
Xanthohumol-Induced Rat Glioma C6 Cells Death by Triggering Mitochondrial Stress
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
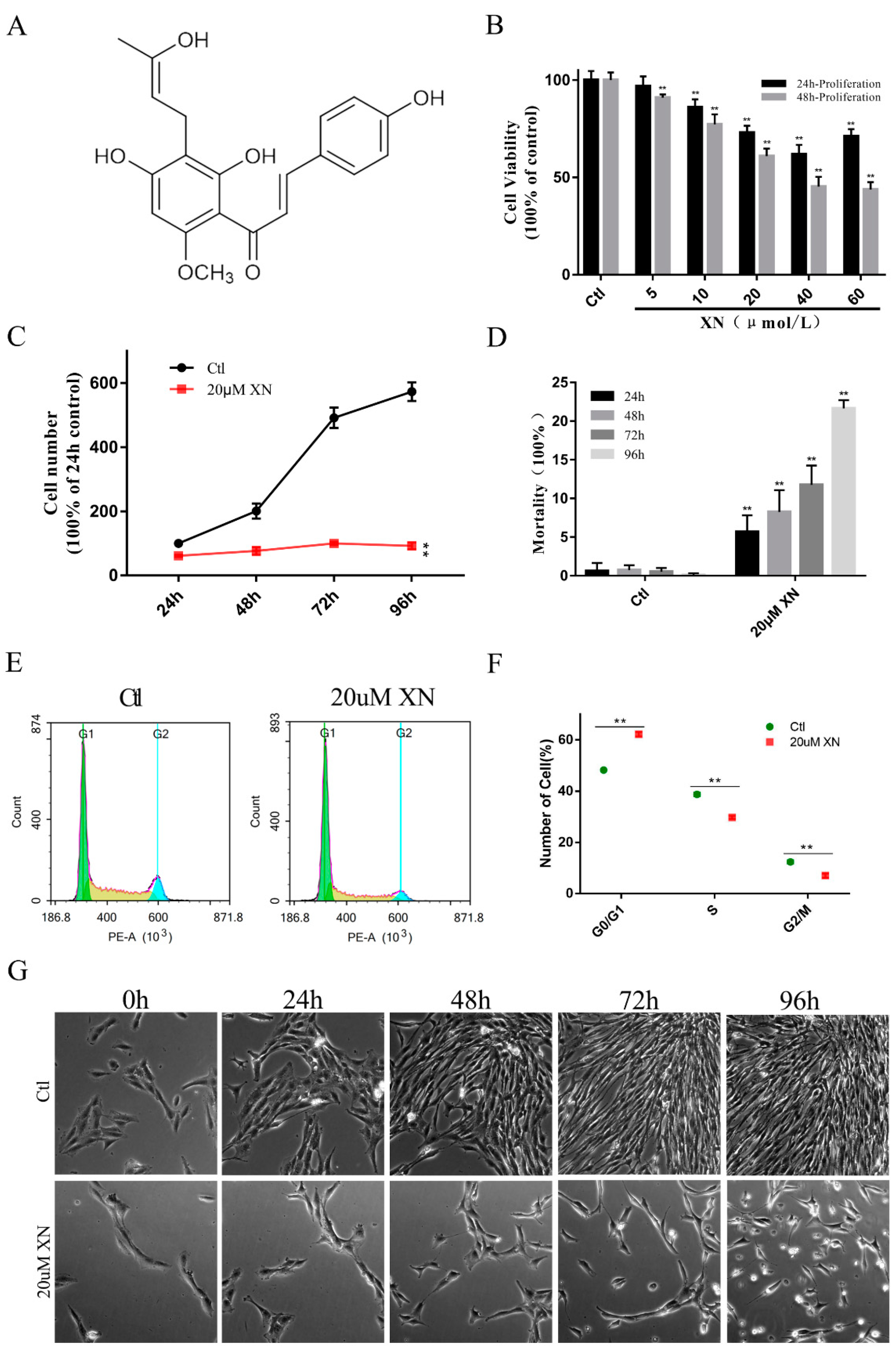
2.1. XN Affected C6 Cell Proliferation in Concentration and Time-Dependent Way
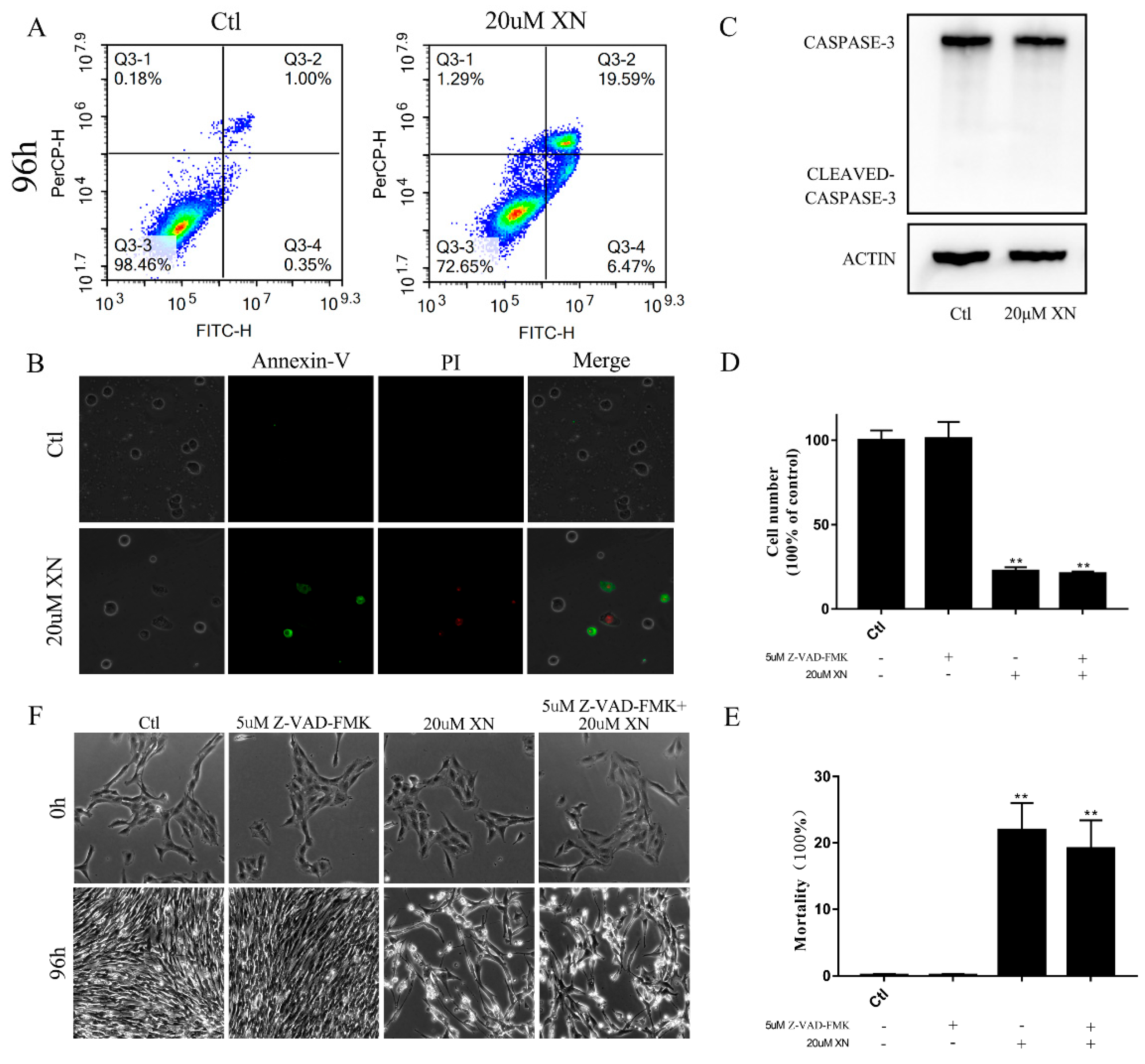
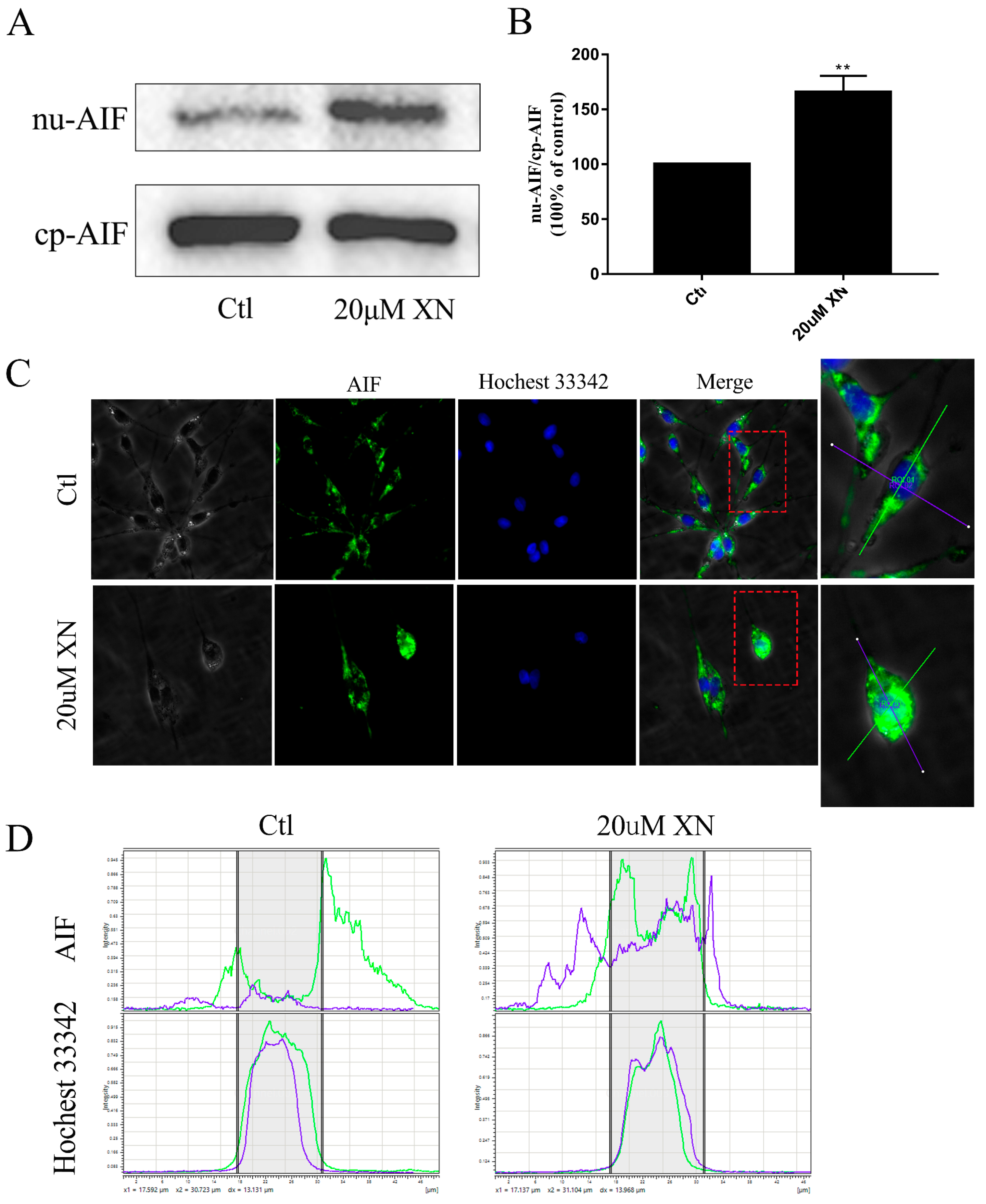
2.2. XN Treatment-Induced AIF-Dependent Apoptosis in C6 Cells
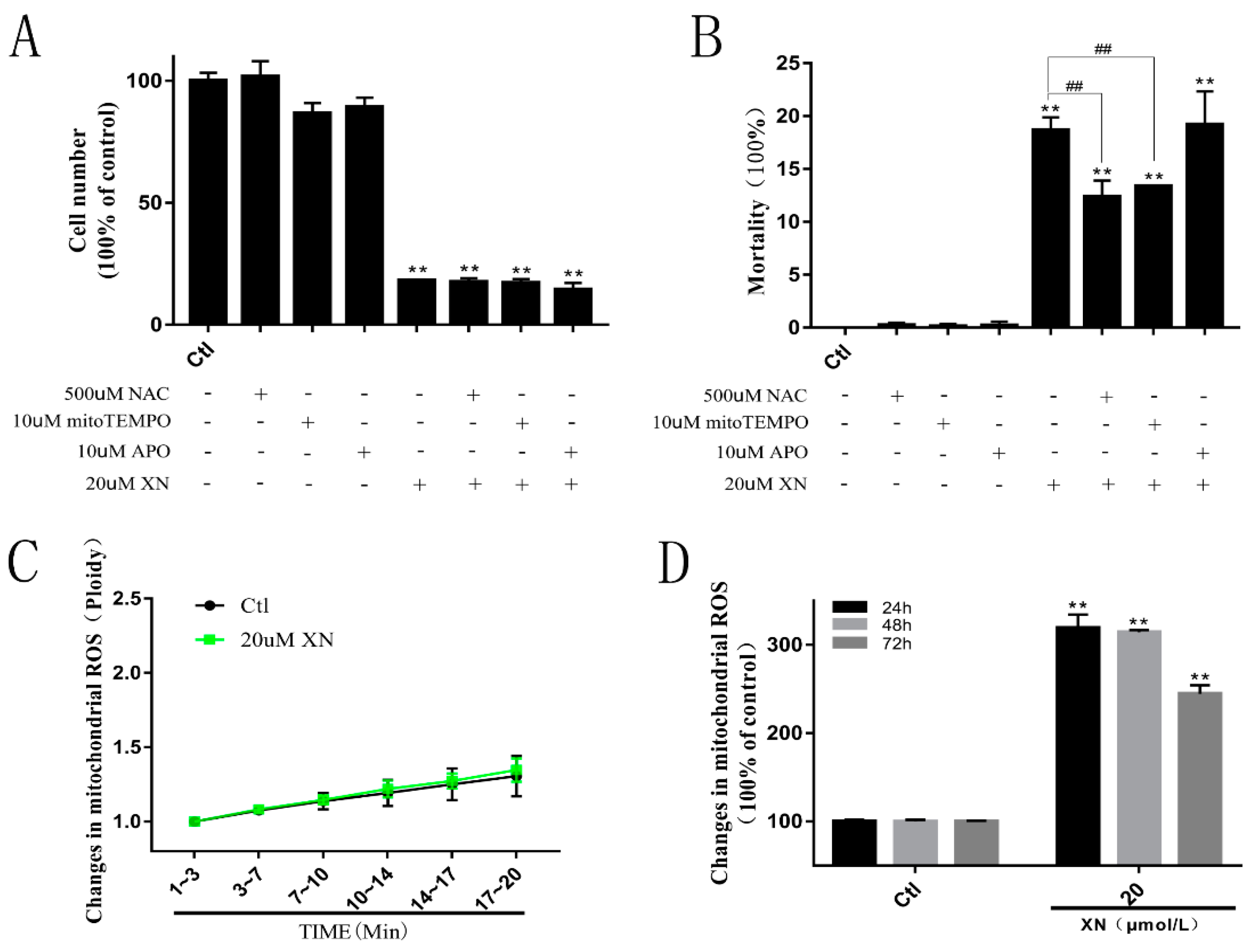
2.3. XN Increased the Amount of Mitochondrial ROS in the Process of Cell Death
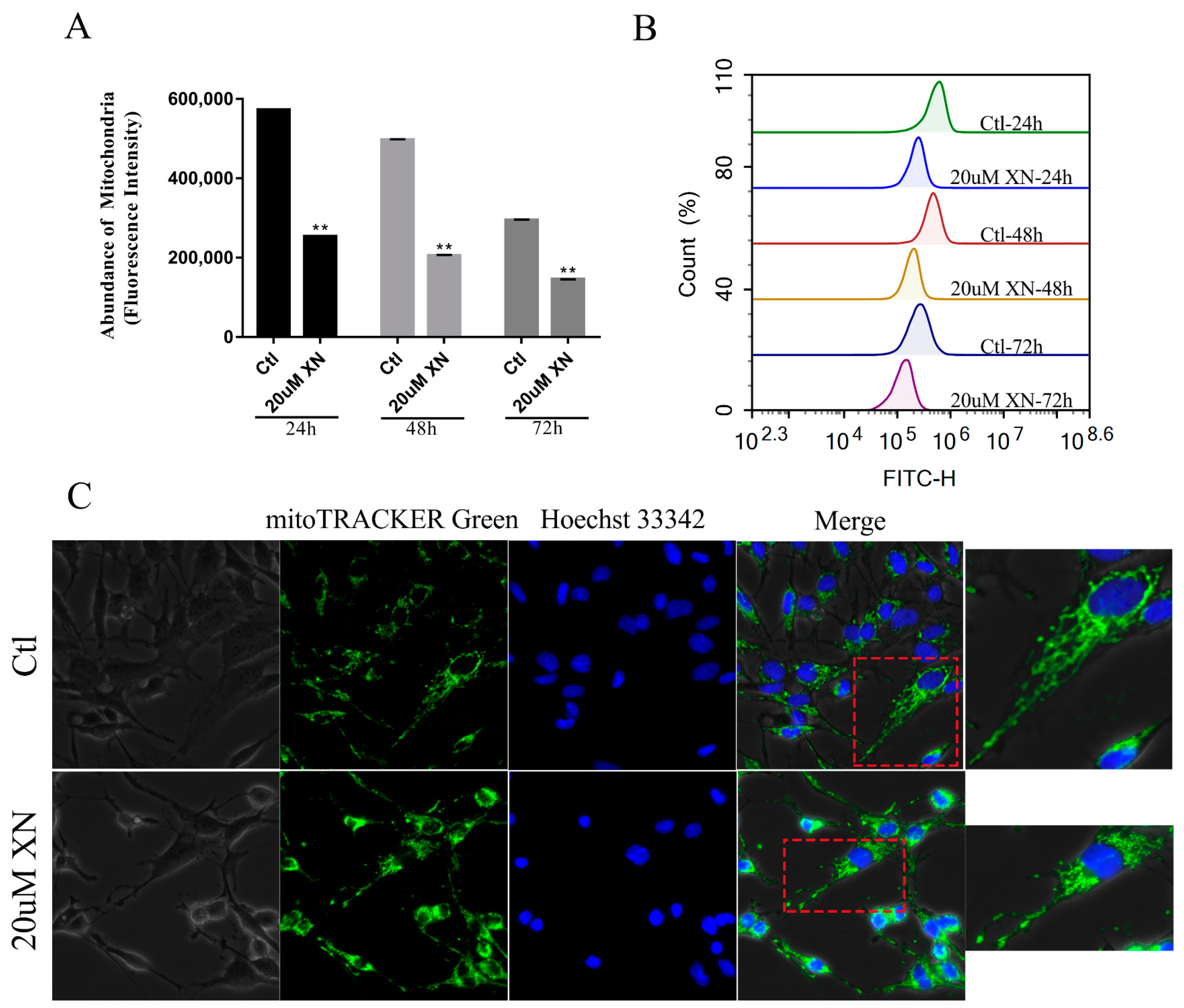
2.4. XN Affected Mitochondrial Metabolism in C6 Cells
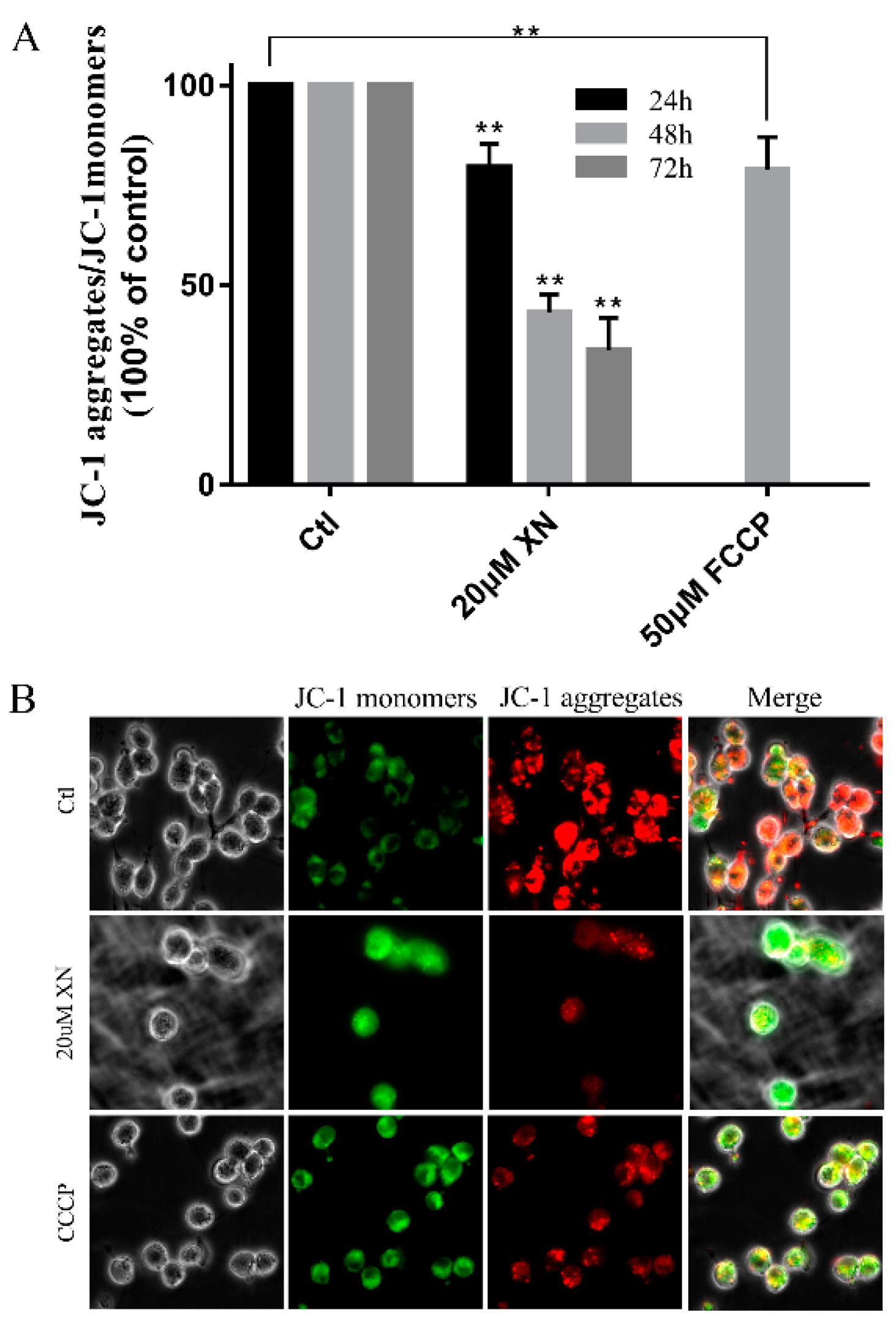
2.5. XN Reduced Mitochondrial Membrane Potential (MMP) in C6 Cell
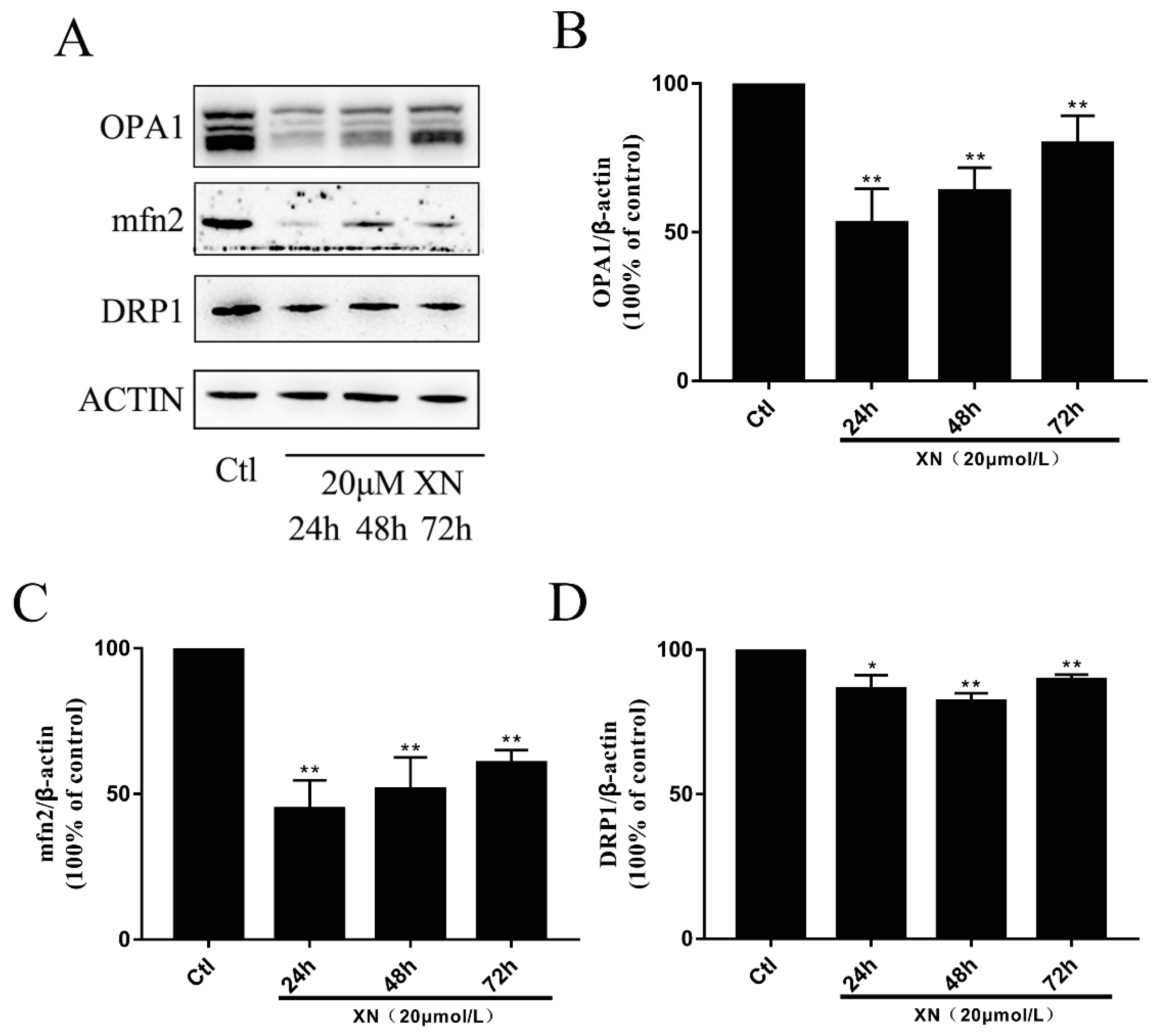
2.6. XN Caused Fusion and Fission Imbalance by Reducing the Mitochondrial Fusion Function in C6 Cells
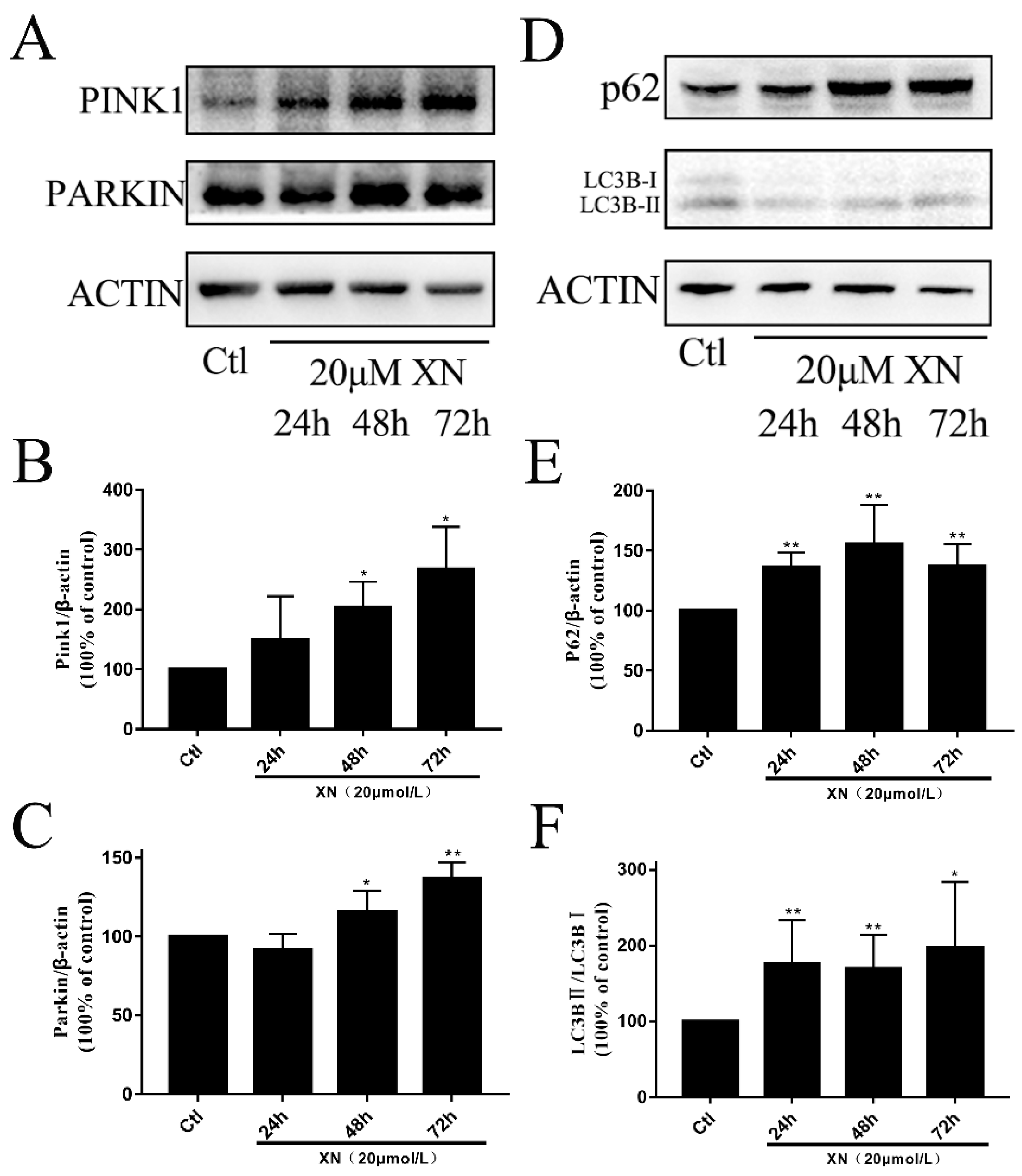
2.7. XN-Induced Mitophagy in C6 Cell
