
Molecular Targeted Therapy for the Bone Loss Secondary to Pyogenic Spondylodiscitis Using Medications for Osteoporosis: A Literature Review

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. The Peculiarity of Bone Loss Secondary to Pyogenic Spondylodiscitis
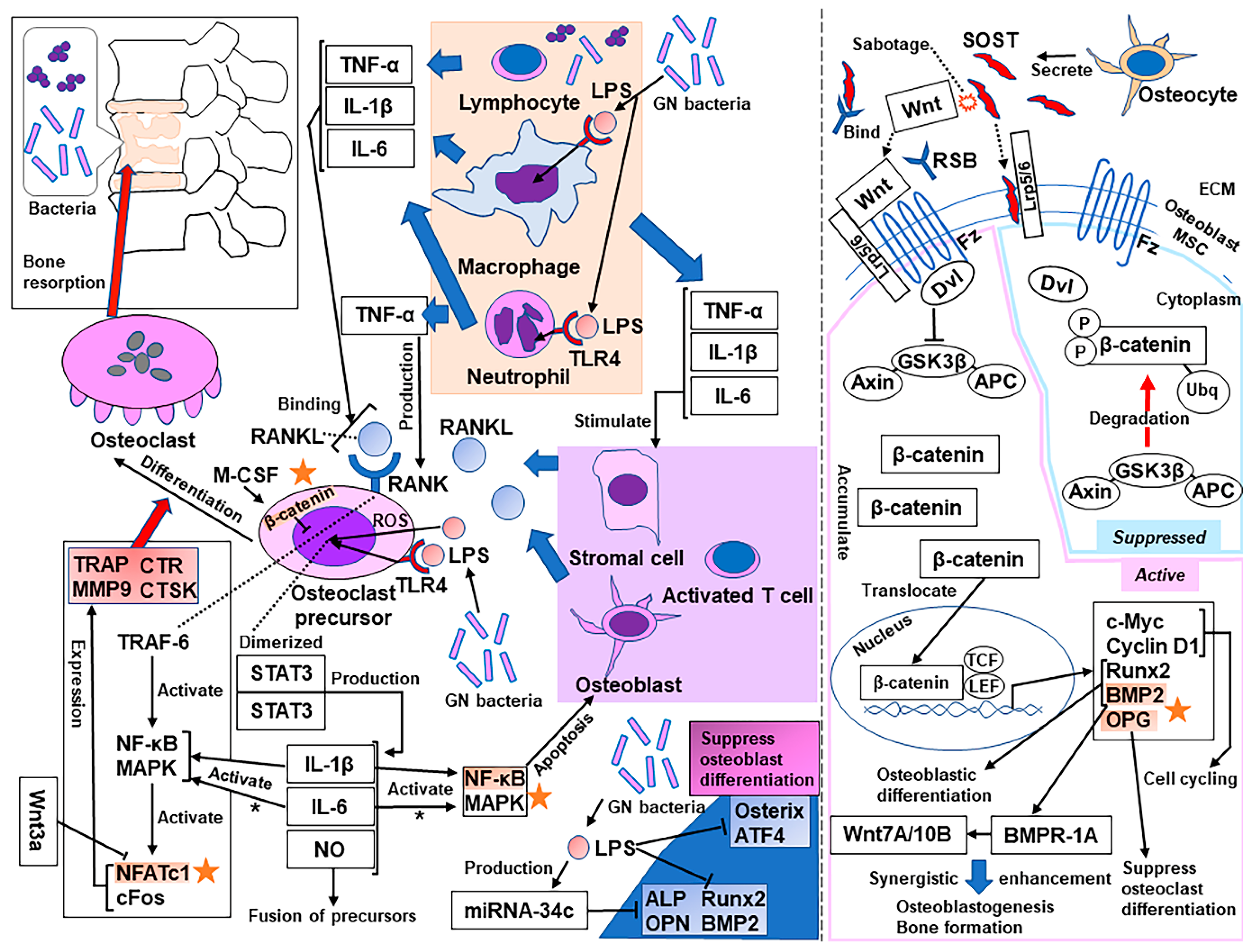
3. Molecular Mechanisms of Bone Resorption Secondary to Infection
4. Osteoporosis Medications as a Treatment for Bone Loss Secondary to Infection
4.1. Anabolic Drug: Romosozumab
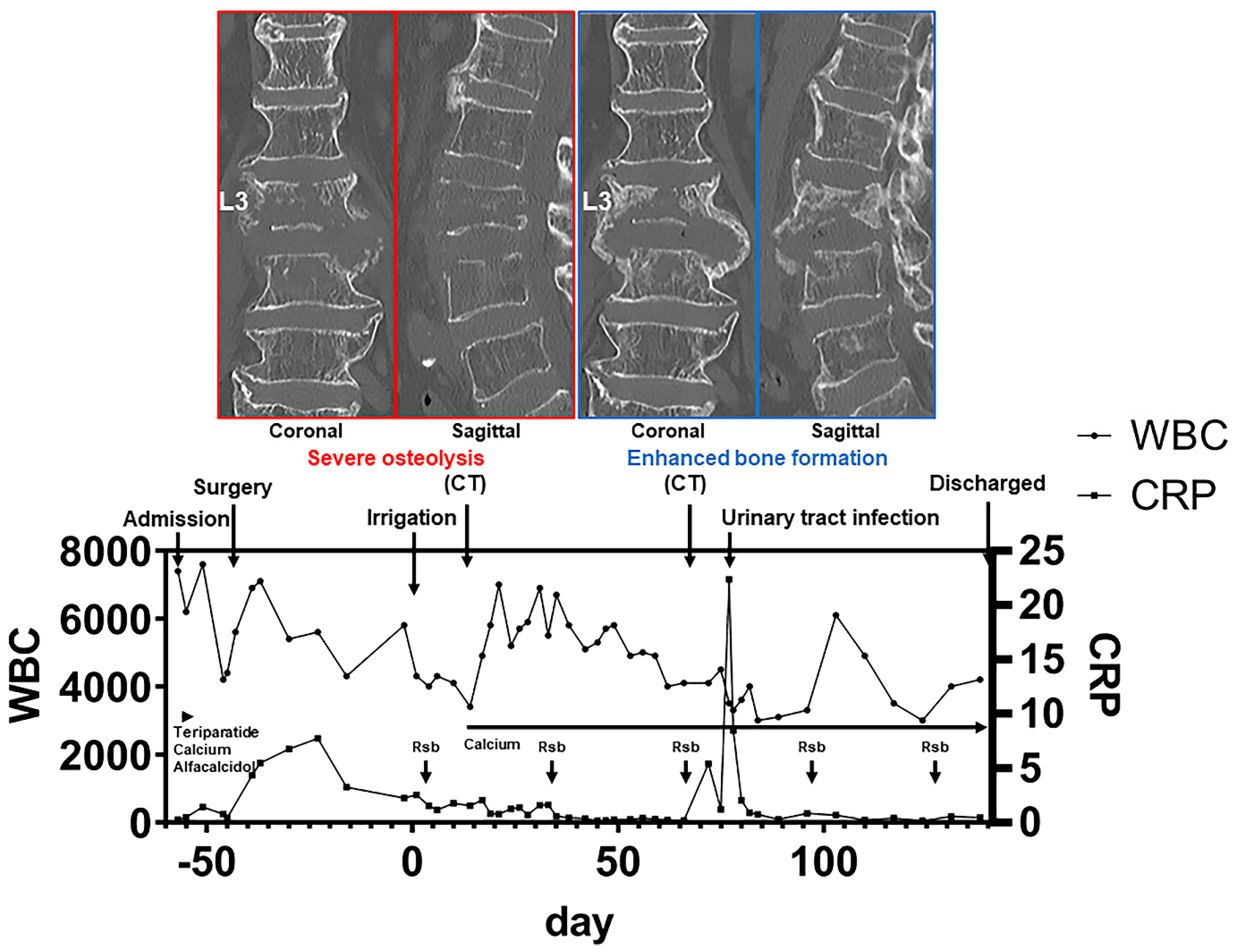
4.2. Anabolic Drug: Teriparatide
4.3. Antiresorptive Drug: Denosumab
4.4. Antiresorptive Drug: BPs
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Pola, E.; Taccari, F.; Autore, G.; Giovannenze, F.; Pambianco, V.; Cauda, R.; Maccauro, G.; Fantoni, M. Multidisciplinary management of pyogenic spondylodiscitis: Epidemiological and clinical features, prognostic factors and long-term outcomes in 207 patients. Eur. Spine J. 2018, 27, 229–236. [Google Scholar] [CrossRef]
- Kehrer, M.; Pedersen, C.; Jensen, T.G.; Lassen, A.T. Increasing incidence of pyogenic spondylodiscitis: A 14-year population-based study. J. Infect. 2014, 68, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Chikuda, H.; Yasunaga, H.; Horiguchi, H.; Fushimi, K.; Saita, K. Incidence and risk factors for mortality of vertebral osteomyelitis: A retrospective analysis using the Japanese diagnosis procedure combination database. BMJ Open 2013, 3. [Google Scholar] [CrossRef]
- Shousha, M.; Boehm, H. Surgical treatment of cervical spondylodiscitis: A review of 30 consecutive patients. Spine 2012, 37, E30–E36. [Google Scholar] [CrossRef]
- Kim, J.; Jang, S.B.; Kim, S.W.; Oh, J.K.; Kim, T.H. Clinical effect of early bisphosphonate treatment for pyogenic vertebral osteomyelitis with osteoporosis: An analysis by the Cox proportional hazard model. Spine J. 2019, 19, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.W.; Hyun, S.J.; Han, S.H.; Kim, K.J.; Jahng, T.A. Pyogenic Vertebral osteomyelitis: Clinical features, diagnosis, and treatment. Korean J. Spine 2017, 14, 27–34. [Google Scholar] [CrossRef]
- Mylona, E.; Samarkos, M.; Kakalou, E.; Fanourgiakis, P.; Skoutelis, A. Pyogenic vertebral osteomyelitis: A systematic review of clinical characteristics. Semin. Arthritis Rheum. 2009, 39, 10–17. [Google Scholar] [CrossRef]
- Sato, K.; Yamada, K.; Yokosuka, K.; Yoshida, T.; Goto, M.; Matsubara, T.; Iwahashi, S.; Shimazaki, T.; Nagata, K.; Shiba, N. Pyogenic spondylitis: Clinical features, diagnosis and treatment. Kurume Med. J. 2019, 65, 83–89. [Google Scholar] [CrossRef]
- Lam, K.S.; Webb, J.K. Discitis. Hosp. Med. 2004, 65, 280–286. [Google Scholar] [CrossRef]
- Yang, J.; Tang, R.; Yi, J.; Chen, Y.; Li, X.; Yu, T.; Fei, J. Diallyl disulfide alleviates inflammatory osteolysis by suppressing osteoclastogenesis via NF-κB-NFATc1 signal pathway. FASEB J. 2019, 33, 7261–7273. [Google Scholar] [CrossRef]
- Komine, M.; Kukita, A.; Kukita, T.; Ogata, Y.; Hotokebuchi, T.; Kohashi, O. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone 2001, 28, 474–483. [Google Scholar] [CrossRef]
