
Impaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
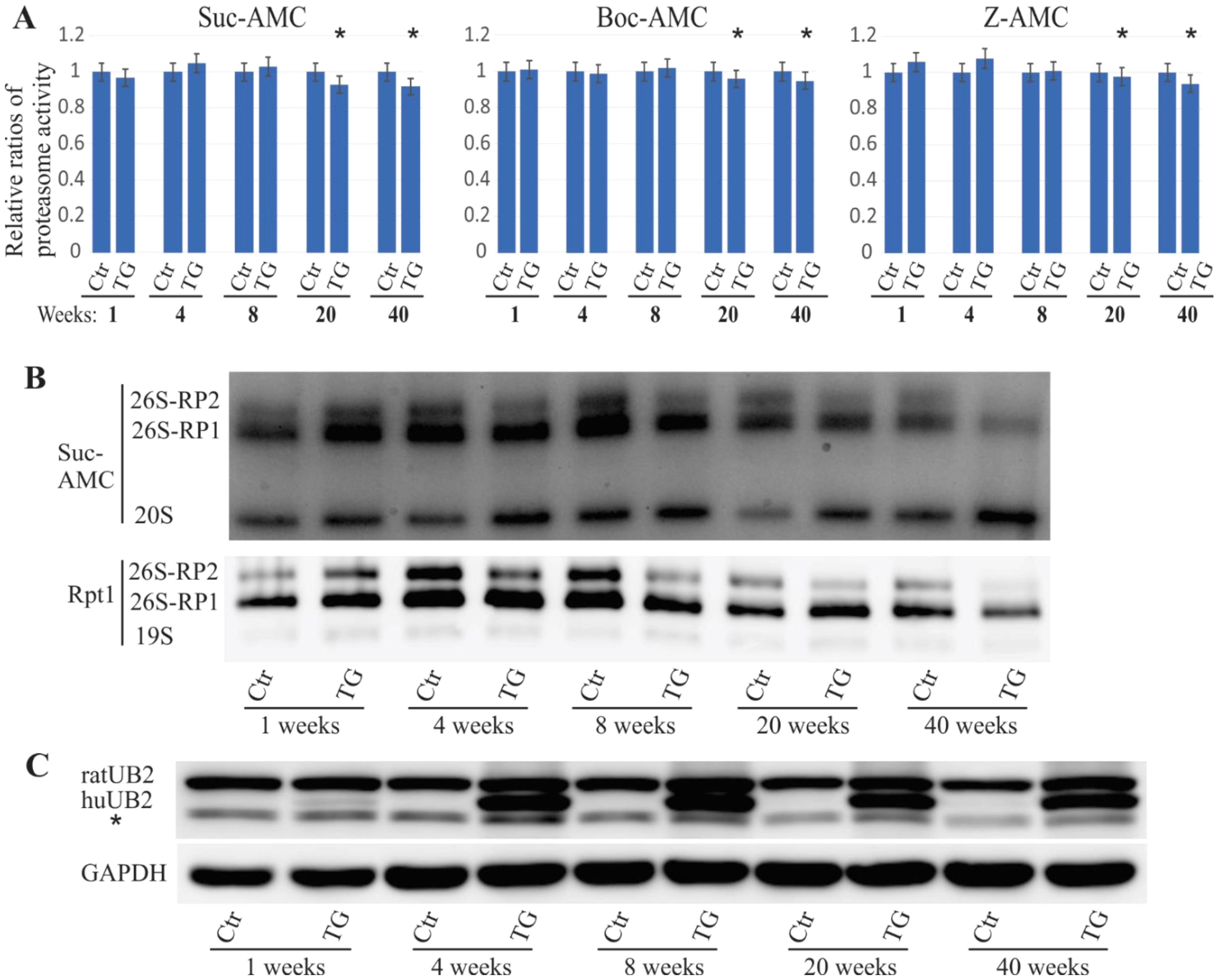
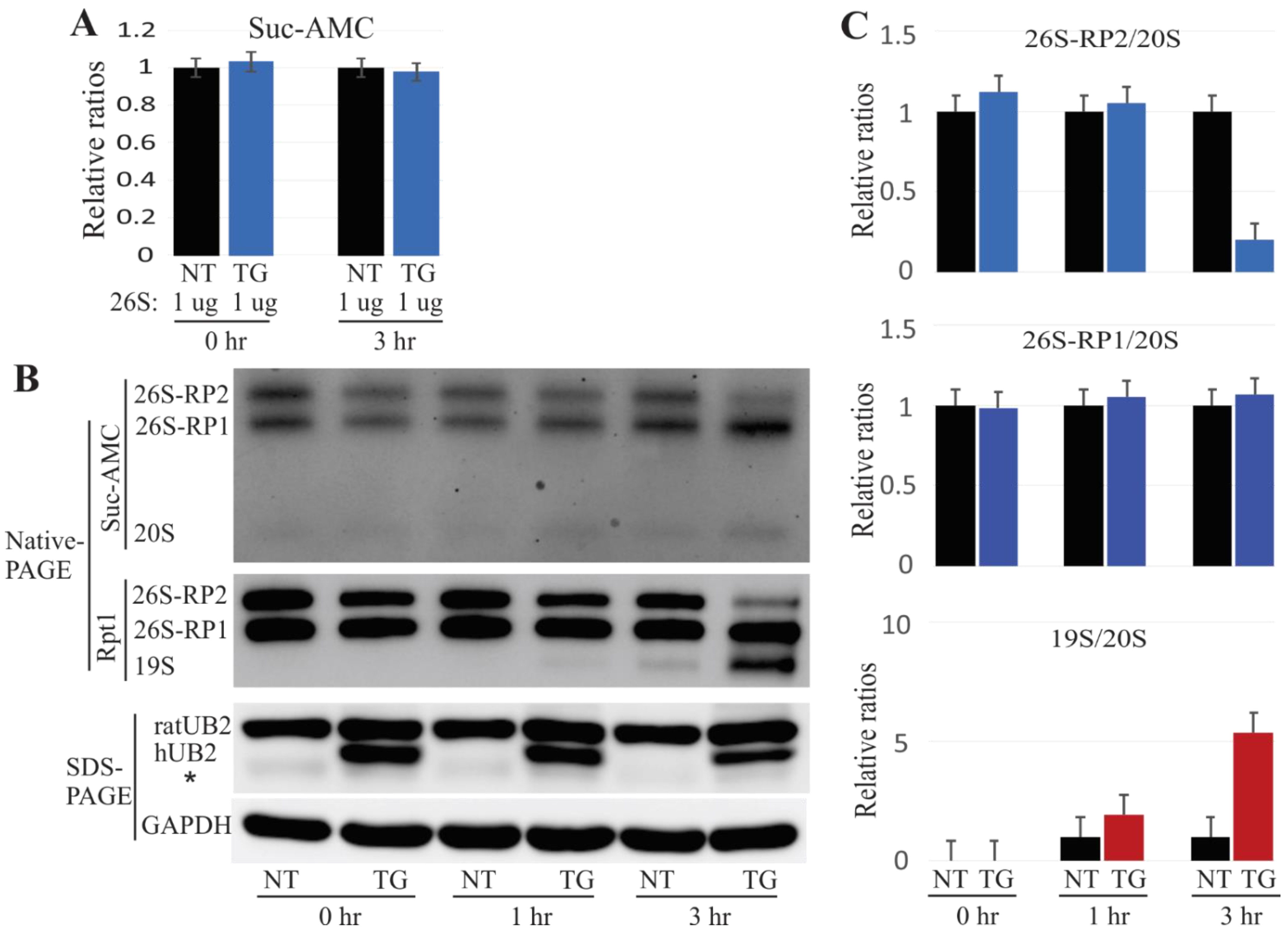
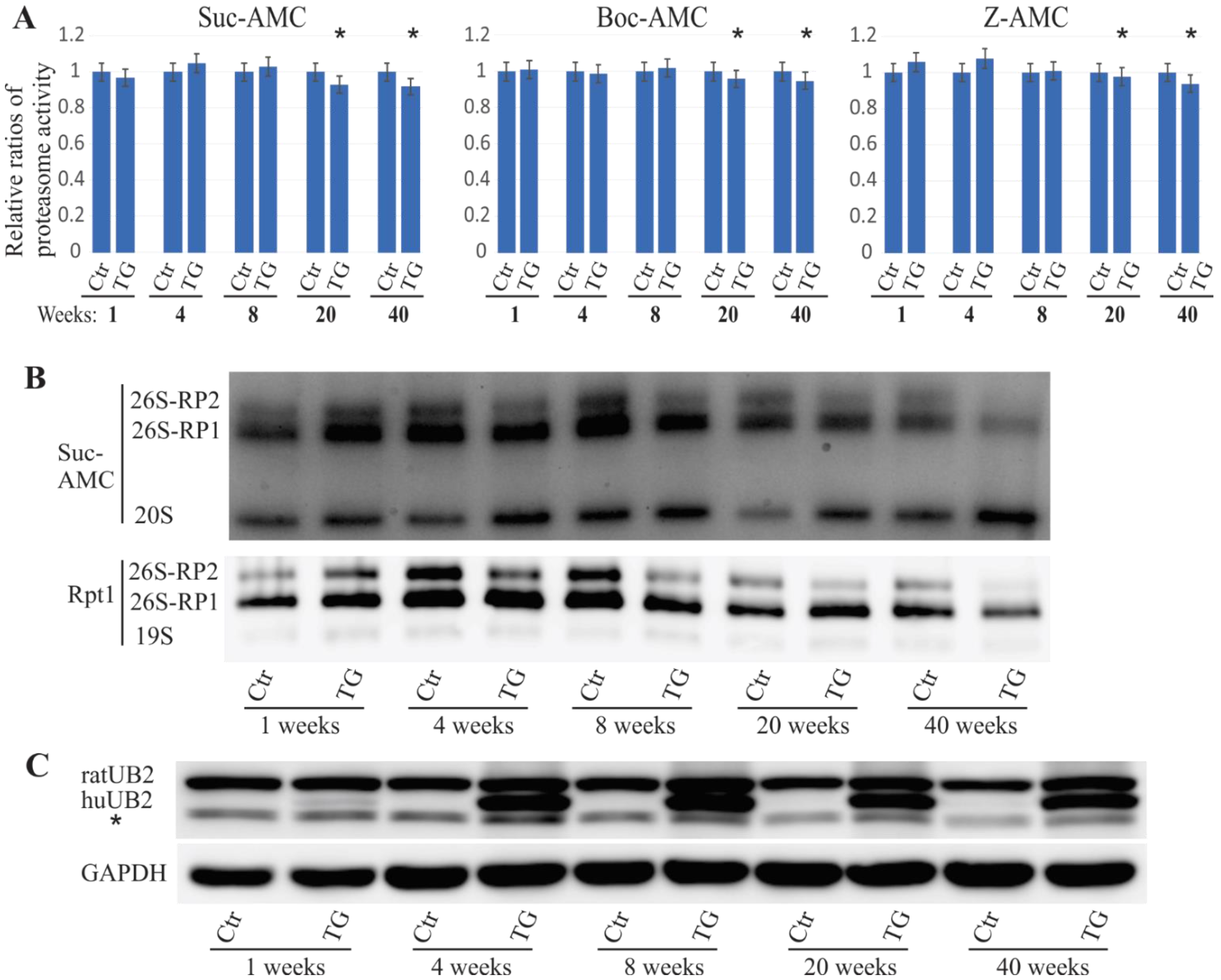
2.1. Proteasome Impairment in Mutant UBQLN2 Transgenic Rats
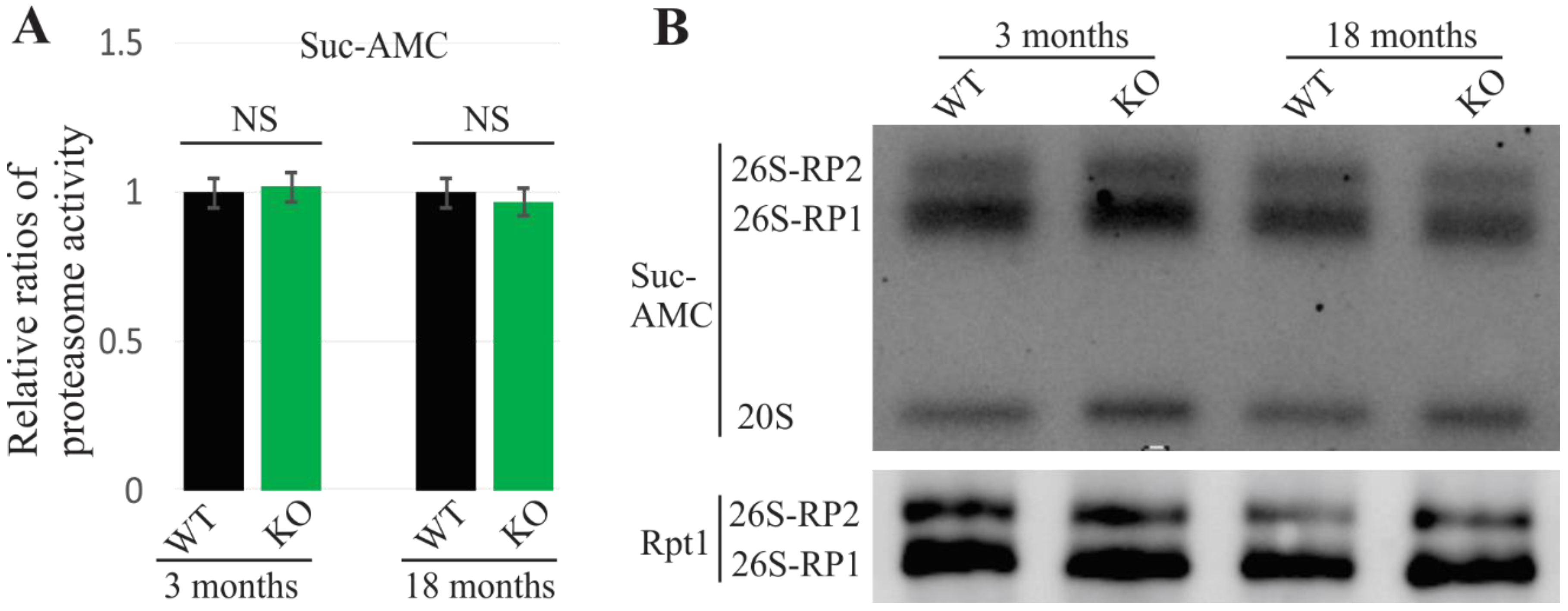
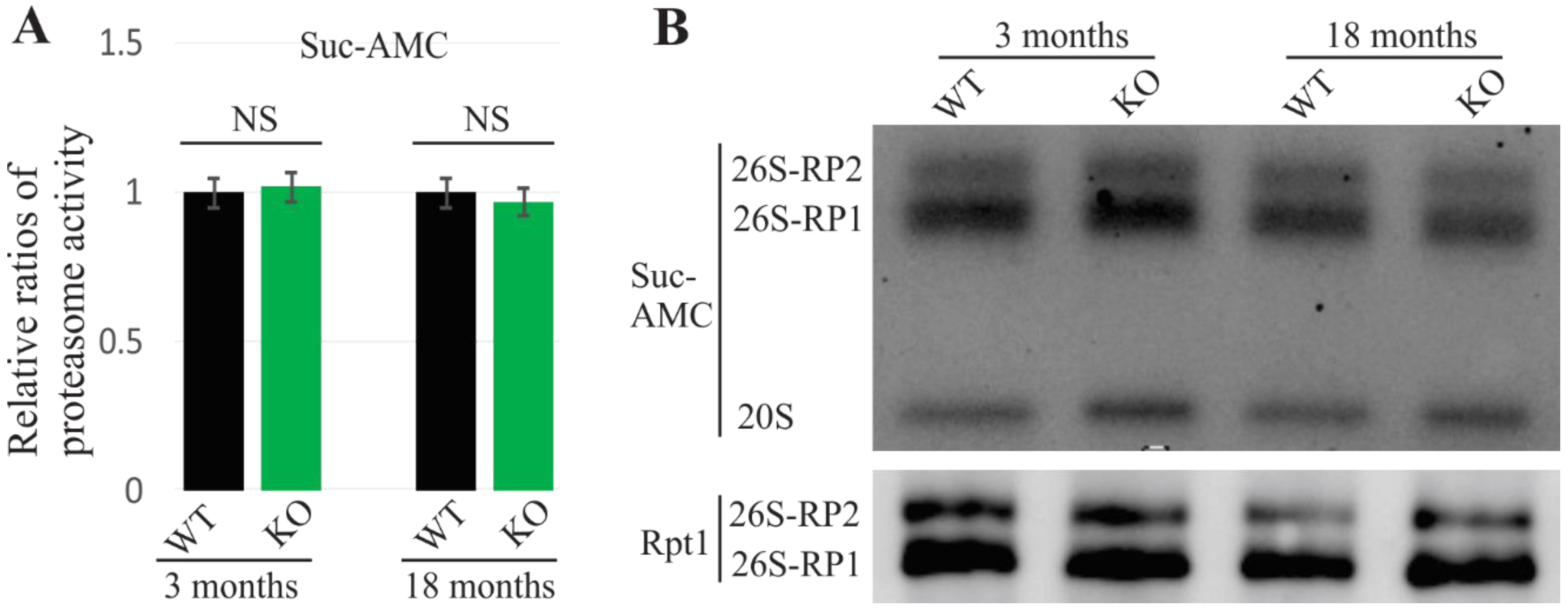
2.2. No Observed Alterations of Proteasome in UBQLN2-Depleted Rats
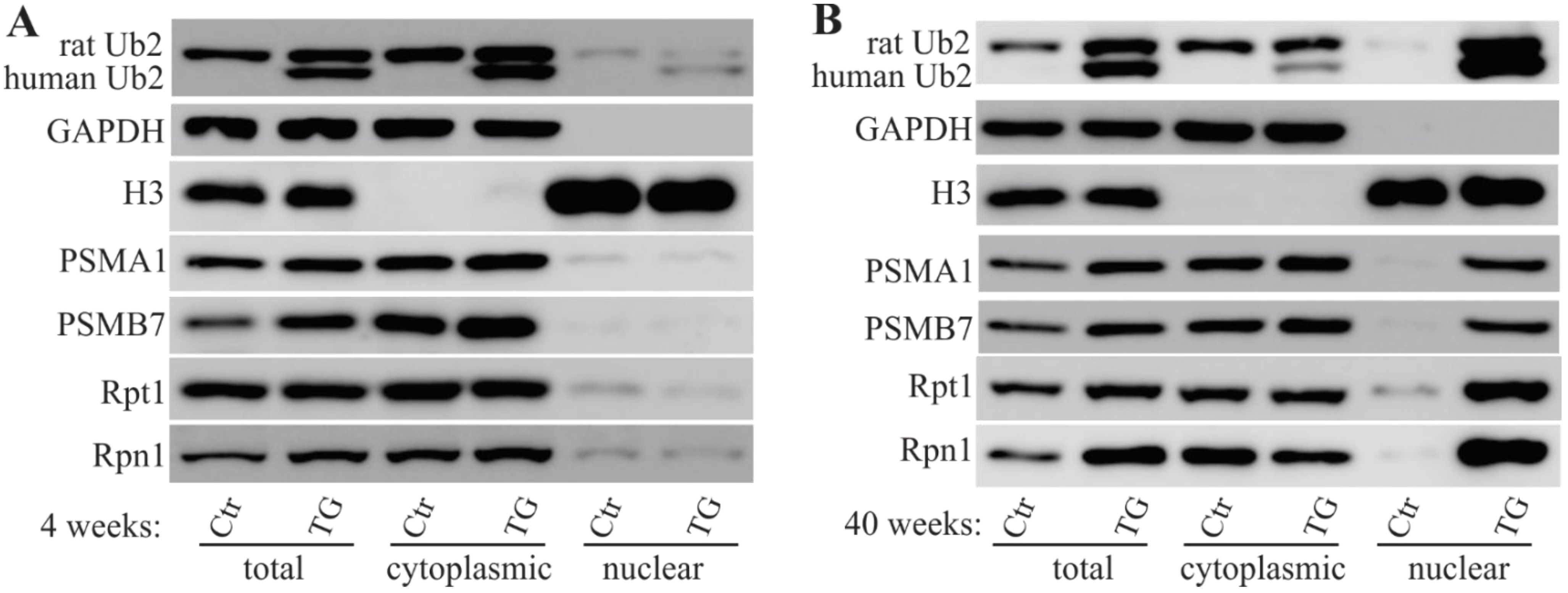
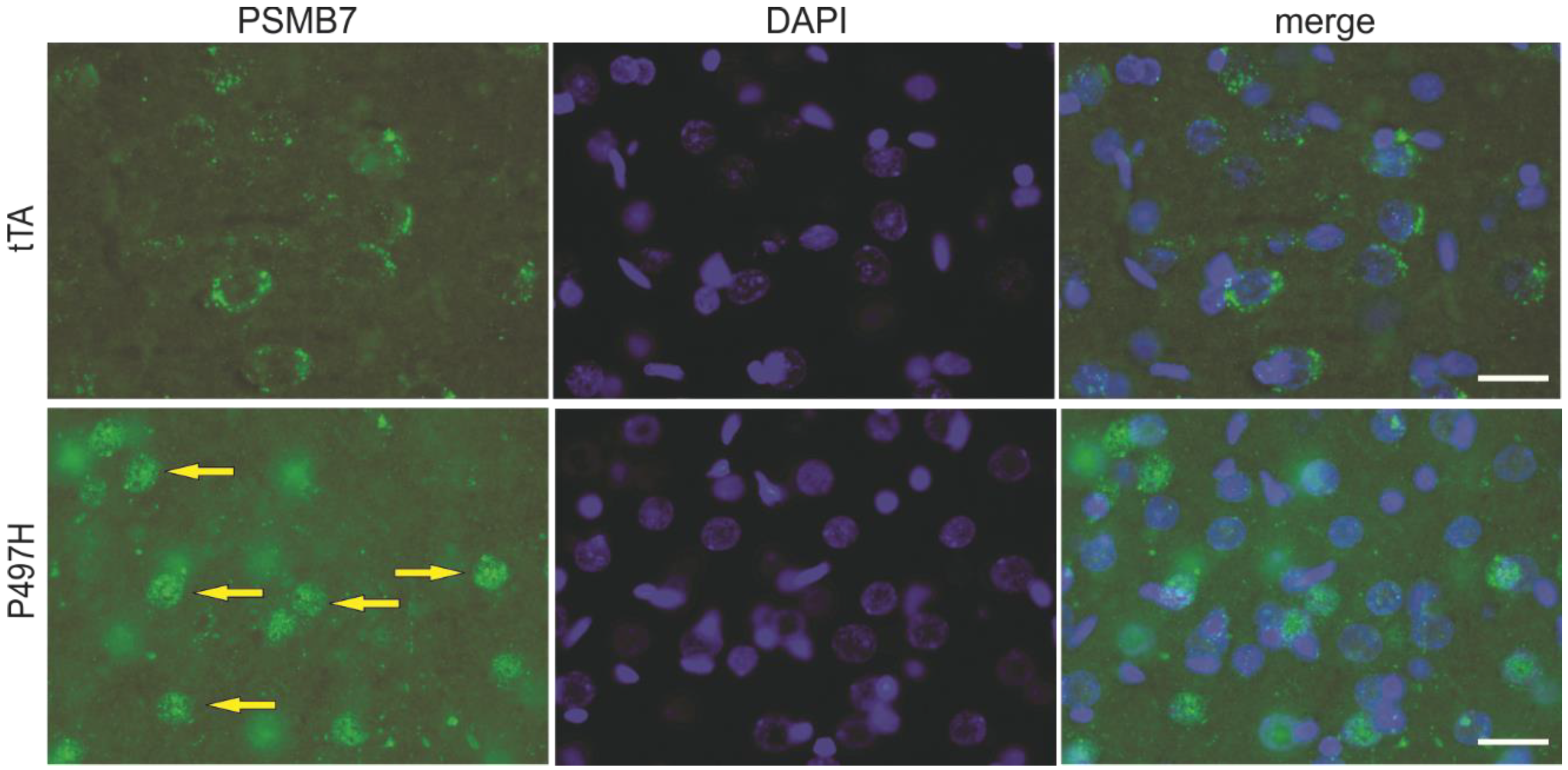
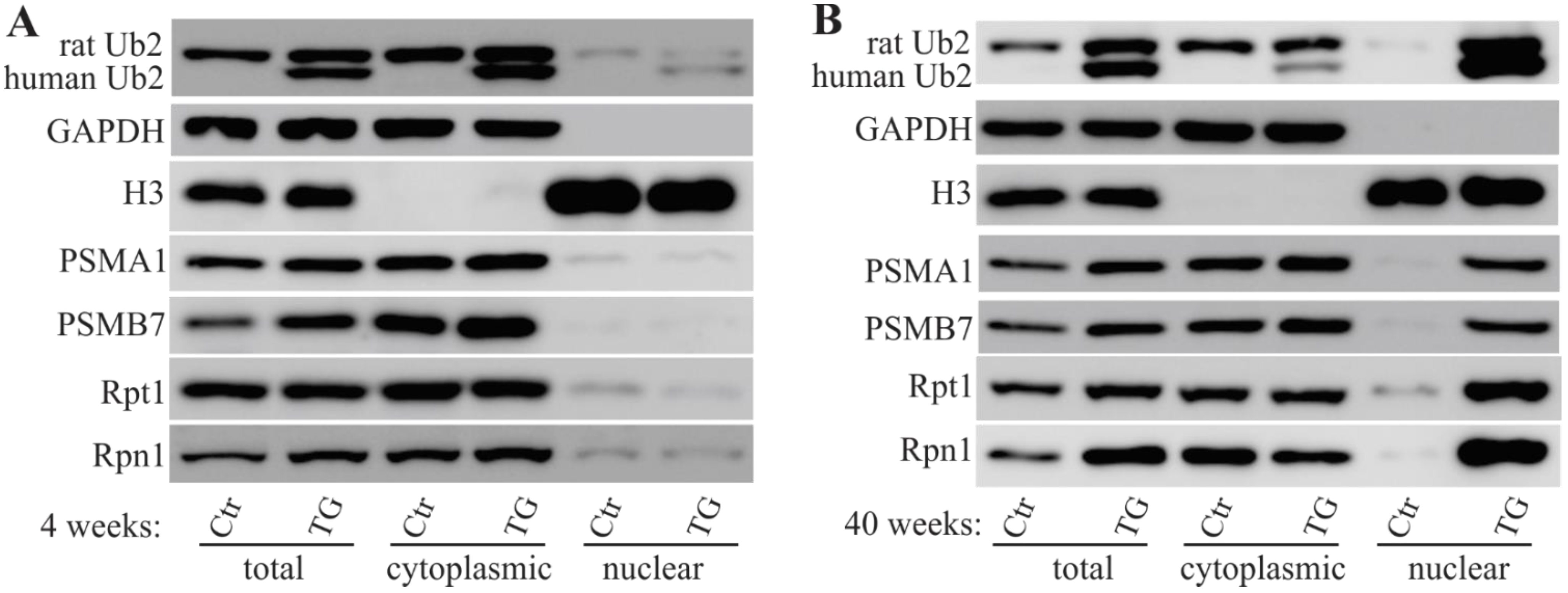
2.3. Mislocalized Proteasome Subunits in Mutant UBQLN2 Transgenic Rats
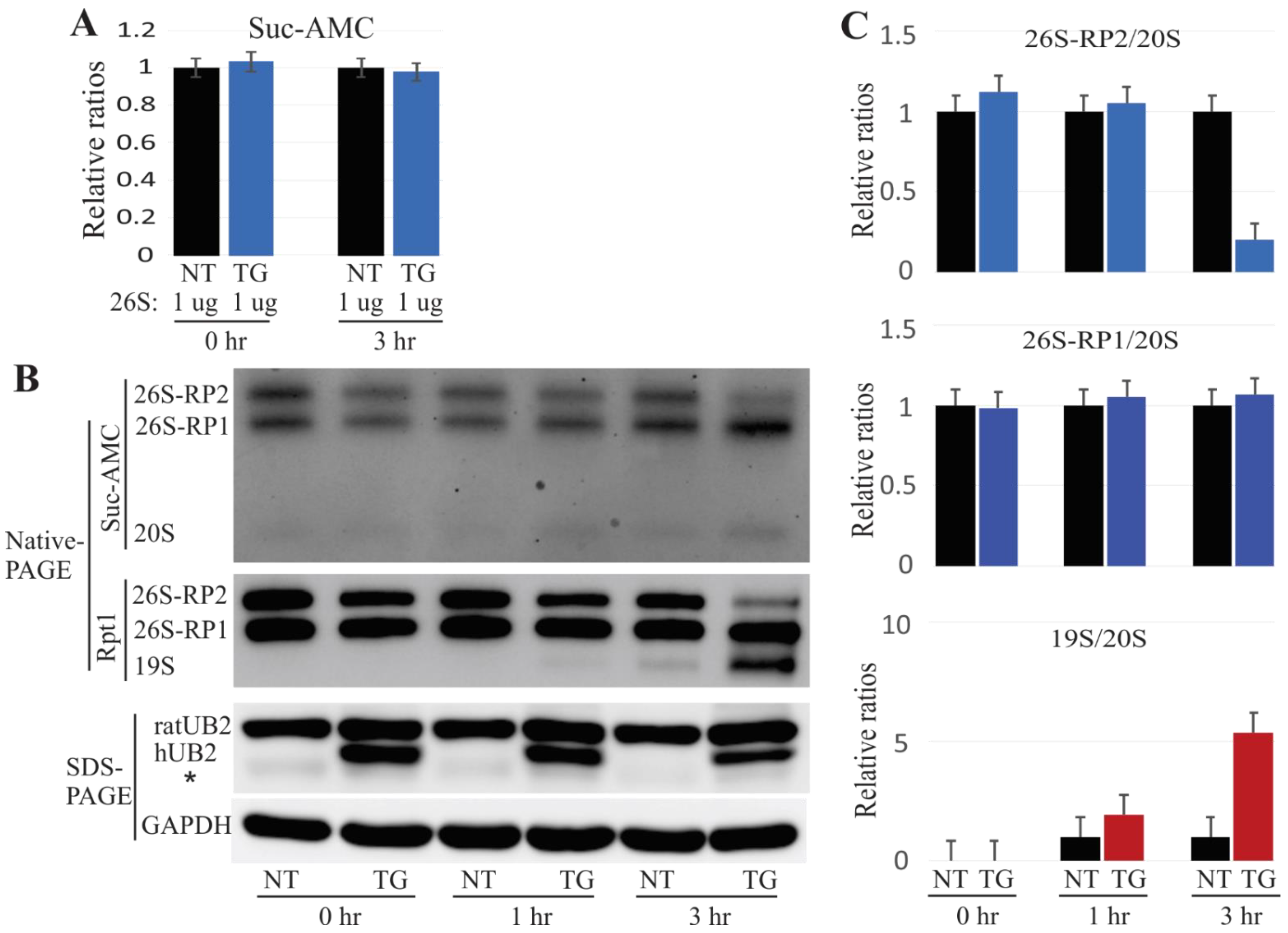
2.4. In Vitro Dissociation of the 26S Proteasome by Mutant UBQLN2
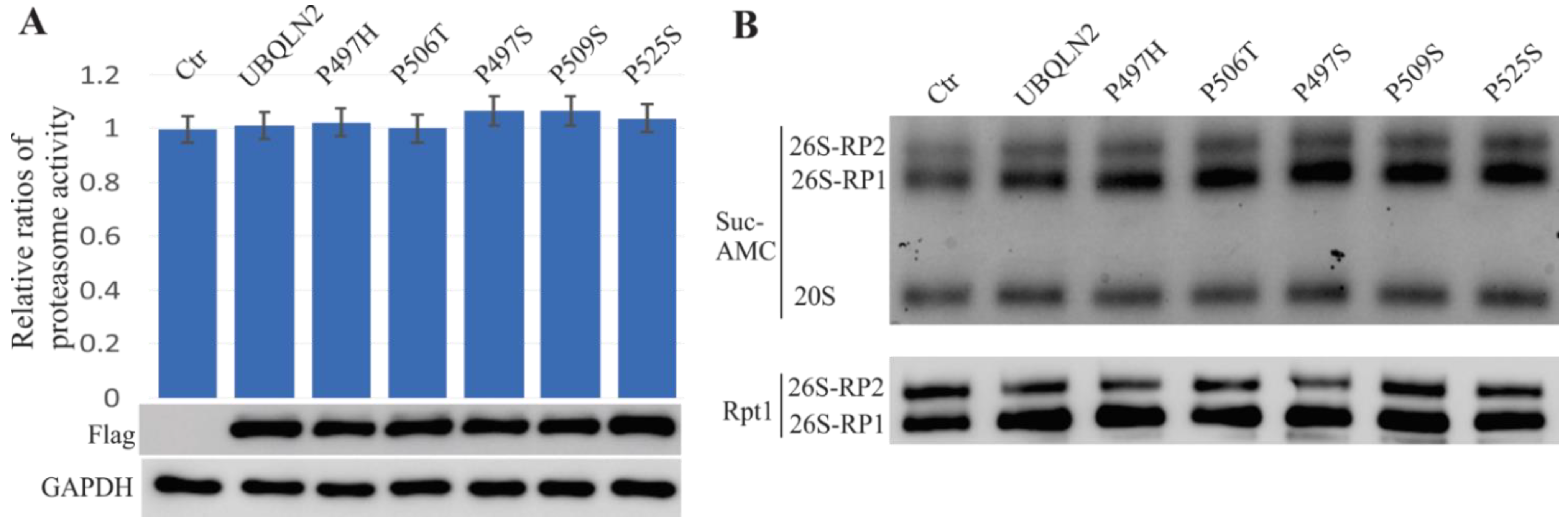
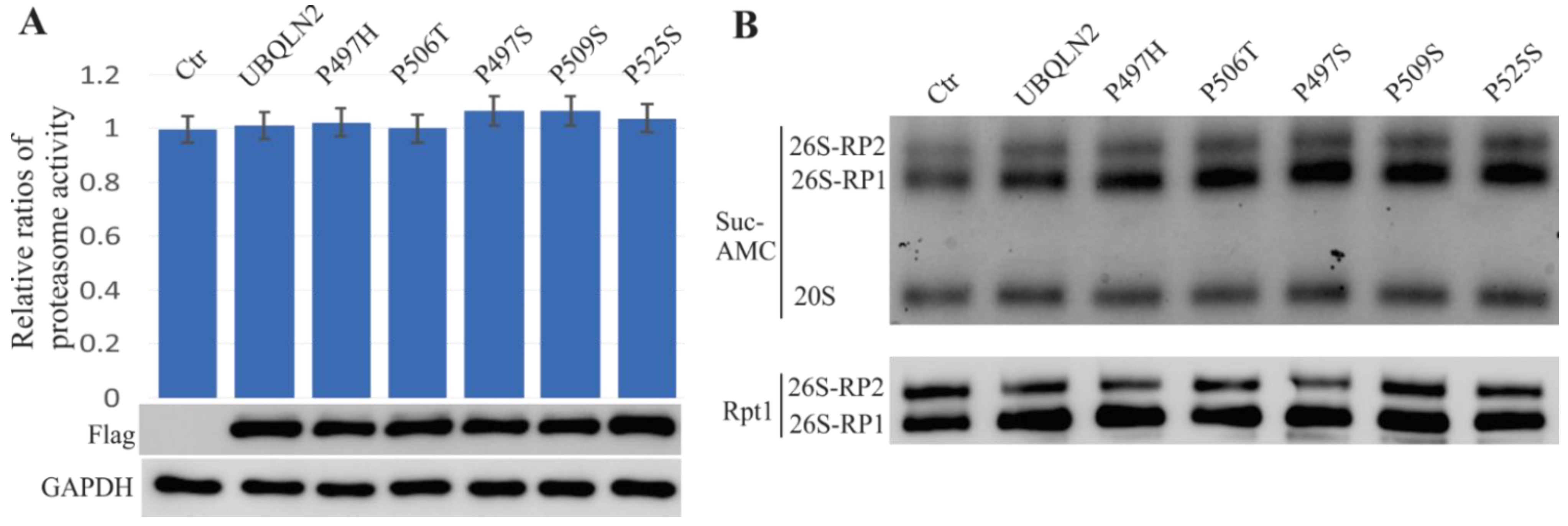
2.5. ALS/FTD-Linked UBQLN2 Mutations Did Not Affect Proteasome Function in HEK-293T Cells
3. Discussion
