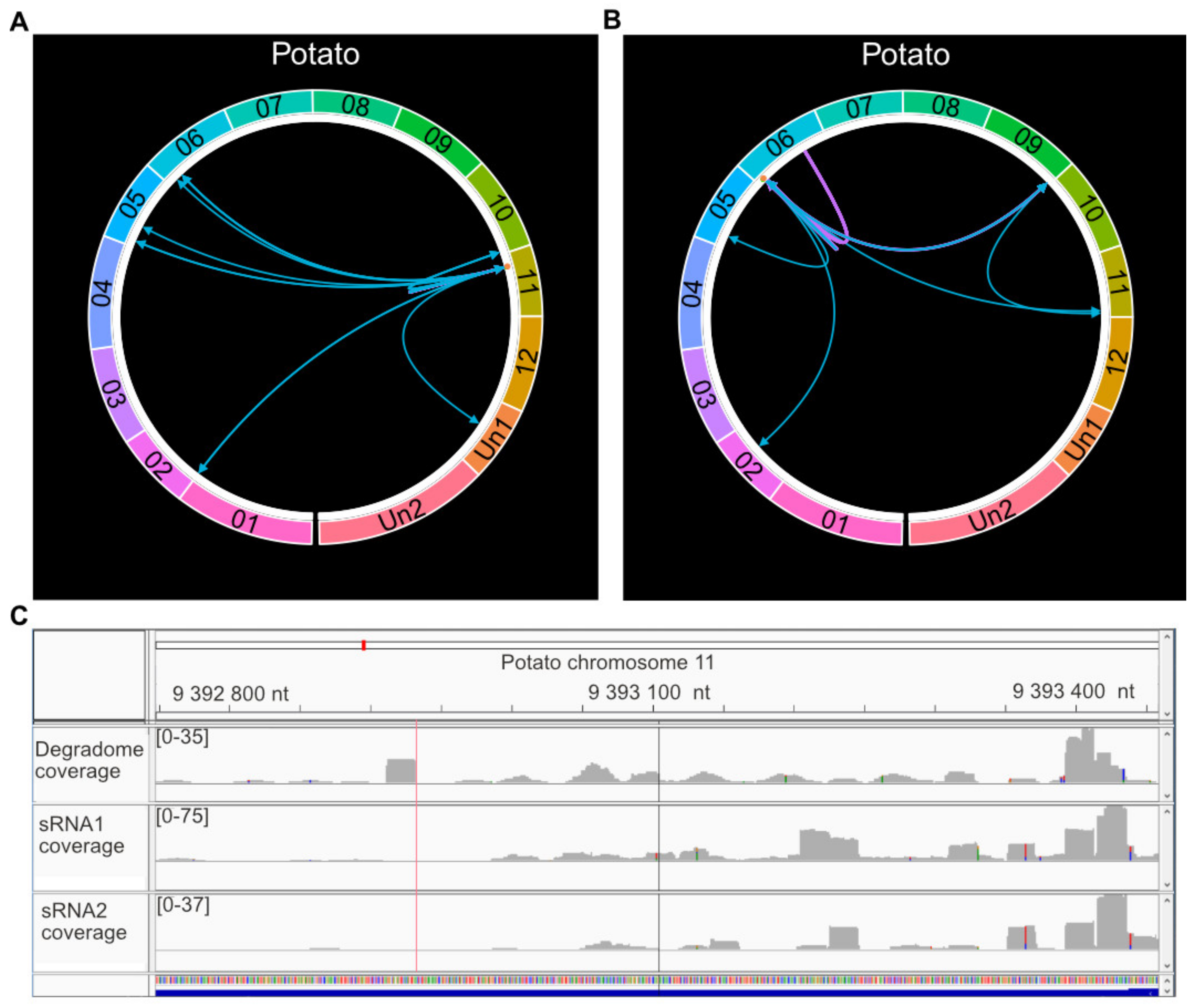
2.6. A cis-Regulating R Gene Induces a Three-Step phasiRNA Cascade
2.7. Resistance Genes Are a Major Target Group in Potato
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Infection, and Growth Conditions
4.2. Library Preparations
4.3. Degradome Data Analysis
4.4. Additional Datasets
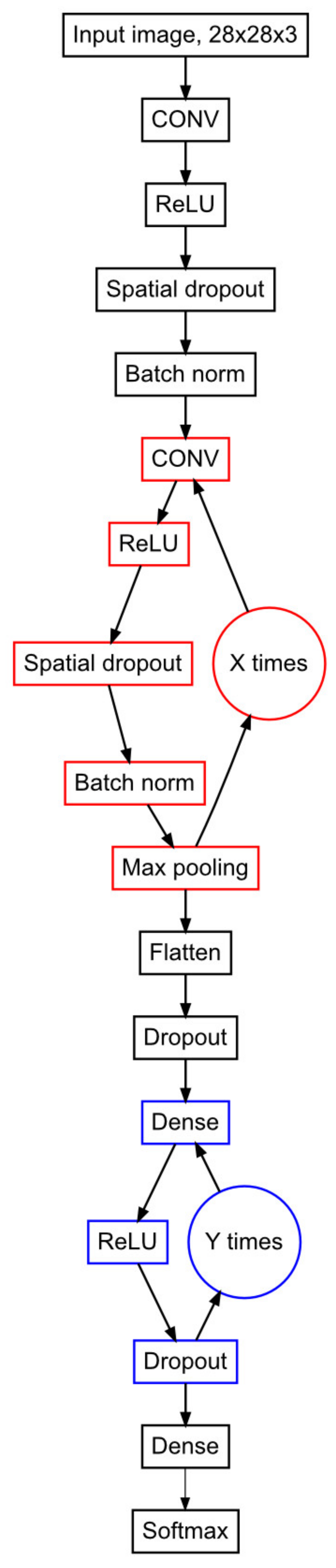
4.5. Design of smartPARE
4.6. Prediction of phasiRNAs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Axtell, M.J. Classification and comparison of small RNAs from plants. Ann. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [Green Version]
- Fei, Q.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Vaucheret, H.; Rajagopalan, R.; Lepers, C.; Gasciolli, V.; Mallory, A.C.; Hilbert, J.L.; Bartel, D.P.; Crété, P. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 2004, 16, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, M.A.; Pillay, M.; Jeong, D.-H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- The Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Zavallo, D.; Crescente, J.M.; Gantuz, M.; Leone, M.; Vanzetti, L.S.; Masuelli, R.W.; Asurmendi, S. Genomic re-assessment of the transposable element landscape of the potato genome. Plant Cell Rep. 2020, 39, 1161–1174. [Google Scholar] [CrossRef]
- Liao, Z.; Persson-Hodén, K.; Singh, R.K.; Dixelius, C. Genome-wide identification of Argonautes in Solanaceae with emphasis on potato. Sci. Rep. 2020, 10, 20577. [Google Scholar] [CrossRef]
- Esposito, S.; Aversao, R.; D’Amella, V.; Villano, C.; Alioto, D.; Mirouze, M.; Carputo, D. Dicer-like and RNA-dependent RNA polymerase gene family identification and annotation in the cultivated Solanum tuberosum and its wild relative S. commersonii. Planta 2018, 248, 729–743. [Google Scholar] [CrossRef]
- Leesutthiphonchai, W.; Vu, A.L.; Ah-Fong, A.M.V.; Judelson, H.S. How does Phytophthora infestans evade control efforts? Modern insight into the late blight disease. Phytopathology 2018, 108, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Vetukuri, R.R.; Avrova, A.O.; Grenville-Briggs, L.J.; van West, P.; Söderbom, F.; Savenkov, E.I.; Whisson, S.E.; Dixelius, C. Evidence for involvement of Dicer-like, Argonaute, and Histone Deacetylase proteins in gene silencing in Phytophthora infestans. Mol. Plant Pathol. 2011, 12, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Bollmann, S.R.; Kasschau, K.D.; Cuperus, J.T.; Press, C.M.; Sullivan, E.J.; Chapman, J.; Hoyer, S.; Kerrigan, B.; Grünwald, N.J.; et al. Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS ONE 2013, 8, e77181. [Google Scholar] [CrossRef]