- Kitaura, H.; Kimura, K.; Ishida, M.; Kohara, H.; Yoshimatsu, M.; Takano-Yamamoto, T. Immunological reaction in TNF-α-mediated osteoclast formation and bone resorption in vitro and in vivo. Clin. Dev. Immunol. 2013, 2013, 181849. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cipriani, P.; Carubbi, F.; Liakouli, V.; Zazzeroni, F.; di Benedetto, P.; Berardicurti, O.; Alesse, E.; Giacomelli, R. The role of IL-1β in the bone loss during rheumatic diseases. Mediators Inflamm. 2015, 2015, 782382. [Google Scholar] [CrossRef]
- Jules, J.; Zhang, P.; Ashley, J.W.; Wei, S.; Shi, Z.; Liu, J.; Michalek, S.M.; Feng, X. Molecular basis of requirement of receptor activator of nuclear factor κB signaling for interleukin 1-mediated osteoclastogenesis. J. Biol. Chem. 2012, 287, 15728–15738. [Google Scholar] [CrossRef]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef]
- Quan, G.H.; Wang, H.; Cao, J.; Zhang, Y.; Wu, D.; Peng, Q.; Liu, N.; Sun, W.C. Calycosin suppresses RANKL-mediated osteoclastogenesis through inhibition of MAPKs and NF-κB. Int. J. Mol. Sci. 2015, 16, 29496–29507. [Google Scholar] [CrossRef]
- Park, H.; Noh, A.L.; Kang, J.H.; Sim, J.S.; Lee, D.S.; Yim, M. Peroxiredoxin II negatively regulates lipopolysaccharide-induced osteoclast formation and bone loss via JNK and STAT3. Antioxid. Redox. Signal. 2015, 22, 63–77. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef]
- Guo, C.; Yuan, L.; Wang, J.G.; Wang, F.; Yang, X.K.; Zhang, F.H.; Song, J.L.; Ma, X.Y.; Cheng, Q.; Song, G.H. Lipopolysaccharide (LPS) induces the apoptosis and inhibits osteoblast differentiation through JNK pathway in MC3T3-E1 cells. Inflammation 2014, 37, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Bandow, K.; Maeda, A.; Kakimoto, K.; Kusuyama, J.; Shamoto, M.; Ohnishi, T.; Matsuguchi, T. Molecular mechanisms of the inhibitory effect of lipopolysaccharide (LPS) on osteoblast differentiation. Biochem. Biophys. Res. Commun. 2010, 402, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, D.; Sun, X.; Wang, Y.; Xiao, Q.; Chen, A. Icariine restores LPS-induced bone loss by downregulating miR-34c level. Inflammation 2016, 39, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zheng, H.; Greasley, A.; Ling, F.; Zhou, Q.; Wang, B.; Ni, T.; Topiwala, I.; Zhu, C.; Mele, T.; et al. The role of miR-711 in cardiac cells in response to oxidative stress and its biogenesis: A study on H9C2 cells. Cell. Mol. Biol. Lett. 2020, 25, 26. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Morrison, A.; Hanley, D.A.; Felsenberg, D.; McCauley, L.K.; O’Ryan, F.; Reid, I.R.; Ruggiero, S.L.; Taguchi, A.; Tetradis, S.; et al. Diagnosis and management of osteonecrosis of the jaw: A systematic review and international consensus. J. Bone Miner. Res. 2015, 30, 3–23. [Google Scholar] [CrossRef]