4. Materials and Methods
4.1. Genotyping of Transgenic Rats
4.2. Plasmid Construction and Cell Culture
4.3. Cell Fractionation
4.4. SDS-PAGE Electrophoresis and Fluorescence Staining
4.5. Proteasome Activity Assays and Native-PAGE Electrophoresis
4.6. Proteasome Dissociation Assay
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef]
- Halloran, M.; Ragagnin, A.M.G.; Vidal, M.; Parakh, S.; Yang, S.; Heng, B.; Grima, N.; Shahheydari, H.; Soo, K.Y.; Blair, I.; et al. Amyotrophic lateral sclerosis-linked UBQLN2 mutants inhibit endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER stress. Cell. Mol. Life Sci. 2020, 77, 3859–3873. [Google Scholar] [CrossRef]
- Gorrie, G.H.; Fecto, F.; Radzicki, D.; Weiss, C.; Shi, Y.; Dong, H.; Zhai, H.; Fu, R.; Liu, E.; Li, S.; et al. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc. Natl. Acad. Sci. USA 2014, 111, 14524–14529. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Huang, B.; Shi, X.; Gao, L.; Huang, C. Mutant UBQLN2(P497H) in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol. Commun. 2018, 6, 122. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, M.; Huang, C.; Liu, X.; Huang, B.; Li, N.; Zhou, H.; Xia, X.G. Pathogenic UBQLN2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015, 129, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Le, N.T.; Chang, L.; Kovlyagina, I.; Georgiou, P.; Safren, N.; Braunstein, K.E.; Kvarta, M.D.; Van Dyke, A.M.; LeGates, T.A.; Philips, T.; et al. Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E7580–E7589. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, L.M.; Sandoval-Pistorius, S.S.; Moore, S.J.; Gerson, J.E.; Komlo, R.; Fischer, S.; Negron-Rios, K.Y.; Crowley, E.V.; Padron, F.; Patel, R.; et al. Modeling UBQLN2-mediated neurodegenerative disease