Abstract
The complement system is an assembly of proteins that collectively participate in the functions of the healthy and diseased brain. The complement system plays an important role in the maintenance of uninjured (healthy) brain homeostasis, contributing to the clearance of invading pathogens and apoptotic cells, and limiting the inflammatory immune response. However, overactivation or underregulation of the entire complement cascade within the brain may lead to neuronal damage and disturbances in brain function. During the last decade, there has been a growing interest in the role that this cascading pathway plays in the neuropathology of a diverse array of brain disorders (e.g., acute neurotraumatic insult, chronic neurodegenerative diseases, and psychiatric disturbances) in which interruption of neuronal homeostasis triggers complement activation. Dysfunction of the complement promotes a disease-specific response that may have either beneficial or detrimental effects. Despite recent advances, the explicit link between complement component regulation and brain disorders remains unclear. Therefore, a comprehensible understanding of such relationships at different stages of diseases could provide new insight into potential therapeutic targets to ameliorate or slow progression of currently intractable disorders in the nervous system. Hence, the aim of this review is to provide a summary of the literature on the emerging role of the complement system in certain brain disorders.
1. Introduction
The complement system is composed of a large family of circulating and membrane-associated proteins that act synergistically in a sequential cascade-like manner to execute and regulate its function. There are three different complement pathways initiated by different stimuli: the classical, alternative, and lectin pathways. All of them converge at component C3, a central molecule in the complement system that ultimately drives complement functions. The cascade involves more than 40 proteins, and the sequence of events was reviewed in great detail elsewhere [1,2,3,4]. Thus, we omit detailed general characteristics and confine ourselves to a brief summary of the current data (Figure 1). Most complement proteins are produced in the liver; however, extrahepatic production of the complement by several organs was also identified. In the brain, complement components are synthesized particularly by resident neurons and glial cells [5,6,7] or are carried by an influx of blood following injury-induced interruption of the BBB [8]. The precise role of the complement in the healthy brain is rather complex. One of the primary actions of complement activation within the central nervous system is a defense of neurons from potentially harmful toxic stimuli, such as aberrant protein aggregates and cell debris, through opsonization by component 3b. As an effector of innate immunity, the complement promotes inflammation via anaphylatoxin (C3a and C5a) and in the final stage initiates assembly to the membrane attack complex (C5b-9, MAC) [9]. In cooperation with other immune and physiological systems, the complement contributes to normal tissue development and to the proliferation, differentiation, and migration of neural progenitors [10,11]. Furthermore, the complement system plays an unexpected role in the elimination (pruning) of inappropriate synapses and mobilization of phagocytes to restore and maintain homeostasis [12,13].
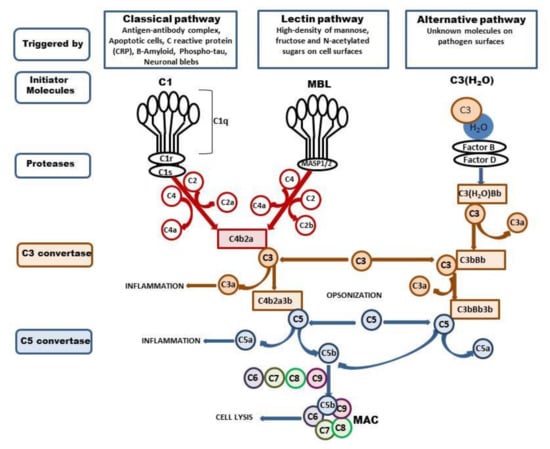
Figure 1.