- Hoefert, S.; Schmitz, I.; Weichert, F.; Gaspar, M.; Eufinger, H. Macrophages and bisphosphonate-related osteonecrosis of the jaw (BRONJ): Evidence of local immunosuppression of macrophages in contrast to other infectious jaw diseases. Clin. Oral Investig. 2015, 19, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Abu-Id, M.H.; Warnke, P.H.; Gottschalk, J.; Springer, I.; Wiltfang, J.; Acil, Y.; Russo, P.A.; Kreusch, T. “Bis-phossy jaws”—High and low risk factors for bisphosphonate-induced osteonecrosis of the jaw. J. Craniomaxillofac. Surg. 2008, 36, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Cristofaro, M.G.; Amantea, M.; Russo, E.; de Fazio, S.; Zuccalà, V.; Conforti, F.; Amorosi, A.; Donato, G.; Tripepi, S.; et al. Jaw osteonecrosis in patients treated with bisphosphonates: An ultrastructural study. Ultrastruct. Pathol. 2010, 34, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Sedghizadeh, P.P.; Kumar, S.K.; Gorur, A.; Schaudinn, C.; Shuler, C.F.; Costerton, J.W. Microbial biofilms in osteomyelitis of the jaw and osteonecrosis of the jaw secondary to bisphosphonate therapy. J. Am. Dent. Assoc. 2009, 140, 1259–1265. [Google Scholar] [CrossRef]
- Hansen, T.; Kunkel, M.; Springer, E.; Walter, C.; Weber, A.; Siegel, E.; Kirkpatrick, C.J. Actinomycosis of the jaws—Histopathological study of 45 patients shows significant involvement in bisphosphonate-associated osteonecrosis and infected osteoradionecrosis. Virchows Arch. 2007, 451, 1009–1017. [Google Scholar] [CrossRef]
- Reid, I.R.; Cornish, J. Epidemiology and pathogenesis of osteonecrosis of the jaw. Nat. Rev. Rheumatol. 2011, 8, 90–96. [Google Scholar] [CrossRef]
- Yoshiga, D.; Yamashita, Y.; Nakamichi, I.; Tanaka, T.; Yamauchi, K.; Yamamoto, N.; Nogami, S.; Kaneuji, T.; Mitsugi, S.; Sakurai, T.; et al. Weekly teriparatide injections successfully treated advanced bisphosphonate-related osteonecrosis of the jaws. Osteoporos. Int. 2013, 24, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, M.; Chen, P.; Miyauchi, A.; Sowa, H.; Krege, J.H. PINP as an aid for monitoring patients treated with teriparatide. Bone 2011, 48, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Giustina, A.; Bilezikian, J.P. Mechanisms of anabolic therapies for osteoporosis. N. Engl. J. Med. 2007, 357, 905–916. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.H.; Jung, H.D.; Jung, Y.S. Treatment of medication-related osteonecrosis of the jaw around the dental implant with a once-weekly teriparatide: A case report and literature review. J. Oral Implantol. 2019, 45, 403–407. [Google Scholar] [CrossRef]
- Chouinard, L.; Felx, M.; Mellal, N.; Varela, A.; Mann, P.; Jolette, J.; Samadfam, R.; Smith, S.Y.; Locher, K.; Buntich, S.; et al. Carcinogenicity risk assessment of romosozumab: A review of scientific weight-of-evidence and findings in a rat lifetime pharmacology study. Regul. Toxicol. Pharmacol. 2016, 81, 212–222. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Khan, A.; Lane, N.E.; Lippuner, K.; Matsumoto, T.; Milmont, C.E.; Libanati, C.; Grauer, A. FRAME study: The foundation effect of building bone with 1 year of romosozumab leads to continued lower fracture risk after transition to denosumab. J. Bone Miner. Res. 2018, 33, 1219–1226. [Google Scholar] [CrossRef]