in mice: Shared and divergent properties of wild type and mutant UBQLN2 in phase separation, subcellular localization, altered proteostasis pathways, and selective cytotoxicity. Neurobiol. Dis. 2020, 143, 105016. [Google Scholar] [CrossRef]
- Gao, L.; Tu, H.; Shi, S.T.; Lee, K.J.; Asanaka, M.; Hwang, S.B.; Lai, M.M. Interaction with a ubiquitin-like protein enhances the ubiquitination and degradation of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003, 77, 4149–4159. [Google Scholar] [CrossRef] [Green Version]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Mikhailov, A.; Kallo-Hosein, H.; Hara, K.; Yonezawa, K.; Avruch, J. Characterization of ubiquilin 1, an mTOR-interacting protein. Biochim. Biophys. Acta BBA Mol. Cell Res. 2002, 1542, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Synofzik, M.; Maetzler, W.; Grehl, T.; Prudlo, J.; Vom Hagen, J.M.; Haack, T.; Rebassoo, P.; Munz, M.; Schols, L.; Biskup, S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol. Aging 2012, 33, 2949.e13–2949.e17. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Monteiro, M.J. Defective Proteasome Delivery of Polyubiquitinated Proteins by Ubiquilin-2 Proteins Containing ALS Mutations. PLoS ONE 2015, 10, e0130162. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Li, X.; Kim, H.M.; Singh, C.R.; Tian, G.; Hoyt, M.A.; Lovell, S.; Battaile, K.P.; Zolkiewski, M.; Coffino, P.; et al. Reconfiguration of the proteasome during chaperone-mediated assembly. Nature. 2013, 497, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef] [Green Version]
- Saeki, Y.; Tanaka, K. Assembly and function of the proteasome. Methods. Mol. Biol. 2012, 832, 315–337. [Google Scholar] [PubMed]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta. Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- Walters, K.J.; Kleijnen, M.F.; Goh, A.M.; Wagner, G.; Howley, P.M. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 2002, 41, 1767–1777. [Google Scholar] [CrossRef]
- Itakura, E.; Zavodszky, E.; Shao, S.; Wohlever, M.L.; Keenan, R.J.; Hegde, R.S. Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol. Cell 2016, 63, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wu, Q.; Zhou, H.; Huang, C.; Xia, X.G. Increased Ubqln2 expression causes neuron death in transgenic rats. J. Neurochem. 2016, 139, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Cai, A.; Greenslade, J.E.; Higgins, N.R.; Fan, C.; Le, N.T.T.; Tatman, M.; Whiteley, A.M.; Prado, M.A.; Dieriks, B.V.; et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc. Natl. Acad. Sci. USA 2020, 117, 15230–15241. [Google Scholar] [CrossRef]
- Ceballos-Diaz, C.; Rosario, A.M.; Park, H.J.; Chakrabarty, P.; Sacino, A.; Cruz, P.E.; Siemienski, Z.; Lara, N.; Moran, C.; Ravelo, N.; et al. Viral expression of ALS-linked ubiquilin-2 mutants causes inclusion pathology and behavioral deficits in mice. Mol. Neurodegener. 2015, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, L.M.; Safren, N.; Pithadia, A.S.; Gerson, J.E.; Dulchavsky, M.; Fischer, S.; Patel, R.; Lantis, G.; Ashraf, N.; Kim, J.H.; et al. Mutant UBQLN2 promotes toxicity by modulating intrinsic self-assembly. Proc. Natl. Acad. Sci. USA 2018, 115, E10495–E10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhang, W.; Huang, B.; Chen, X.; Huang, C. UBQLN2 Promotes the Production of Type I Interferon via the TBK1-IRF3 Pathway. Cells 2020, 9, 1205. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Tong, J.; Bi, F.; Zhou, H.; Xia, X.G. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J. Clin. Investig. 2012, 122, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Huang, B.; Gao, L.; Huang, C. Impaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats. Int. J. Mol. Sci. 2021, 22, 4319. https://doi.org/10.3390/ijms22094319
Zhang W, Huang B, Gao L, Huang C. Impaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats. International Journal of Molecular Sciences. 2021; 22(9):4319. https://doi.org/10.3390/ijms22094319
Chicago/Turabian StyleZhang, Wenjuan, Bo Huang, Limo Gao, and Cao Huang. 2021. "Impaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats" International Journal of Molecular Sciences 22, no. 9: 4319. https://doi.org/10.3390/ijms22094319
APA StyleZhang, W., Huang, B., Gao, L., & Huang, C. (2021). Impaired 26S Proteasome Assembly Precedes Neuronal Loss in Mutant UBQLN2 Rats. International Journal of Molecular Sciences, 22(9), 4319. https://doi.org/10.3390/ijms22094319