
High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
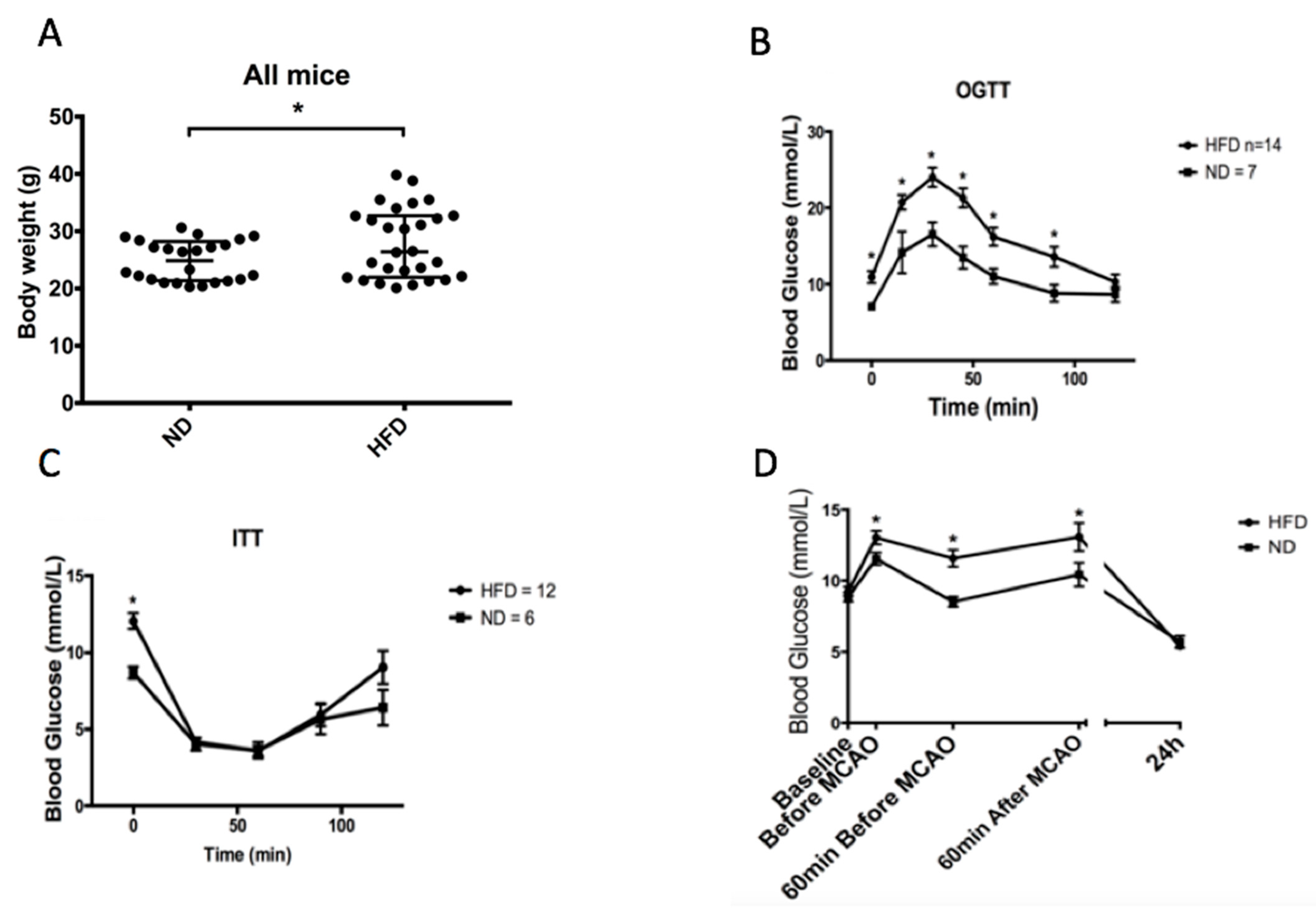
2.1. High-Fat Diet Induces Weight Gain and Impairs Glucose Tolerance
2.2. HFD Alters Metabolic and Inflammatory Plasma Markers
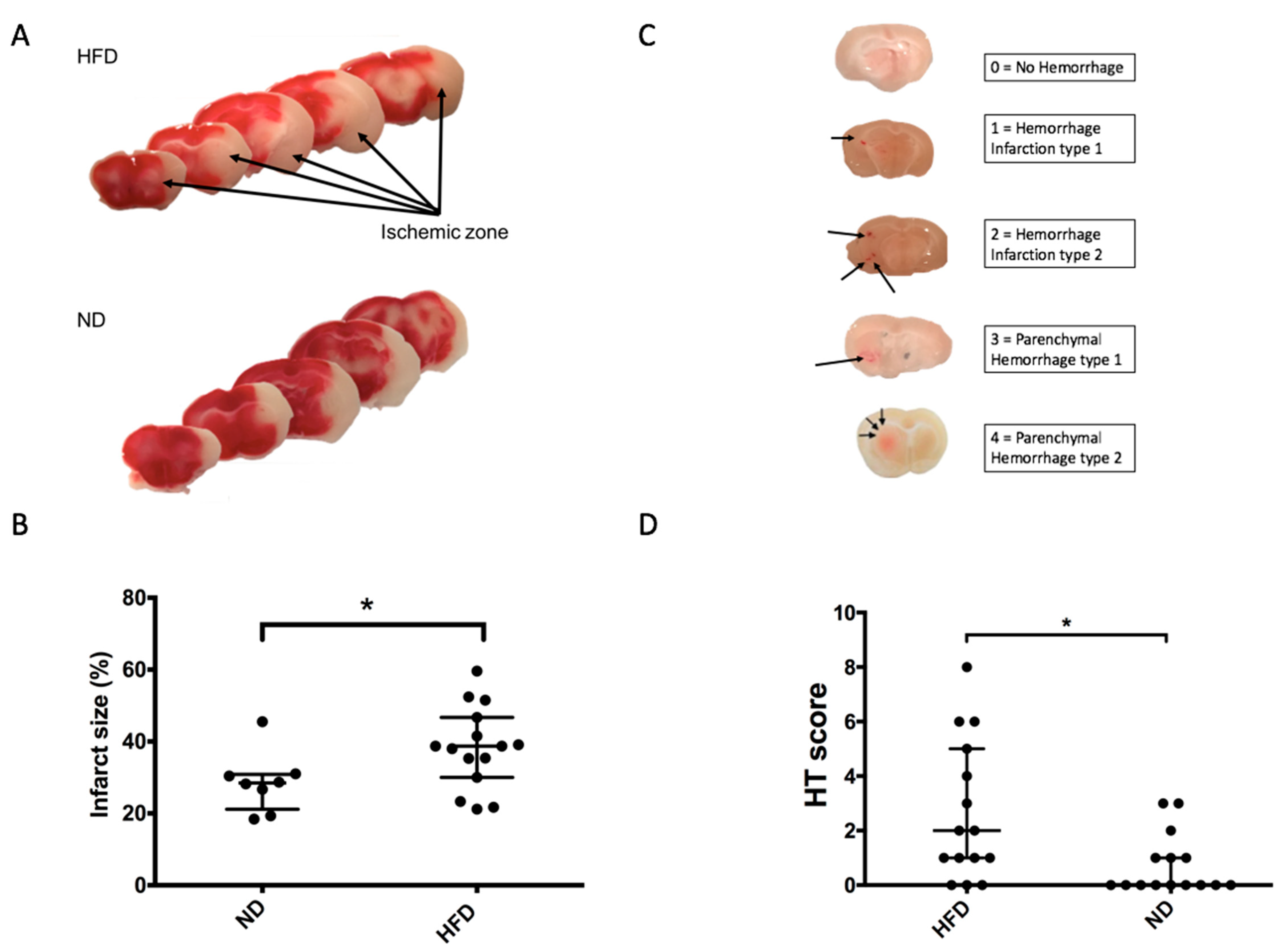
2.3. Impaired Glucose Tolerance Induced by HFD Is Associated with Increased Brain Damage in a 90-Min MCAO Model
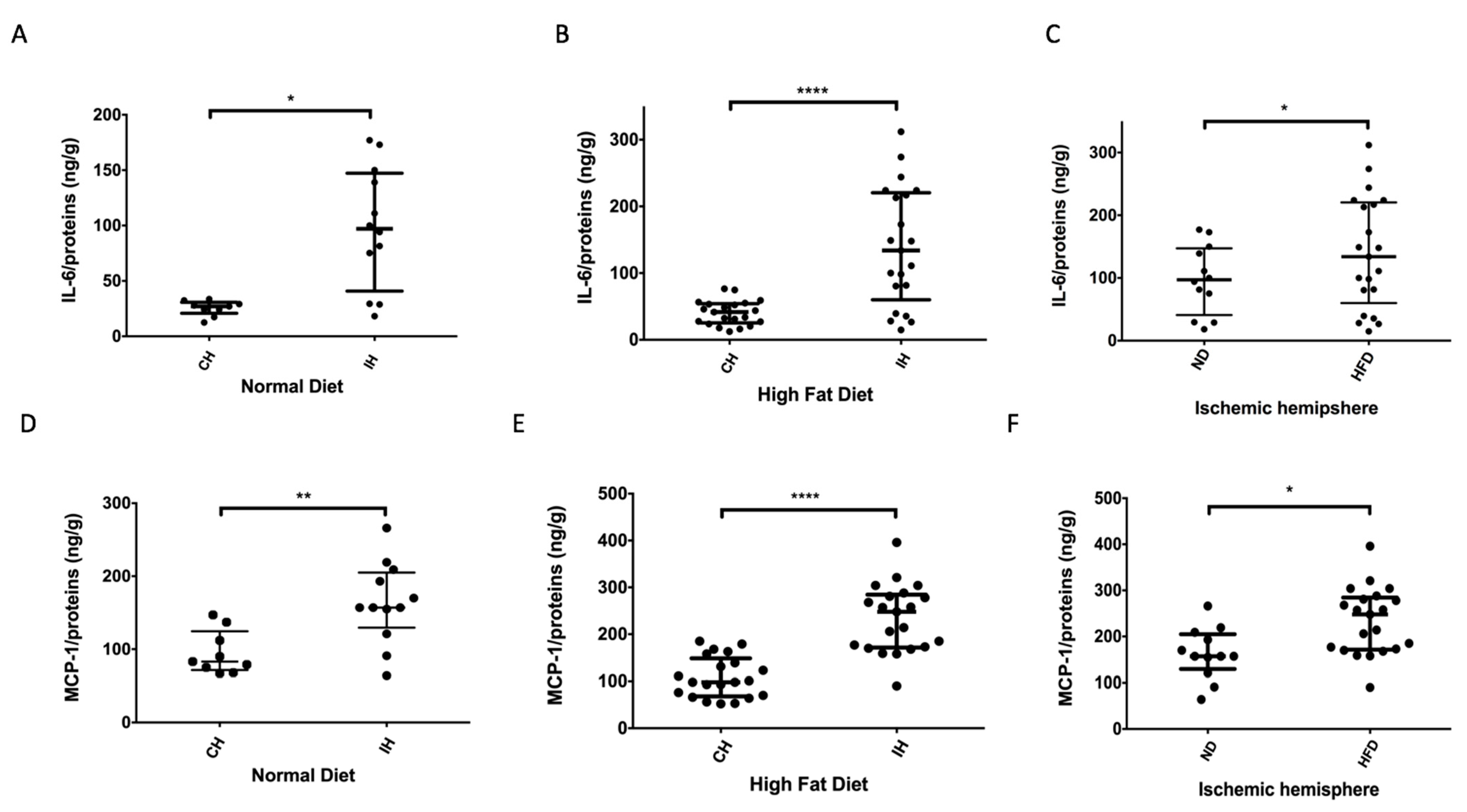
2.4. HFD Promotes Local Brain Inflammation
3. Discussion
4. Materials and Methods
4.1. Animals and Diet
4.2. Body Weight and Glycemia Evolution
4.3. Oral Glucose Tolerance and Insulin Tolerance Tests
4.4. Stroke Model
4.5. Neurological Evaluation
4.6. Evaluation of Infarct Volume and Intracerebral Hemorrhage
4.6.1. Infarct Volume Assessment
4.6.2. Hemorrhagic Score Assessment
4.7. Evaluation of Brain and Systemic Inflammation
4.7.1. Enzyme-Linked Immunosorbent Assay for Interleukin-6 (IL-6) and Leptin
4.7.2. Enzyme-Linked Immunosorbent Assay for IL-6 and MCP-1 in Brain
4.7.3. Colorimetric Enzymatic Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Adams, R.; Alberts, M.J.; Appel, L.J.; Brass, L.M.; Bushnell, C.D.; Culebras, A.; Degraba, T.J.; Gorelick, P.B.; Guyton, J.R.; et al. Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline. Stroke 2006, 37, 1583–1633. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Jauch, E.C.; Saver, J.L.; Adams, H.P., Jr.; Bruno, A.; Connors, J.J.; Demaerschalk, B.M.; Khatri, P.; McMullan, P.W., Jr.; Qureshi, A.I.; Rosenfield, K.; et al. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 870–947. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N. Engl. J. Med. 2015, 372, 2285–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovin, T.G.; Chamorro, A.; Cobo, E.; de Miquel, M.A.; Molina, C.A.; Rovira, A.; San Roman, L.; Serena, J.; Abilleira, S.; Ribo, M.; et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N. Engl. J. Med. 2015, 372, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Albers, G.W.; Marks, M.P.; Kemp, S.; Christensen, S.; Tsai, J.P.; Ortega-Gutierrez, S.; McTaggart, R.A.; Torbey, M.T.; Kim-Tenser, M.; Leslie-Mazwi, T.; et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N. Engl. J. Med. 2018, 378, 708–718. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef]