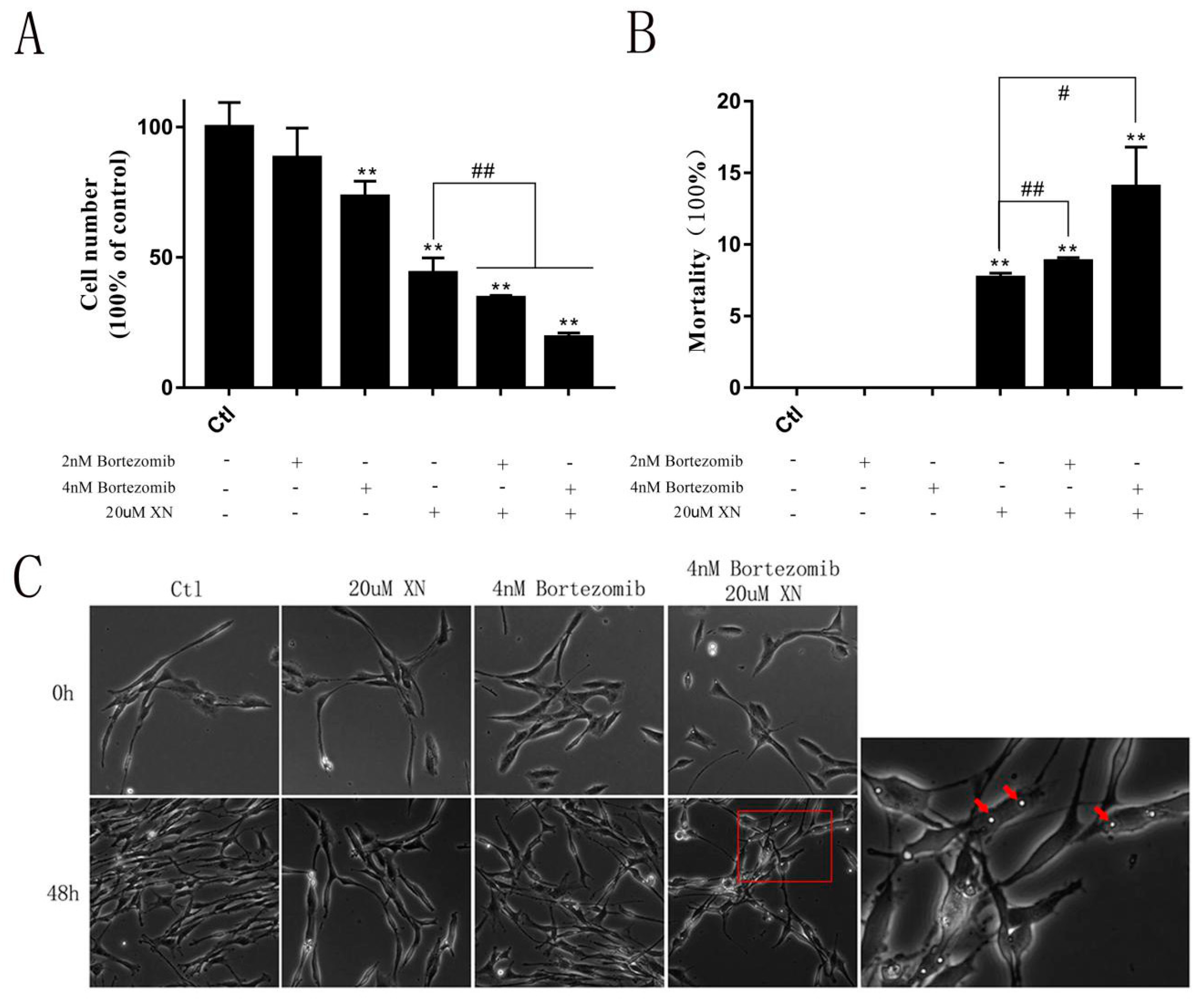
2.8. LONP1 Alleviated the Injury of XN
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Viability Assay and Morphological Observation
4.4. Trypan Blue Assay
4.5. Cell Cycle Assay
4.6. Apoptosis Assay
4.7. Measurement of Mitochondrial ROS
4.8. Detection of the Mitochondrial Membrane Potential
4.9. Western Blot Analysis
4.10. Immunocytochemistry
4.11. Detection of ATP, ADP, and AMP
4.12. Detection of Content of Succinic Dehydrogenase
4.13. Immunofluorescence staining
4.14. Detection of Mitochondrial Abundance
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurother. J. Am. Soc. Exp. NeuroTher. 2017, 14, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Shi, X.B.; Xu, B.; Yuan, C.S.; Zheng, W.; Li, G.; Li, J.; Wang, Z.H. Endoplasmic reticulum stress mediated the xanthohumol induced murine melanoma B16-F10 cell death. J. Asian Nat. Prod. Res. 2020, 22, 850–863. [Google Scholar] [CrossRef]