Activation of complement system. The complement system can be activated via three ways: classical, lectin, and alternative. The classical pathway is triggered by binding of the antibody-antigen complexes, apoptotic cells, CRP, Β-Amyloid, phospho-tau, or neuronal blebs to C1. The C1 is composed with C1q, C1r, and C1s, and binding of Cq1 with enumerated components changes the C1 conformation and activates C1s protease. The active C1s cleaves the C4 to C4a and C4b and C2 to C2a and C2, and forms C3 convertase—C4b2a which splits C3 into two fragments, C3b, which can opsonize microbial pathogens, and C3a, which activates mast cells and macrophages and promotes inflammation. The lectin pathway is activated by the binding of mannose-binding lectin (MBL) to mannose residues on the pathogen surface. This in turn activates the MBL-associated serine proteases, MASP-1/2, which activate C4 and C2 and form C4b2a. The C3b can bind to C4b2b to form the C5 convertase (C4b2a3b). C5 is cleaved to form C5a, which promotes inflammation, and C5b, which binds to C6, C7, C8, and C9 and forms the membrane attack complex (MAC) which enables cell lysis. The alternative pathway is triggered by hydrolysis of C3 to C3-H2O; then, factors B and D generate the C3 convertase—C3(H2O)Bb, which cleaves other C3 molecules to C3a and C3b. This in turn forms C3bBb complex which continues cleavage of the C3 and enables formation of the C5 convertase—C3bBb3b, which cleaves C5 and leads to the formation of the MAC.
Underregulated or overactivated systems may lead to unwarranted neuronal damage and disturbances in brain function. However, the molecular events that lead from the alteration of this initial complement cleavage to brain insult are not completely understood. Based on recent studies, alterations in complement-mediated synaptic remodeling are hypothesized to contribute to synapse dysfunction and decline in cognitive functions in several neurodegenerative and psychiatric disorders. As another option, complement activation fragments can induce local stimulation of microglia/astrocytes that together with other proinflammatory cascades accelerate pathogenesis and neuronal damage. However, despite recent advances, several aspects of the participation of the complement system in the pathogenesis of brain disorders remain unclear. The understanding of component functions at different stages of diseases could provide new insight into potential therapeutic targets.
Hence, the aim of this review is to provide a brief summary of the current knowledge about the functional role of complement regulation in selected neuropathologies, including ischemia, traumatic brain injury, spinal cord injury, neurodevelopmental impairments (schizophrenia and autism spectrum disorder), neurodegenerative diseases, and chronic disorders such as Parkinson disease (PD), Huntington disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and epilepsy.
2. Complement in Acute Brain Trauma
2.1. Brain Ischemia
Brain ischemia triggers a widespread inflammatory reaction that, in conjunction with other induced responses, such as excitotoxicity and oxidative stress, contributes to neuronal death and neurological deficits. Inflammation, the key response to brain injury, is driven primarily by activation of inflammatory glial cells residing in the CNS together with infiltrating cells of the peripheral immune system to produce several proinflammatory factors, which lead to disruption of cellular homeostasis and structural damage to brain tissue. Based on several studies, it was postulated that the complement system represents one of the major effectors of the innate immune system, and it was assumed to play a prominent role as one of the putative mechanisms in the pathophysiology of ischemic stroke.
The well-established neurobiological role of the complement was defined in large part by clinical data. Clinical studies documented elevated levels of certain components, specifically C3, its conversion product C3a, and C5b-9, in plasma/serum samples taken from ischemic patients. Moreover, the presence of these factors likely predicts the risk of outcome [14,15,16,17,18]. It is worth noting that the presence of C1q, C4d, C3c, C9, and MAC/C5 was detected in the ischemic regions of stroke patients postmortem [15,19,20,21,22]. These findings, in conjunction with the reduced levels of the system regulators CD55 and CD59, indicate impairment of the whole system.
A functional role of the complement in experimental ischemia/reperfusion was demonstrated early by Huang et al. [23], who found that complement inhibitory depletion induced by sLex-glycosylated protein results in neuronal protection. Similarly, a protective effect was observed after cobra venous factor (CVF) pretreatment before the induction of transient ischemia in rodents. These animals present better outcomes in terms of somatosensory-evoked potential and reduced infarct volume [24]. Beneficial effects were also observed in neonatal hypoxia [25,26]. In contrast, another set of data revealed that CVF did not effectively reduce infarct volume in permanent ischemia or thromboembolic stroke [27,28].