- Hacke, W.; Kaste, M.; Fieschi, C.; von Kummer, R.; Davalos, A.; Meier, D.; Larrue, V.; Bluhmki, E.; Davis, S.; Donnan, G.; et al. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Lancet 1998, 352, 1245–1251. [Google Scholar] [CrossRef]
- Chamorro, A.; Vila, N.; Ascaso, C.; Saiz, A.; Montalvo, J.; Alonso, P.; Tolosa, E. Early prediction of stroke severity. Role of the erythrocyte sedimentation rate. Stroke 1995, 26, 573–576. [Google Scholar] [CrossRef]
- Mazya, M.; Egido, J.A.; Ford, G.A.; Lees, K.R.; Mikulik, R.; Toni, D.; Wahlgren, N.; Ahmed, N.; Investigators, S. Predicting the risk of symptomatic intracerebral hemorrhage in ischemic stroke treated with intravenous alteplase: Safe Implementation of Treatments in Stroke (SITS) symptomatic intracerebral hemorrhage risk score. Stroke 2012, 43, 1524–1531. [Google Scholar] [CrossRef] [Green Version]
- Dzialowski, I.; Pexman, J.H.; Barber, P.A.; Demchuk, A.M.; Buchan, A.M.; Hill, M.D.; Investigators, C. Asymptomatic hemorrhage after thrombolysis may not be benign: Prognosis by hemorrhage type in the Canadian alteplase for stroke effectiveness study registry. Stroke 2007, 38, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Desilles, J.P.; Syvannarath, V.; Ollivier, V.; Journe, C.; Delbosc, S.; Ducroux, C.; Boisseau, W.; Louedec, L.; Di Meglio, L.; Loyau, S.; et al. Exacerbation of Thromboinflammation by Hyperglycemia Precipitates Cerebral Infarct Growth and Hemorrhagic Transformation. Stroke 2017, 48, 1932–1940. [Google Scholar] [CrossRef]
- McColl, B.W.; Rose, N.; Robson, F.H.; Rothwell, N.J.; Lawrence, C.B. Increased brain microvascular MMP-9 and incidence of haemorrhagic transformation in obese mice after experimental stroke. J. Cereb. Blood Flow Metab. 2010, 30, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef]