- Vetukuri, R.R.; Åsman, A.K.M.; Tellgren-Roth, C.; Jahan, S.N.; Reimegård, J.; Savenkov, E.I.; Söderbom, F.; Avrova, A.O.; Whisson, S.C.; Dixelius, C. Evidence for small RNAs homologous to effector-encoding genes and transposable elements in the oomycete, Phytophthora infestans. PLoS ONE 2012, 7, e51399. [Google Scholar]
- Åsman, A.K.M.; Vetukuri, R.R.; Jahan, S.N.; Fogelqvist, J.; Corcoran, P.; Avrova, A.O.; Whisson, S.; Dixelius, C. Fragmentation of tRNA in Phytophthora infestans asexual life cycle stages and during host plant infection. BMC Microbiol. 2014, 14, 308. [Google Scholar] [CrossRef] [Green Version]
- Åsman, A.K.M.; Fogelqvist, J.; Vetukuri, R.R.; Dixelius, C. Phytophthora infestans Argonaute 1 binds microRNA and small RNAs from effector genes and transposable elements. New Phytol. 2016, 211, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Sharma, N.; Muthamilarasan, M.; Rana, S.; Prasad, M. Recent advances in small RNA mediated plant-virus interactions. Crit. Rev. Biotechnol. 2019, 39, 587–601. [Google Scholar] [CrossRef]
- Yan, Y.; Ham, B.-K.; Chong, Y.H.; Yeh, S.-D.; Lukas, W.J. A plant small RNA-binding protein 1 family mediates cell-to-cell trafficking of RNAi signals. Mol. Plant. 2020, 13, 321–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, C.C.; Dean, R.A. Host-induced gene silencing: A tool for understanding fungal host interaction and for developing novel disease control strategies. Mol. Plant Pathol. 2012, 13, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ma, W. Natural host-induced gene silencing offers new opportunities to engineer disease resistance. Trends Microbiol. 2020, 28, 109–117. [Google Scholar] [CrossRef]
- Jahan, S.; Åsman, A.; Corcoran, P.; Fogelqvist, J.; Vetukuri, R.R.; Dixelius, C. Plant-mediated gene silencing restricts growth of the potato late blight pathogen Phytophthora infestans. J. Exp. Bot. 2015, 66, 2785–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Persson-Hodén, K.; Liao, Z.; Dölfors, F.; Åsman, A.; Dixelius, C. Phytophthora infestans Ago1-associate miRNA promotes potato late blight disease. BioRxiv 2020. [Google Scholar] [CrossRef]
- Gulli, A.; Pal, S. Deep Learning with Keras; Packt Publishing Ltd.: Birmingham, UK, 2017. [Google Scholar]
- Thody, J.; Folkes, L.; Medina-Calzada, Z.; Xu, P.; Dalmay, T.; Moulton, V. PAREsnip2: A tool for high-throughput prediction of small RNA targets from degradome sequencing data using configurable targeting rules. Nuclear Acids Res. 2018, 46, 8730–8739. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.N. Cyclical learning rates for training neural networks. In Proceedings of the 2017 IEEE Winter Conference on Applications of Computer Vision, WACV 2017, Santa Rosa, CA, USA, 24–31 March 2017; pp. 464–472. [Google Scholar]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical Bayesian optimization of machine learning algorithms. Int. Conf. Neural Info. Proc. Syst. 2012, 2, 2951–2959. [Google Scholar]