The impact of the complement on the pathophysiology of ischemia was confirmed using transgenic mice deficient in specific complement proteins C1q, C3, and C5. Mice devoid of the C1q gene did not show any protection after ischemia induced in adult animals [20,29], despite the strongly upregulated expression of mRNA-encoding C1q in the brain following focal and global ischemia [30,31]. Simply stated, the lack of beneficial effects was not coupled with the activity of the classic pathway. The elevation of this component on neuronal cell bodies may imply its involvement in the effective clearance of damaged neurons or cellular debris [23,32,33].
Soon after the discovery of the lack of neuroprotection in C1q−/− mice, it became clear that other components respond to complement action in ischemia/reperfusion. Subsequent works performed on this matter focused on activation of C3, the central player in activation pathways. The level of component C3 is elevated after stroke and might participate in reperfusion injury [12,34,35]. Deletion of the C3 gene resulted in smaller stroke volume, diminished neutrophil infiltration, suppression of oxidative stress, and improvement of locomotor score outcomes after transient ischemia in mice. Furthermore, reconstitution with C3 protein before ischemia eliminated the effect [20]. These findings correspond with those of an in vitro study that showed that inhibition of C3 expression via small interfering RNA enhanced the viability of cultured neurons under OGD conditions [20,36].
The neuroprotective mechanism is dependent on C3a, the cleavage product of C3, which mediates downstream responses by interacting with its cognitive receptor C3aR. The generation of C3a is known to have proinflammatory properties, and its expression appears to be detrimental in several models of CNS injury. However, in some cases, e.g., following LPS administration, C3a presents anti-inflammatory action exhibited by decreasing LPS-mediated cytokine release [37].
Component C3a binds to its canonical receptor C3aR. Activation of C3aR in a mouse model of transient and permanent ischemia led to neutrophil recruitment to the ischemic zone and worsened the ischemia-induced tissue injury [38,39]. Furthermore, deletion or pharmacological inhibition of C3aR was found to improve functional and morphological outcomes following transient but not permanent ischemia in adult mouse models. This effect may be connected with restoration of post-ischemic blood flow to the brain [39]. Additional data showed that absence of the CR3 receptor protects against intracerebral hemorrhage induced by tPA treatment after MCAO. It is quite possible that reduced BBB permeability does not allow for massive neutrophil infiltration and suppresses infarction [40,41].
However, although an antagonist of CR3 presents a potentially protective action following ischemic injury, it has become clear that C3 and C3aR deficiency impairs ischemia-induced neurogenesis and consequently suppresses tissue repair following the insult [12,42,43,44]. Surprisingly, controversial data indicated improvements in functional recovery after intranasal administration of C3 in the post-acute phase of photothrombotic stroke [45]. It can thus be concluded that both C3 and C3aR have dual roles, and therapeutic manipulation of C3 must be carefully timed. An improved understanding of the time course of complement involvement may identify a therapeutic window during which complement deficiency will improve outcomes. Temporal distinction between the processes of brain injury and repair allows for the rational design of strategies.
Surprisingly, the role of the second convertase cleavage product, C3b, in ischemic stroke also remains largely undetermined. Research implies that C3b is involved in the elimination of pathogens or injured tissue [46], which may contribute to tissue repair and recovery after injury. Additionally, the function of one particular C3b receptor, CR1, is not well established in the human CNS [47,48,49,50,51]. Similarly, the role of CR2 in the brain was scarcely investigated until now.