- Bray, G.A. Medical consequences of obesity. J. Clin. Endocrinol. Metab. 2004, 89, 2583–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, T.; Gaziano, J.M.; Berger, K.; Kase, C.S.; Rexrode, K.M.; Cook, N.R.; Buring, J.E.; Manson, J.E. Body mass index and the risk of stroke in men. Arch. Intern. Med. 2002, 162, 2557–2562. [Google Scholar] [CrossRef] [PubMed]
- Maysami, S.; Haley, M.J.; Gorenkova, N.; Krishnan, S.; McColl, B.W.; Lawrence, C.B. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J. Neuroinflamm. 2015, 12, 140. [Google Scholar] [CrossRef] [Green Version]
- Haley, M.J.; Lawrence, C.B. Obesity and stroke: Can we translate from rodents to patients? J. Cereb. Blood Flow Metab. 2016, 36, 2007–2021. [Google Scholar] [CrossRef]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Nagai, N.; Van Hoef, B.; Lijnen, H.R. Plasminogen activator inhibitor-1 contributes to the deleterious effect of obesity on the outcome of thrombotic ischemic stroke in mice. J. Thromb. Haemost. 2007, 5, 1726–1731. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S. Monogenic human obesity. Front Horm. Res. 2008, 36, 1–11. [Google Scholar] [CrossRef]
- Denes, A.; Thornton, P.; Rothwell, N.J.; Allan, S.M. Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav. Immun. 2010, 24, 708–723. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Rundek, T.; Gardener, H.; Xu, Q.; Goldberg, R.B.; Wright, C.B.; Boden-Albala, B.; Disla, N.; Paik, M.C.; Elkind, M.S.; Sacco, R.L. Insulin resistance and risk of ischemic stroke among nondiabetic individuals from the northern Manhattan study. Arch. Neurol. 2010, 67, 1195–1200. [Google Scholar] [CrossRef]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke J. Cereb. Circ. 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, N.Y.; Chechneva, O.V.; Chen, Y.J.; Tsai, Y.C.; Little, L.K.; Dang, J.; Tancredi, D.J.; Conston, J.; Anderson, S.E.; O’Donnell, M.E. Exacerbated brain edema in a rat streptozotocin model of hyperglycemic ischemic stroke: Evidence for involvement of blood-brain barrier Na-K-Cl cotransport and Na/H exchange. J. Cereb. Blood Flow Metab. 2019, 39, 1678–1692. [Google Scholar] [CrossRef] [PubMed]