- Derrien, B.; Clavel, M.; Baumberger, N.; Iki, T.; Sarazin, A.; Hacquard, T.; Ponce, M.R.; Ziegler-Graff, V.; Vaucheret, H.; Micol, J.L. A suppressor screen for AGO1 degradation by the viral F-Box PO proteins uncovers a role for AGO DUF1785 in sRNA duplex unwinding. Plant Cell 2018, 30, 1353–1374. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Peral, M.M.; Li, J.; Li, Y.; Allen, R.S.; Schnippenkoetter, W.; Ohms, S.; White, R.G.; Millar, A.A. The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010, 154, 757–771. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Shen, J.; Hou, X.; Yao, J.; Li, X.; Xiao, J.; Xiong, L. Gradual increase of miR156 regulates temporal expression changes of numerous genes during leaf development in rice. Plant Physiol. 2012, 158, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.; Kumar, P.; Baker, B. MicroRNA regulation of plant innate immune receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790–1795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xian, Z.; Huang, W.; Li, Z. Evidence for the biological function of miR403 in tomato development. Sci. Hortic. 2015, 197, 619–626. [Google Scholar] [CrossRef]
- Jupé, F.; Witek, K.; Verweij, W.; Śliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, P.; Muhammad, S.; Cao, M.; Wu, L. Biogenesis and regulatory hierarchy of phased small interfering RNAs in plants. Plant Biotechn. J. 2018, 16, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Teng, C.; Xia, R.; Mayers, B.C. PhasiRNAs in plants: Their biogenesis, genic sources, and roles in stress responses, development and reproduction. Plant Cell 2020, 32, 3059–3080. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Jeong, D.-H.; de Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; González, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [Green Version]
- Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R.P.; de Freitas, N. Taking the human out of the loop: A review of Bayesian optimization. Proc. IEEE 2016, 104, 148–175. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Arikit, S.; Simon, S.A.; Kingham, B.F.; Meyers, B.C. Rapid construction of parallel analysis of RNA end (PARE) libraries for Illumina sequencing. Methods 2014, 67, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Suzuki, T.; Murata, S.; Nakamura, S.; Hino, T.; Maeo, K.; Tabata, R.; Kawai, T.; Tanaka, K.; Niwa, Y.; et al. Improved Gateway binary vectors: High-performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci. Biotechnol. Biochem. 2007, 71, 2095–2100. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Carbonell, A.; Marques, C.M.; Martinez, G.; Gomez, G. Dynamic architecture and regulatory implications of the miRNA network underlying the response to stress in melon. RNA Biol. 2020, 17, 292–308. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Fahlgren, N.; Carrington, J.C. miRNA target prediction in plants. Plant MicroRNAs Methods Protoc. 2010, 592, 51–57. [Google Scholar]