The other intensively investigated component is C5, which is involved in a final step of complement system activation. C5 exerts activities after being cleaved by convertase 5 to soluble factor C5a and membrane-bound C5b [12,52,53]. C5a signals through the intracellular receptor C5aR1 (known as CD88). In general, the functional role of C5 in ischemic injury is unclear and strongly depends on the time and severity of injury [4]. One particularly illuminating experiment conducted by Mocco et al. [20] showed that the deletion of C5 did not offer protection after transient focal ischemia (acute ischemic injury), but another group demonstrated a reduction in infarct size and attenuated neurological deficits in a model of permanent stroke associated with long-term activation of C5 [54]. Studies revealed that the action of C5a protects against glutamate-mediated neurotoxicity in mice [55,56]. In addition, a follow-up experiment showed that neuronal apoptosis induced by C5a under OGD conditions was prevented by its absence.
Finally, the functional role of MAC/C5b was also investigated. Deficiency of CD59, an inhibitor of MAC (a pore-forming complex that mediates cell lysis), increased infarct volumes, brain swelling, and greater neurological deficits in a model of mild stroke but not in a model of severe stroke in mice [57]. In summary, despite several inconsistencies, deficiencies of individual components of the classical pathway improve outcomes after adult brain ischemia.
In an effort to identify which activation pathway might also participate in complement system activity after brain ischemia, a few studies focused on the potential involvement of lectins and alternative pathways. However, the exact pathway has not been clarified, and this subject remains rather controversial. Pharmacological deficiency of MBL or neutralizing antibodies reduced infarct volume and neurological impairment [58,59,60]. Another set of data showed that there was no significant difference in infarct volume, brain oedema, or complement deposition between wild-type and MBL-deficient mice [61]. A more detailed study indicated that protection in MBL-deficient mice was pronounced only in the acute phase and not sustained in the subacute phase [62]. Thus, it is reasonable to speculate that the lectin pathway is insufficient to initiate complement activation. Moreover, inhibition of the alternative pathway through deletion of factor B in knockout mice and administration of CR2-factor H produced significant improvements in neurological scores and reduced ischemic volume and neutrophil infiltration [63]. These findings support the concept that the lectin and alternate pathways are significant contributors to the pathogenesis of stroke.
2.2. Neonatal Hypoxic-Ischemic Encephalopathy
Hypoxic-ischemic encephalopathy (HIE) in newborn infants remains one of the most important causes of death and/or long-term neurobehavioral and cognitive dysfunction. The development of injury to the neonatal brain is a complex process with multiple contributing mechanisms and pathways resulting in both early and delayed disturbances. Inflammation has long been implicated in the pathogenesis of HIE. In the past decade, new research focused on the functions of the complement system, an essential component of the inflammatory response.
Complement cascade proteins are widely expressed in the healthy developing brain and are responsible for synapse elimination, a key developmental process [64]. The abnormal synaptic morphology of neurons differentiated during the inflammatory state may lead to serious consequences with interruption of brain survival [65]. Thus, the complement system has assumed a prominent and unique role in processes associated with hypoxic-ischemic injury. Notable advances from clinical data showed reduced levels of the complement component C3 in the blood of neonates who subsequently developed cerebral palsy [66] and increased levels of peptides C3a and C5a after fetal acidosis [67]. In addition, a diminished mean C9 concentration in CSF samples was also detected. Moreover, the analysis of postmortem brain tissue 4–5 days after severe HIE revealed that activated C9 was deposited on neurons [68].
To evaluate the role of complement in neonatal HIE models further, two groups of investigators observed the effect of CVF treatment. However, the results obtained are inconclusive. One series of experiments revealed that CVF diminished post-ischemic cerebral infarct volume and atrophy in neonatal rats and decreased C3 component deposition [25]. In contrast, two years later, Figueroa et al. [26] did not observe a beneficial effect from administration of this factor. A main point of confusion is that investigators applied different experimental paradigms. These studies demonstrated increased expression of genes related to complement activation [69,70]. Insight into the mechanisms mediating inflammatory neonatal brain damage after HI came from experimental studies showing the deposition of C1q and C3 and the generation of proinflammatory mediators such as C5a [22,71,72].