- Couret, D.; Bourane, S.; Catan, A.; Nativel, B.; Planesse, C.; Dorsemans, A.C.; Ait-Arsa, I.; Cournot, M.; Rondeau, P.; Patche, J.; et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J. Comp. Neurol. 2018, 526, 1006–1016. [Google Scholar] [CrossRef]
- Langdon, K.D.; Clarke, J.; Corbett, D. Long-term exposure to high fat diet is bad for your brain: Exacerbation of focal ischemic brain injury. Neuroscience 2011, 182, 82–87. [Google Scholar] [CrossRef]
- Haley, M.J.; Krishnan, S.; Burrows, D.; de Hoog, L.; Thakrar, J.; Schiessl, I.; Allan, S.M.; Lawrence, C.B. Acute high-fat feeding leads to disruptions in glucose homeostasis and worsens stroke outcome. J. Cereb. Blood Flow Metab. 2019, 39, 1026–1037. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Lonnqvist, F.; Arner, P.; Nordfors, L.; Schalling, M. Overexpression of the obese (ob) gene in adipose tissue of human obese subjects. Nat. Med. 1995, 1, 950–953. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The gut microbiome as therapeutic target. Pharmacol. Ther. 2011, 130, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Dali-Youcef, N.; Mecili, M.; Ricci, R.; Andres, E. Metabolic inflammation: Connecting obesity and insulin resistance. Ann. Med. 2013, 45, 242–253. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [Green Version]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.Z.; Dale, V.Q.; Denes, A.; Bennett, G.; Rothwell, N.J.; Allan, S.M.; McColl, B.W. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J. Cereb. Blood Flow Metab. 2009, 29, 1764–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruyt, N.D.; Biessels, G.J.; Devries, J.H.; Roos, Y.B. Hyperglycemia in acute ischemic stroke: Pathophysiology and clinical management. Nat. Rev. Neurol. 2010, 6, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bozzao, L.; Angeloni, U.; Bastianello, S.; Fantozzi, L.M.; Pierallini, A.; Fieschi, C. Early angiographic and CT findings in patients with hemorrhagic infarction in the distribution of the middle cerebral artery. AJNR Am. J. Neuroradiol. 1991, 12, 1115–1121. [Google Scholar] [PubMed]