- Han, Q.; Han, L.; Tie, F.; Wang, Z.; Ma, C.; Li, J.; Wang, H.; Li, G. (20S)-Protopanaxadiol Ginsenosides Induced Cytotoxicity via Blockade of Autophagic Flux in HGC-27 Cells. Chem. Biodivers. 2020, 17, e2000187. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, H.; Ma, X.F.; Zhou, X.; Gan, L.; Liu, Y.Y.; Wang, Z.H. Isoliquiritigenin enhances radiosensitivity of HepG2 cells via disturbance of redox status. Cell Biochem. Biophys. 2013, 65, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hansen, P.E.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef]
- Chen, P.H.; Chang, C.K.; Shih, C.M.; Cheng, C.H.; Lin, C.W.; Lee, C.C.; Liu, A.J.; Ho, K.H.; Chen, K.C. The miR-204-3p-targeted IGFBP2 pathway is involved in xanthohumol-induced glioma cell apoptotic death. Neuropharmacology 2016, 110 Pt A, 362–375. [Google Scholar] [CrossRef]
- Festa, M.; Capasso, A.; D’Acunto, C.W.; Masullo, M.; Rossi, A.G.; Pizza, C.; Piacente, S. Xanthohumol induces apoptosis in human malignant glioblastoma cells by increasing reactive oxygen species and activating MAPK pathways. J. Nat. Prod. 2011, 74, 2505–2513. [Google Scholar] [CrossRef]
- Ho, K.H.; Kuo, T.C.; Lee, Y.T.; Chen, P.H.; Shih, C.M.; Cheng, C.H.; Liu, A.J.; Lee, C.C.; Chen, K.C. Xanthohumol regulates miR-4749-5p-inhibited RFC2 signaling in enhancing temozolomide cytotoxicity to glioblastoma. Life Sci. 2020, 254, 117807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Y.; Li, Y.W.; Tse, A.K.; Hau, D.K.; Leung, C.H.; Yu, Z.L.; Fong, W.F. 20(S)-Protopanaxadiol, a metabolite of ginsenosides, induced cell apoptosis through endoplasmic reticulum stress in human hepatocarcinoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 88–98. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nature Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 2008, 7, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef]