A more precise study performed by Ten [73] revealed that deletion of C1q in mice conferred significant and long-lasting neuroprotection as expressed by diminished brain infarction and improvement of functional impairments compared to wild-type controls. The same group showed that neurons of C1q−/− mice are resistant to HI with preservation of brain mitochondrial respiration and reduced production of reactive oxygen species. The results highlight the importance of the classical C1q-dependent activation pathway after HI.
It is worth noting that in neonatal mice, unlike in adult rodents, the nonclassical pathway is underdeveloped. In this case, the genetic deletion of C1q results in inhibition of the terminal C cascade response to HI and is sufficient to confer neuroprotective effects. Indeed, decreased deposition of C3 in the infarcted brain coupled with a lesser degree of brain damage in C1q−/− mice compared with WT mice suggests that C3 activation plays an important role in the exacerbation of brain damage. However, the findings of Jarlestedt et al. [74] provide evidence that the C3a peptide is neuroprotective against HI-induced injury to the immature brain. It ameliorates memory impairment after neonatal HI. The beneficial effect of C3a was also supported by results published by Moran et al. [75]. The data described above are consistent with the suggestion that increased interactions of C3a–C3aR during hypothermia induced in hypoxic neonates contribute to decreased inflammation and tissue damage [72]. The beneficial effect of C3a was supported strongly by results described by Moran et al. [75]. They found that intranasal treatment with C3a demonstrated neuroprotective action against HI. In addition, this approach ameliorated injury and induced reactive gliosis in the hippocampus but was not able to reduce the extent of hippocampal tissue loss.
The last component to be studied was proinflammatory C5a, generated by classical complement pathway activation. The elevation of C5a demonstrates deleterious consequences in the early phase. The function of C5a is mediated by its receptor C5aR, which is expressed predominantly in microglia. Mice with C5aR deletion presented only short- but not long-term improvement. In addition, therapeutic hypothermia combined with inhibition of the expression of C5aR1 was able to reduce brain infarct within three days of brain ischemia [72]. The effects in the later stages were not yet explored.
Analysis of the C9 component, a marker for C5b-9 assembly, showed that deposition of C9 in neurons in the infant brain appeared to be neurotoxic [76]. Thus, C9 deficiency reduced the brain infarct volume, whereas the reverse action was induced after C9 administration [77].
In summary, the results presented illustrate the different roles of complement proteins in ischemic injury between immature and adult brains. Therefore, the findings obtained in adults cannot be extrapolated to HI-induced injury in the immature brain.
2.3. Traumatic Brain Injury
Traumatic brain injury (TBI) is a consequence of an external mechanical force that induces disruption of the normal structure and function of the brain tissue and blood vessels. Pathophysiology following TBI is classified into the primary (initial) injury and secondary injury that develops subsequently, a determinant of outcomes. Secondary injury is generally associated with a cascade of pathophysiological processes, including glutamatergic excitotoxicity, calcium overload, and neuroinflammation, which are thought to be some of the leading factors inducing damage [1,78,79].
Increasing evidence indicates the involvement of complement-driven networks in inflammatory processes; thus, the complement system is thought to play a particularly significant role in the subsequent injury that occurs in the context of TBI. One piece of the evidence collected showed increased immunoreactivity for certain complement components (C1q, C9, C3 C5b-9, MAC, and FB) in CSF, blood plasma, and in the immediate vicinity of neurons in the penumbra of TBI patients [80,81,82,83,84].