- Graeff, C.; Campbell, G.M.; Peña, J.; Borggrefe, J.; Padhi, D.; Kaufman, A.; Chang, S.; Libanati, C.; Glüer, C.C. Administration of romosozumab improves vertebral trabecular and cortical bone as assessed with quantitative computed tomography and finite element analysis. Bone 2015, 81, 364–369. [Google Scholar] [CrossRef]
- Lv, F.; Cai, X.; Yang, W.; Gao, L.; Chen, L.; Wu, J.; Ji, L. Denosumab or romosozumab therapy and risk of cardiovascular events in patients with primary osteoporosis: Systematic review and meta-analysis. Bone 2020, 130, 115121. [Google Scholar] [CrossRef]
- Matheny, J.B.; Torres, A.M.; Ominsky, M.S.; Hernandez, C.J. Romosozumab treatment converts trabecular rods into trabecular plates in male cynomolgus monkeys. Calcif. Tissue Int. 2017, 101, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Raterman, H.G.; Lems, W.F. Pharmacological management of osteoporosis in rheumatoid arthritis patients: A review of the literature and practical guide. Drugs Aging 2019, 36, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Gül, G.; Sendur, M.A.; Aksoy, S.; Sever, A.R.; Altundag, K. A comprehensive review of denosumab for bone metastasis in patients with solid tumors. Curr. Med. Res. Opin. 2016, 32, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damião, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Mattei, T.A.; Ramos, E.; Rehman, A.A.; Shaw, A.; Patel, S.R.; Mendel, E. Sustained long-term complete regression of a giant cell tumor of the spine after treatment with denosumab. Spine J. 2014, 14, e15–e21. [Google Scholar] [CrossRef]
- Chen, Y.; Alman, B.A. Wnt pathway, an essential role in bone regeneration. J. Cell. Biochem. 2009, 106, 353–362. [Google Scholar] [CrossRef]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef]
- Boyce, R.W.; Niu, Q.T.; Ominsky, M.S. Kinetic reconstruction reveals time-dependent effects of romosozumab on bone formation and osteoblast function in vertebral cancellous and cortical bone in cynomolgus monkeys. Bone 2017, 101, 77–87. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther. Adv. Musculoskelet. Dis. 2014, 6, 48–57. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The regulation of bone metabolism and disorders by wnt signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Karantalis, V.; Hare, J.M. Use of mesenchymal stem cells for therapy of cardiac disease. Circ. Res. 2015, 116, 1413–1430. [Google Scholar] [CrossRef]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef]
- Zhang, R.; Oyajobi, B.O.; Harris, S.E.; Chen, D.; Tsao, C.; Deng, H.W.; Zhao, M. Wnt/β-catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone 2013, 52, 145–156. [Google Scholar] [CrossRef]
- Weivoda, M.M.; Ruan, M.; Hachfeld, C.M.; Pederson, L.; Howe, A.; Davey, R.A.; Zajac, J.D.; Kobayashi, Y.; Williams, B.O.; Westendorf, J.J.; et al. Wnt signaling inhibits osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways. J. Bone Miner. Res. 2016, 31, 65–75. [Google Scholar] [CrossRef]