- Kramer, P.; Bressan, P. Our (Mother’s) Mitochondria and Our Mind. Perspect. Psychol. Sci. J. Associ. Psychol. Sci. 2018, 13, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, 469–471. [Google Scholar] [CrossRef]
- Grela, E.; Kozłowska, J.; Grabowiecka, A. Current methodology of MTT assay in bacteria—A review. Acta Histochem. 2018, 120, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.J.; Gao, H.Y. Effect of propofol on mitochondrial ATP content and ATPase activity in hippocampus of rats with cerebral ischemia-reperfusion injury. Saudi J. Biolog. Sci. 2017, 24, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodoro, J.S.; Palmeira, C.M.; Rolo, A.P. Mitochondrial Membrane Potential (ΔΨ) Fluctuations Associated with the Metabolic States of Mitochondria. Methods Mol. Biol. 2018, 1782, 109–119. [Google Scholar]
- Chang, S.Y.; Lee, M.Y.; Chung, P.S.; Kim, S.; Choi, B.; Suh, M.W.; Rhee, C.K.; Jung, J.Y. Enhanced mitochondrial membrane potential and ATP synthesis by photobiomodulation increases viability of the auditory cell line after gentamicin-induced intrinsic apoptosis. Sci. Rep. 2019, 9, 19248. [Google Scholar] [CrossRef]
- Davila, M.P.; Muñoz, P.M.; Bolaños, J.M.; Stout, T.A.; Gadella, B.M.; Tapia, J.A.; da Silva, C.B.; Ferrusola, C.O.; Peña, F.J. Mitochondrial ATP is required for the maintenance of membrane integrity in stallion spermatozoa, whereas motility requires both glycolysis and oxidative phosphorylation. Reproduction 2016, 152, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Sakthivel, R.; Malar, D.S.; Devi, K.P. Phytol shows anti-angiogenic activity and induces apoptosis in A549 cells by depolarizing the mitochondrial membrane potential. Biomed. Pharmacother. 2018, 105, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Michel, S.; Wanet, A.; De Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell. Physiol. 