Evidence for the impact of the complement in TBI neuropathology come from a variety of experience-based animal models (cryoinjury, controlled cortical impact, or weight drop). The results, despite several inconsistencies, indicate that certain alterations of complement proteins could improve outcomes after TBI. Early studies by Kaczorowski et al. [85] revealed that suppression of C3 convertase formation by administration of the soluble complement receptor sCR1 reduced neutrophil filtration [85]. After this discovery, it became clear that the complement plays a pivotal role in the neuroinflammatory response. The beneficial role of the inhibition of C3 convertase formation was further confirmed by C3 deletion and/or C3 systemic inhibition via administration of recombinant chimeric Crry-Ig. Both procedures resulted in reduction of microglial activation/infiltration, improved cognitive and functional recovery, and diminished extent of neuronal cell death. Likewise, decreased neuronal death post-TBI was also noted after targeted deletion of the cfb gene, which stimulates C3 convertase [86,87]. Altogether, a beneficial effect of C3 inhibition could be proposed as an attractive drug target [88,89,90,91,92,93].
Other research groups working at the same time demonstrated the participation of two components, C4 and C5, in the pathogenesis of TBI, as deficiency of these components and the presence of their antagonists reduced secondary damage in some models of TBI [89,90,92]. Within a few years, additional work performed by Stahel et al. [94] indicated the detrimental role of the terminal complement pathway, MAC (C5b-9). They found that overexuberant MAC formation is an important predominant factor implicated in secondary injury following TBI. Consistent with this hypothesis is the beneficial effect of the complement inhibitor OmCI, which binds C5 and blocks MAC formation. This paradigm decreased neuropathology and protected recovery [95]. A similar neuroprotective effect was noticed by another inhibitor of MAC formation, the CD59-CR1 hybrid (which localized to areas of C3b/iCb deposition in the injured brain) [96]. Therefore, it is concluded that the final pathway may function as a therapeutic target because its inhibition prevents the amplification of C3 and C5 convertase generation required for MAC formation.
One further notable advance is the recognition of the important role of factor B. The targeted deletion of the factor B gene extended the survival of neuronal cells in mice. This response may indicate the importance of the alternative complement pathway in the pathophysiology of TBI [86]. Thus, site-targeted alternative pathways may represent a novel therapeutic avenue [91].
Finally, by analyzing the lectin-dependent pathway, surprisingly, the pathogenic role of MBL was identified. However, data reported by different groups are truly conflicting. According to De Blasio et al. [97] and Longhi et al. [98], inactivation of the lectin pathway using a multivalent MBL ligand improved functional and pathological outcomes and decreased cortical cell death in mouse TBI. In contrast, MBL deficiency increased the number of degenerating neurons and exacerbated neurological disturbances [99]. In addition, one study showed that there is no correlation between lectin complement pathway activation and mortality/consciousness after severe TBI [100]. These conflicting findings may be related to different experimental paradigms (severity of TBI and time analysis). It is postulated that this pathway may play a dual role. The specific neuroprotective capacity may be demonstrated in the early phase of TBI secondary injury before switching to a deleterious phenotype in the late phase.
2.4. Spinal Cord Injury
Spinal cord injury (SCI) is caused by sudden traumatic insult damaging neural tissue. It results in dysfunction and sometimes loss of function below the lesion sites. Several mechanisms may contribute to secondary pathology caused by SCI, including axonal injury, demyelination, excitotoxicity, oxidative damage, and inflammation frequently associated with disruption of the blood-spinal cord barrier and recruitment of immune cells [101]. It was assumed that complement activation plays an important role in the inflammatory response [102]. The involvement of components is described in detail in a review published within the last year by Lee et al. [11]. Therefore, we will only briefly report the current data.
The detection of elevated levels of certain components, C3, C4, and C5, in the plasma of patients post-SCI provided insightful views on their role in pathology [103,104]. Furthermore, analysis performed in several animal models of SCI showed that complement proteins, including C1q, C4, FB, C3, MAC-C5b9, and complement regulator factor H, were deposited in neurons and oligodendrocytes at injured sites [105,106,107,108]. In addition, the ability to visualize C1q and factor B in axons provided views into the role of complement activation in demyelination or axonal degeneration [105]. The participation of the complement system in SCI was confirmed using knockout mice. In fact, C1q-, FB-, C9-, and C3-deficient animals exhibited reduced lesion sites at the injury epicenters, reduced infiltration of neutrophils and macrophages, and, to some degree, improved functional recovery. Moreover, administration of the complement inhibitor CR2-Crry or a factor B antibody also improved neurological deficits [109,110,111]. It is worth noting that the injured spinal cord in SCI is vulnerable to complement overactivation, for example, in the absence of the complement regulator CD59. The lack of regulators exacerbates neuropathology in SCI and increases MAC-C5b-9 deposition [111].