- Fujita, K.; Janz, S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol. Cancer 2007, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhou, H.; Pan, B.; Lu, L.; Liu, J.; Kang, Y.; Yao, X.; Feng, S. Effectiveness of teriparatide on fracture healing: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0168691. [Google Scholar] [CrossRef] [PubMed]
- Dempster, D.W.; Cosman, F.; Parisien, M.; Shen, V.; Lindsay, R. Anabolic actions of parathyroid hormone on bone. Endocr. Rev. 1993, 14, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Ali, A.A.; Gubrij, I.; Plotkin, L.I.; Fu, Q.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: A novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 2005, 146, 4577–4583. [Google Scholar] [CrossRef] [PubMed]
- Mouyis, M.; Fitz-Clarence, H.; Manson, J.; Ciurtin, C. Teriparatide: An unexpected adjunct for the treatment of a long-standing infected elbow prosthesis prevented arm amputation. Clin. Rheumatol. 2015, 34, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Kaneshiro, S.; Takami, K.; Owaki, H.; Fuji, T. Bone stock reconstruction for huge bone loss using allograft-bones, bone marrow, and teriparatide in an infected total knee arthroplasty. J. Clin. Orthop. Trauma 2019, 10, 329–333. [Google Scholar] [CrossRef]
- Rollo, G.; Luceri, F.; Falzarano, G.; Salomone, C.; Bonura, E.M.; Popkov, D.; Ronga, M.; Pica, G.; Bisaccia, M.; Russi, V.; et al. Effectiveness of teriparatide combined with the Ilizarov technique in septic tibial non-union. Med. Glas. 2021, 18. [Google Scholar] [CrossRef]
- Shinohara, A.; Ueno, Y.; Marumo, K. Weekly teriparatide therapy rapidly accelerates bone healing in pyogenic spondylitis with severe osteoporosis. Asian Spine J. 2014, 8, 498–501. [Google Scholar] [CrossRef]
- Terashima, A.; Okamoto, K.; Nakashima, T.; Akira, S.; Ikuta, K.; Takayanagi, H. Sepsis-induced osteoblast ablation causes immunodeficiency. Immunity 2016, 44, 1434–1443. [Google Scholar] [CrossRef]
- Unsinger, J.; McGlynn, M.; Kasten, K.R.; Hoekzema, A.S.; Watanabe, E.; Muenzer, J.T.; McDonough, J.S.; Tschoep, J.; Ferguson, T.A.; McDunn, J.E.; et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J. Immunol. 2010, 184, 3768–3779. [Google Scholar] [CrossRef]
- Venet, F.; Foray, A.P.; Villars-Méchin, A.; Malcus, C.; Poitevin-Later, F.; Lepape, A.; Monneret, G. IL-7 restores lymphocyte functions in septic patients. J. Immunol. 2012, 189, 5073–5081. [Google Scholar] [CrossRef]
- Ferrari-Lacraz, S.; Ferrari, S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2011, 22, 435–446. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; de Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Yamaguchi, N.; Abe, M.; Hirose, S.; Shirai, T.; Okumura, K.; Yagita, H. Suppression of antibody production by TNF-related apoptosis-inducing ligand (TRAIL). Cell. Immunol. 2002, 219, 82–91. [Google Scholar] [CrossRef]