- Taile, J.; Patche, J.; Veeren, B.; Gonthier, M.P. Hyperglycemic Condition Causes Pro-Inflammatory and Permeability Alterations Associated with Monocyte Recruitment and Deregulated NFkappaB/PPARgamma Pathways on Cerebral Endothelial Cells: Evidence for Polyphenols Uptake and Protective Effect. Int. J. Mol. Sci. 2021, 22, 1385. [Google Scholar] [CrossRef] [PubMed]
- Arcambal, A.; Taile, J.; Couret, D.; Planesse, C.; Veeren, B.; Diotel, N.; Gauvin-Bialecki, A.; Meilhac, O.; Gonthier, M.P. Protective Effects of Antioxidant Polyphenols against Hyperglycemia-Mediated Alterations in Cerebral Endothelial Cells and a Mouse Stroke Model. Mol. Nutr. Food Res. 2020, 64, e1900779. [Google Scholar] [CrossRef] [PubMed]
- Gidday, J.M.; Gasche, Y.G.; Copin, J.C.; Shah, A.R.; Perez, R.S.; Shapiro, S.D.; Chan, P.H.; Park, T.S. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H558–H568. [Google Scholar] [CrossRef]
- Justicia, C.; Panes, J.; Sole, S.; Cervera, A.; Deulofeu, R.; Chamorro, A.; Planas, A.M. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J. Cereb. Blood Flow Metab. 2003, 23, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Rosell, A.; Cuadrado, E.; Ortega-Aznar, A.; Hernandez-Guillamon, M.; Lo, E.H.; Montaner, J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008, 39, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgebaly, M.M.; Prakash, R.; Li, W.; Ogbi, S.; Johnson, M.H.; Mezzetti, E.M.; Fagan, S.C.; Ergul, A. Vascular protection in diabetic stroke: Role of matrix metalloprotease-dependent vascular remodeling. J. Cereb. Blood Flow Metab. 2010, 30, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Bellolio, M.F.; Gilmore, R.M.; Ganti, L. Insulin for glycaemic control in acute ischaemic stroke. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Johnston, K.C.; Bruno, A.; Pauls, Q.; Hall, C.E.; Barrett, K.M.; Barsan, W.; Fansler, A.; Van de Bruinhorst, K.; Janis, S.; Durkalski-Mauldin, V.L.; et al. Intensive vs Standard Treatment of Hyperglycemia and Functional Outcome in Patients with Acute Ischemic Stroke: The SHINE Randomized Clinical Trial. JAMA 2019, 322, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Menzies, S.A.; Hoff, J.T.; Betz, A.L. Middle cerebral artery occlusion in rats: A neurological and pathological evaluation of a reproducible model. Neurosurgery 1992, 31, 100–106, discussion 106–107. [Google Scholar] [CrossRef] [PubMed]