Recent findings indicate a more complex function for the complement cascade. For example, the role of C5aR1 evolves with time after SCI. The initial pathogenic role demonstrated a later delayed neuroprotection [11,112]. In addition, Lee et al. [11] revealed the protective function of other complement receptors, C5aR2 and C3aR. However, until now, data relating to the pathology of SCI were insufficient, and there are no proven therapies. There have been only a few studies using anti-complement treatment. However, the timing of intervention may be crucial to avoid impacting the deleterious effect of the complement.
3. Neurodegenerative Diseases
Neurodegenerative diseases constitute a heterogeneous group of disorders that increase in incidence as the population ages. These disorders are characterized by a progressive decline in cognitive ability and memory formation, leading to profound dementia. A common feature of these diseases is neuroinflammation, and it is logical to suggest that complement components contribute to pathogenesis [102]. Indeed, several studies showed that dysregulated systems and excessive complement-mediated synapse loss are associated with neurological disturbances. The most common diseases include AD, PD, HD, ALS, and MS. In this review, we do not consider AD because this was recently discussed elsewhere [113,114]. We present the key findings for complement participation in selected neurodegenerative diseases.
3.1. Parkinson Disease
Parkinson disease (PD) is a progressive, degenerative disease that leads to the loss of dopaminergic neurons in the substantia nigra (SN), causing depletion of dopamine in striatal projections [115,116]. The main feature of this pathology is progressive motor dysfunction. The neuropathological hallmarks required for a PD diagnosis are intracellular protein aggregates called Lewy bodies, consisting predominantly of alpha-synuclein and ubiquitin. Although the mechanism that triggers brain degeneration in PD is unknown, several factors (such as altered mitochondrial activity, loss of trophic factors, abnormal kinase activity, proteosomal and lysosomal dysfunction, and neuroinflammation) are thought to be the key components in the pathogenesis of disease [117,118]. In recent decades, the complement system received considerable attention as an important mediator of inflammatory responses in several neurological disorders. Hence, the possible linkage of the complement with PD was postulated. The first observations of activated complement products C3d and C4d within the postmortem brains of PD patients were reported in 1980 by McGeer and McGeer [119]. A few years later, the same group of researchers found a marked elevation in the mRNA levels of complement components in regions affected by PD [120]. Then, a subsequent series of publications identified several components—C1q, C3d, C4d, C7, and C9—in neuronal Lewy bodies of PD patients [121,122,123]. In addition, alteration of complement factors within the blood of PD patients was also detected [124]. Further systemic in vitro studies showed that activation of the complement is caused by the disease-associated splice variant of alpha-synuclein-112 but not the full-length protein [125]. In the same year, Wang et al. [126] demonstrated that C5a synergized with IgG isolated from the serum of PD patients led to selective death of dopaminergic neurons in rat mesencephalic neuron-glia cultures. Nevertheless, despite the continued insufficiency of data, it is postulated that the complement system is involved in PD pathology. Furthermore, the exact functional roles of complement components in this disorder are not yet clarified.
The absence of C1q and C3 did not protect against the depletion of dopaminergic neurons in a toxin-induced MPTP mouse model [121,127]. The beneficial effect expressed by the protection of dopaminergic neuron loss and motor dysfunction was observed after deletion of receptor CR3 in mice. This observation clearly suggests that CR3 contributes to the disease process [128]. The mode of CR3 action is associated with the activation of microglial NADPH oxidase and subsequent degeneration in a toxin-induced PD mouse model. Finally, the relevance of these observations to PD pathology remains notable. However, the available data are largely insufficient, in contrast to information on other CNS diseases, and do not allow us to define the role of the complement system in PD.