- Kim, D.; Mebius, R.E.; MacMicking, J.D.; Jung, S.; Cupedo, T.; Castellanos, Y.; Rho, J.; Wong, B.R.; Josien, R.; Kim, N.; et al. Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J. Exp. Med. 2000, 192, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.J.; Tallquist, M.D.; Aicher, A.; Rafferty, K.L.; Marshall, A.J.; Moon, J.J.; Ewings, M.E.; Mohaupt, M.; Herring, S.W.; Clark, E.A. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J. Immunol. 2001, 166, 1482–1491. [Google Scholar] [CrossRef]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Anastasilakis, A.D.; Toulis, K.A.; Goulis, D.G.; Polyzos, S.A.; Delaroudis, S.; Giomisi, A.; Terpos, E. Efficacy and safety of denosumab in postmenopausal women with osteopenia or osteoporosis: A systematic review and a meta-analysis. Horm. Metab. Res. 2009, 41, 721–729. [Google Scholar] [CrossRef]
- Ellis, G.K.; Bone, H.G.; Chlebowski, R.; Paul, D.; Spadafora, S.; Smith, J.; Fan, M.; Jun, S. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J. Clin. Oncol. 2008, 26, 4875–4882. [Google Scholar] [CrossRef]
- Watts, N.B.; Roux, C.; Modlin, J.F.; Brown, J.P.; Daniels, A.; Jackson, S.; Smith, S.; Zack, D.J.; Zhou, L.; Grauer, A.; et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: Coincidence or causal association? Osteoporos. Int. 2012, 23, 327–337. [Google Scholar] [CrossRef]
- Toulis, K.A.; Anastasilakis, A.D. Increased risk of serious infections in women with osteopenia or osteoporosis treated with denosumab. Osteoporos. Int. 2010, 21, 1963–1964. [Google Scholar] [CrossRef]
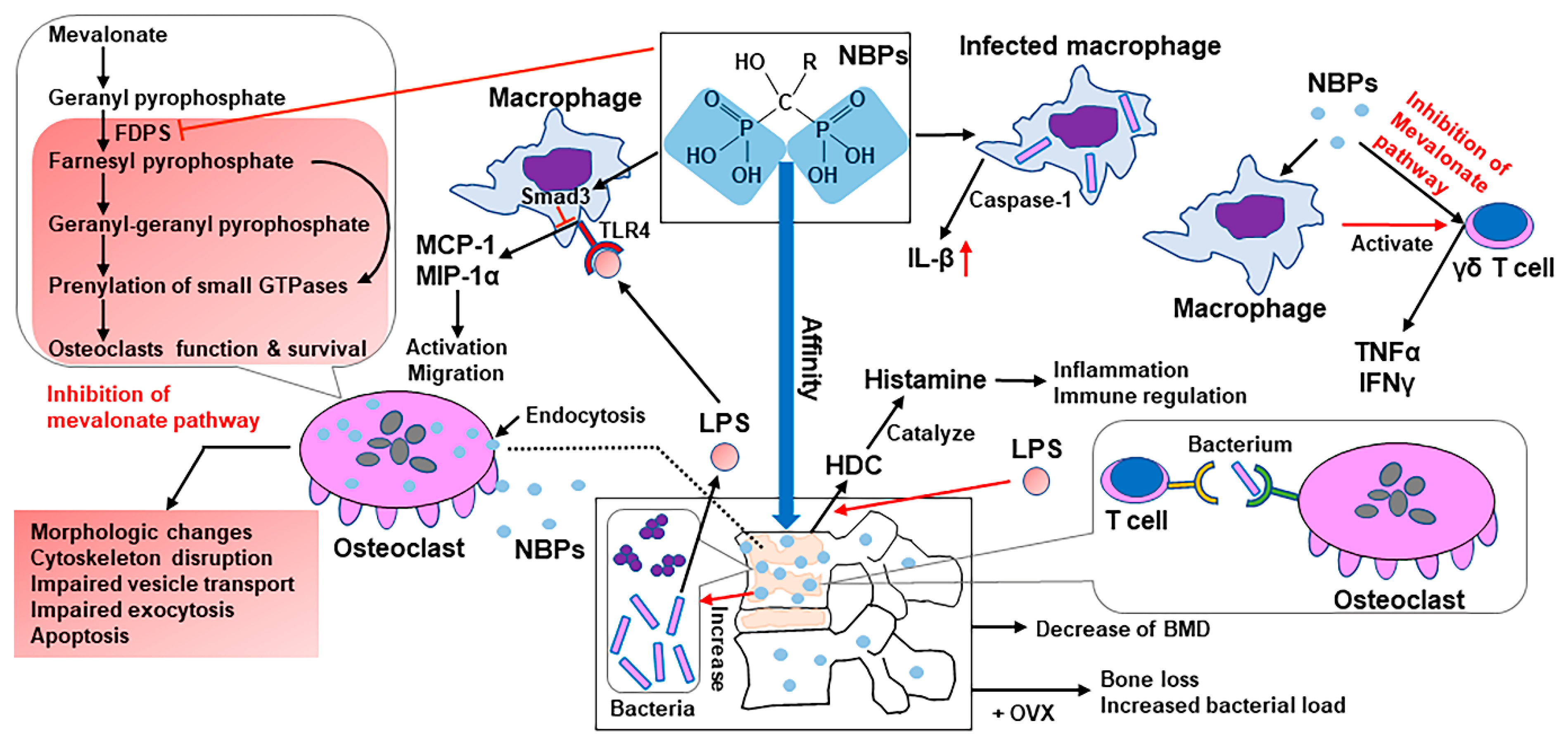
- Gong, L.; Altman, R.B.; Klein, T.E. Bisphosphonates pathway. Pharmacogenet. Genom. 2011, 21, 50–53. [Google Scholar] [CrossRef]
- Shikama, Y.; Nagai, Y.; Okada, S.; Oizumi, T.; Shimauchi, H.; Sugawara, S.; Endo, Y. Pro-IL-1β accumulation in macrophages by alendronate and its prevention by clodronate. Toxicol. Lett. 2010, 199, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.C.; Mönkkönen, J.; Blackburn, G.M.; Russell, R.G.; Rogers, M.J. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5′-(beta, gamma-dichloromethylene) triphosphate, by mammalian cells in vitro. J. Bone Miner. Res. 1997, 12, 1358–1367. [Google Scholar] [CrossRef]
- Lehenkari, P.P.; Kellinsalmi, M.; Näpänkangas, J.P.; Ylitalo, K.V.; Mönkkönen, J.; Rogers, M.J.; Azhayev, A.; Väänänen, H.K.; Hassinen, I.E. Further insight into mechanism of action of clodronate: Inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol. Pharmacol. 2002, 61, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Schindeler, A.; Little, D.G. Bisphosphonate action: Revelations and deceptions from in vitro studies. J. Pharm. Sci. 2007, 96, 1872–1878. [Google Scholar] [CrossRef]
- Reszka, A.A.; Rodan, G.A. Bisphosphonate mechanism of action. Curr. Rheumatol. Rep. 2003, 5, 65–74. [Google Scholar] [CrossRef]
- Rodan, G.A.; Reszka, A.A. Bisphosphonate mechanism of action. Curr. Mol. Med. 2002, 2, 571–577. [Google Scholar] [CrossRef]
- Luckman, S.P.; Hughes, D.E.; Coxon, F.P.; Graham, R.; Russell, G.; Rogers, M.J. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J. Bone Miner. Res. 1998, 13, 581–589. [Google Scholar] [CrossRef]
- Dunford, J.E.; Thompson, K.; Coxon, F.P.; Luckman, S.P.; Hahn, F.M.; Poulter, C.D.; Ebetino, F.H.; Rogers, M.J. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 2001, 296, 235–242. [Google Scholar]