- Fiorelli, M.; Bastianello, S.; von Kummer, R.; del Zoppo, G.J.; Larrue, V.; Lesaffre, E.; Ringleb, A.P.; Lorenzano, S.; Manelfe, C.; Bozzao, L. Hemorrhagic transformation within 36 hours of a cerebral infarct: Relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke 1999, 30, 2280–2284. [Google Scholar] [CrossRef]
- Lapergue, B.; Dang, B.Q.; Desilles, J.P.; Ortiz-Munoz, G.; Delbosc, S.; Loyau, S.; Louedec, L.; Couraud, P.O.; Mazighi, M.; Michel, J.B.; et al. High-density lipoprotein-based therapy reduces the hemorrhagic complications associated with tissue plasminogen activator treatment in experimental stroke. Stroke J. Cereb. Circ. 2013, 44, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.; Adams, R.D. Observations on brain embolism with special reference to the mechanism of hemorrhagic infarction. J. Neuropathol. Exp. Neurol. 1951, 10, 92–94. [Google Scholar]
- Hornig, C.R.; Dorndorf, W.; Agnoli, A.L. Hemorrhagic cerebral infarction—A prospective study. Stroke 1986, 17, 179–185. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grisotto, C.; Taïlé, J.; Planesse, C.; Diotel, N.; Gonthier, M.-P.; Meilhac, O.; Couret, D. High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model. Int. J. Mol. Sci. 2021, 22, 4571. https://doi.org/10.3390/ijms22094571
Grisotto C, Taïlé J, Planesse C, Diotel N, Gonthier M-P, Meilhac O, Couret D. High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model. International Journal of Molecular Sciences. 2021; 22(9):4571. https://doi.org/10.3390/ijms22094571
Chicago/Turabian StyleGrisotto, Coline, Janice Taïlé, Cynthia Planesse, Nicolas Diotel, Marie-Paule Gonthier, Olivier Meilhac, and David Couret. 2021. "High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model" International Journal of Molecular Sciences 22, no. 9: 4571. https://doi.org/10.3390/ijms22094571