- Hardigan, M.A.; Crisovan, E.; Hamilton, J.P.; Kim, J.; Laimbeer, P.; Leisner, C.P.; Manrique-Carpintero, N.C.; Newton, L.; Pham, G.M.; Vaillancour, B.; et al. Genome reduction uncovers a large dispensable genome and adaptive role for copy number variation in asexually propagated Solanum tuberosum. Plant Cell 2016, 28, 388–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2017. Available online: https://www.R-project.org/ (accessed on 30 June 2020).
- Cano, L.; Kamoun, S.; Win, J. The Effector Secretome of the Irish Potato Famine Pathogen Phytophthora infestans, Data Set. 2019. Available online: https://zenodo.org/record/3574589#.X81dqrN7mUk (accessed on 13 January 2020).
- Kondhare, K.R.; Malankar, N.N.; Devani, R.S.; Banerjee, A.K. Genome-wide transcriptome analysis reveals small RNA profiles involved in early stages of stolon-to-tuber transitions in potato under photoperiodic conditions. BMC Plant Biol. 2018, 18, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chollet, F. Keras. 2015. Available online: https://github.com/fchollet/keras.
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-scale machine learning on heterogeneous systems. arXiv 2015, arXiv:1603.04467v2. [Google Scholar]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2019. [Google Scholar]
- Guo, Q.; Qu, X.; Jin, W. PhaseTank: Genome-wide computational identification of phasiRNAs and their regulatory cascades. Bioinformatics 2015, 31, 284–286. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persson Hodén, K.; Hu, X.; Martinez, G.; Dixelius, C. smartPARE: An R Package for Efficient Identification of True mRNA Cleavage Sites. Int. J. Mol. Sci. 2021, 22, 4267. https://doi.org/10.3390/ijms22084267
Persson Hodén K, Hu X, Martinez G, Dixelius C. smartPARE: An R Package for Efficient Identification of True mRNA Cleavage Sites. International Journal of Molecular Sciences. 2021; 22(8):4267. https://doi.org/10.3390/ijms22084267
Chicago/Turabian StylePersson Hodén, Kristian, Xinyi Hu, German Martinez, and Christina Dixelius. 2021. "smartPARE: An R Package for Efficient Identification of True mRNA Cleavage Sites" International Journal of Molecular Sciences 22, no. 8: 4267. https://doi.org/10.3390/ijms22084267
APA StylePersson Hodén, K., Hu, X., Martinez, G., & Dixelius, C. (2021). smartPARE: An R Package for Efficient Identification of True mRNA Cleavage Sites. International Journal of Molecular Sciences, 22(8), 4267. https://doi.org/10.3390/ijms22084267