- Luckman, S.P.; Coxon, F.P.; Ebetino, F.H.; Russell, R.G.; Rogers, M.J. Heterocycle-containing bisphosphonates cause apoptosis and inhibit bone resorption by preventing protein prenylation: Evidence from structure-activity relationships in J774 macrophages. J. Bone Miner. Res. 1998, 13, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- Coxon, F.P.; Rogers, M.J. The role of prenylated small GTP-binding proteins in the regulation of osteoclast function. Calcif. Tissue Int. 2003, 72, 80–84. [Google Scholar] [CrossRef]
- Coxon, F.P.; Helfrich, M.H.; Van’t Hof, R.; Sebti, S.; Ralston, S.H.; Hamilton, A.; Rogers, M.J. Protein geranylgeranylation is required for osteoclast formation, function, and survival: Inhibition by bisphosphonates and GGTI-298. J. Bone Miner. Res. 2000, 15, 1467–1476. [Google Scholar] [CrossRef]
- Deng, X.; Tamai, R.; Endo, Y.; Kiyoura, Y. Alendronate augments interleukin-1beta release from macrophages infected with periodontal pathogenic bacteria through activation of caspase-1. Toxicol. Appl. Pharmacol. 2009, 235, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Yu, Z.; Funayama, H.; Shoji, N.; Sasano, T.; Iwakura, Y.; Sugawara, S.; Endo, Y. Mutual augmentation of the induction of the histamine-forming enzyme, histidine decarboxylase, between alendronate and immuno-stimulants (IL-1, TNF, and LPS), and its prevention by clodronate. Toxicol. Appl. Pharmacol. 2006, 213, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Rolli-Derkinderen, M.; Arock, M.; Dy, M. Trends in histamine research: New functions during immune responses and hematopoiesis. Trends Immunol. 2002, 23, 255–263. [Google Scholar] [CrossRef]
- Miyagawa, F.; Tanaka, Y.; Yamashita, S.; Minato, N. Essential requirement of antigen presentation by monocyte lineage cells for the activation of primary human gamma delta T cells by aminobisphosphonate antigen. J. Immunol. 2001, 166, 5508–5514. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Weissinger, F.; Tony, H.P.; Wilhelm, M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Thompson, K.; Rogers, M.J. Statins prevent bisphosphonate-induced gamma, delta-T-cell proliferation and activation in vitro. J. Bone Miner. Res. 2004, 19, 278–288. [Google Scholar] [CrossRef]
- Masuda, T.; Deng, X.; Tamai, R. Mouse macrophages primed with alendronate down-regulate monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1alpha (MIP-1alpha) production in response to Toll-like receptor (TLR) 2 and TLR4 agonist via Smad3 activation. Int. Immunopharmacol. 2009, 9, 1115–1121. [Google Scholar] [CrossRef]
- Li, X.; Qin, L.; Bergenstock, M.; Bevelock, L.M.; Novack, D.V.; Partridge, N.C. Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J. Biol. Chem. 2007, 282, 33098–33106. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Cruz, J.C.; Craig, F.; Chung, H.; Devlin, R.D.; Roodman, G.D.; Alsina, M. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood 2000, 96, 671–675. [Google Scholar] [CrossRef]