2012, 227, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, F.; Chen, D.; Chen, L.; Yan, C.; Lu, B.; Zhu, Y.; Chen, S.; Shi, A.; Chan, D.C.; Song, Z. Sam50 Regulates PINK1-Parkin-Mediated Mitophagy by Controlling PINK1 Stability and Mitochondrial Morphology. Cell Rep. 2018, 23, 2989–3005. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Yu, H.; Gu, S.; Xu, Y.; Su, J.; Li, H.; Kang, J.; Cui, M. p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int. J. Oncol. 2014, 45, 2341–2348. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, G.; Thomas, R.E.; Vincow, E.S.; Morris, D.R.; Pallanck, L.J. Lon protease inactivation in Drosophila causes unfolded protein stress and inhibition of mitochondrial translation. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Pareek, G.; Pallanck, L.J. Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis. 2018, 9, 1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. CB 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Juwono, J.; Martinus, R.D. Does Hsp60 Provide a Link between Mitochondrial Stress and Inflammation in Diabetes Mellitus? J. Diabetes Res. 2016, 2016, 8017571. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, S.; Song, Y.; Sun, D.; Zhu, S.; Wang, Z. Xanthohumol-Induced Rat Glioma C6 Cells Death by Triggering Mitochondrial Stress. Int. J. Mol. Sci. 2021, 22, 4506. https://doi.org/10.3390/ijms22094506
Hou S, Song Y, Sun D, Zhu S, Wang Z. Xanthohumol-Induced Rat Glioma C6 Cells Death by Triggering Mitochondrial Stress. International Journal of Molecular Sciences. 2021; 22(9):4506. https://doi.org/10.3390/ijms22094506
Chicago/Turabian StyleHou, Shaozhi, Yang Song, Di Sun, Shujun Zhu, and Zhenhua Wang. 2021. "Xanthohumol-Induced Rat Glioma C6 Cells Death by Triggering Mitochondrial Stress" International Journal of Molecular Sciences 22, no. 9: 4506. https://doi.org/10.3390/ijms22094506