- Ganguli, A.; Steward, C.; Butler, S.L.; Philips, G.J.; Meikle, S.T.; Lloyd, A.W.; Grant, M.H. Bacterial adhesion to bisphosphonate coated hydroxyapatite. J. Mater. Sci. Mater. Med. 2005, 16, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Freitag, L.; Styger, U.; Camenisch, K.; Zeiter, S.; Arens, D.; Richards, R.G.; Moriarty, T.F.; Stadelmann, V.A. Impact of low bone mass and antiresorptive therapy on antibiotic efficacy in a rat model of orthopedic device-related infection. J. Orthop. Res. 2020. [Google Scholar] [CrossRef]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, Q. Cross talk between the bone and immune systems: Osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Thillemann, T.M.; Pedersen, A.B.; Mehnert, F.; Johnsen, S.P.; Søballe, K. Postoperative use of bisphosphonates and risk of revision after primary total hip arthroplasty: A nationwide population-based study. Bone 2010, 46, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Gromov, K.; Proulx, S.T.; Xie, C.; Li, J.; Crane, D.P.; Søballe, K.; O’Keefe, R.J.; Awad, H.A.; Xing, L.; et al. Effects of antiresorptive agents on osteomyelitis: Novel insights into the pathogenesis of osteonecrosis of the jaw. Ann. N. Y. Acad. Sci. 2010, 1192, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Sawa, N.; Sawa, Y.; Miyazono, S. Effect of bisphosphonates on healing of tooth extraction wounds in infectious osteomyelitis of the jaw. Bone 2021, 143, 115611. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drugs | Advantages | Disadvantages |
|---|---|---|
| Romosozumab * |
|
|
| Teriparatide |
|
|
| Denosumab |
|
|
| NBPs |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohnishi, T.; Ogawa, Y.; Suda, K.; Komatsu, M.; Harmon, S.M.; Asukai, M.; Takahata, M.; Iwasaki, N.; Minami, A. Molecular Targeted Therapy for the Bone Loss Secondary to Pyogenic Spondylodiscitis Using Medications for Osteoporosis: A Literature Review. Int. J. Mol. Sci. 2021, 22, 4453. https://doi.org/10.3390/ijms22094453
Ohnishi T, Ogawa Y, Suda K, Komatsu M, Harmon SM, Asukai M, Takahata M, Iwasaki N, Minami A. Molecular Targeted Therapy for the Bone Loss Secondary to Pyogenic Spondylodiscitis Using Medications for Osteoporosis: A Literature Review. International Journal of Molecular Sciences. 2021; 22(9):4453. https://doi.org/10.3390/ijms22094453
Chicago/Turabian StyleOhnishi, Takashi, Yuki Ogawa, Kota Suda, Miki Komatsu, Satoko Matsumoto Harmon, Mitsuru Asukai, Masahiko Takahata, Norimasa Iwasaki, and Akio Minami. 2021. "Molecular Targeted Therapy for the Bone Loss Secondary to Pyogenic Spondylodiscitis Using Medications for Osteoporosis: A Literature Review" International Journal of Molecular Sciences 22, no. 9: 4453. https://doi.org/10.3390/ijms22094453
APA StyleOhnishi, T., Ogawa, Y., Suda, K., Komatsu, M., Harmon, S. M., Asukai, M., Takahata, M., Iwasaki, N., & Minami, A. (2021). Molecular Targeted Therapy for the Bone Loss Secondary to Pyogenic Spondylodiscitis Using Medications for Osteoporosis: A Literature Review. International Journal of Molecular Sciences, 22(9), 4453. https://doi.org/10.3390/ijms22094453