3.2. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease associated with progressive degeneration of upper and lower motor neurons. Damage to motor neurons and denervation of neuromuscular synapses in the peripheral nervous system result in the loss of control of voluntary muscle movements, spasticity, respiratory failure, and ultimately paralysis and death within 2–5 years of diagnosis [129,130]. Although the exact mechanism precipitating motor neuronal death is not yet precisely defined, subsequent analyses of animal models and human patients identified a plethora of pathological events in the course of ALS, including increased glutamate-mediated excitotoxicity, oxidative stress, mitochondrial dysfunction, and sustained upregulated immune responses, accompanying the pathogenesis of ALS [131,132]. However, the primary process leading to ALS pathogenicity remains a matter of discussion. New evidence indicates the contribution of complement-driven mechanisms in the promotion and development of both familial and sporadic forms of the disease [114,133]. The increased expression of upstream complement components C1q, C3, and C4 in the peripheral blood and spinal cerebral fluid (SCF) in living ALS patients and in the postmortem motor cortex and spinal cord in proximity to motor neurons found in ALS individuals suggests an ongoing complement activation process [134,135,136,137]. In addition, the increased deposition of the terminal component MAC/C5b-9 in motor end-plates in muscle biopsies from ALS patients further supports the engagement of the system in the disease. Moreover, it was hypothesized that the complement components may affect outcomes [102,138,139,140]. Another set of supporting information came from examining animal models, which recapitulated the onset and progression of ALS-hSOD1G93A and TDP43Q31k. These studies have revealed increased expression of complement genes and proteins C1q, C4, C3, C5, and factor B detected early in the spinal cord and skeletal muscle in the disease process in transgenic mice overexpressing mutant SOD1 [134,141,142,143,144,145,146,147,148]. The detailed study of Bahia et al. [134] showed deposition of C1q and C3/C3b at the motor end-plate before neurological symptoms. Notably, complement deposition contributes to nerve terminal destruction in ALS [149]. The described alteration of the complement was closely linked to the reduced expression of the regulatory proteins CD55 and CD59a, regulating C3 and MAC, respectively [144,145,147]. The next dataset published by these authors suggested that complement dysregulation could play a role in motor neuron loss and denervation of neuromuscular junctions.
Considering these results, it seemed surprising that, despite the upregulation of C1q, C3, and C4 components, genetic deletion of the upregulated components failed to prevent or at least delayed the onset of disease in hSOD1G93A transgenic mice [149,150]. Importantly, deletion of the gene encoding C4 significantly reduced numbers of activated macrophages found in the sciatic nerves of mSOD1G93A mice but, despite this effect, also failed to influence disease course. Therefore, all the data described above provoked the suggestion that the complement does not contribute to ALS progression.
This proposal contrasted somewhat with results showing that genetic deletion or pharmacologic blocking of the downstream C5aR1 with antagonist PMX205 ameliorated motor deficits and extended survival in mutant mouse models (hSOD1G43A, TDP-43Q331K) [144,148,151]. Thus, it was concluded that C5a-C5aR1 signaling may affect disease progression through the activation of local immune cells and infiltration by macrophages, leading to an overall increase in inflammation/neuroinflammation and then neurodegeneration [147,148,152,153]. The contribution of strengthened C5aR1 signaling to motor neuron death was further supported by studying cell cultures [136,154].
The reasons for the conflicting results among C1q, C3, and C4 knockout, indicating lack of effect and beneficial C5aR1 deletion/antagonism, currently remain unclear, but it seems logical to suppose that in the absence of upstream complement proteins, there is downstream pathway compensation, potentially allowing for pathological terminal complement activation and C5a/C5AR1 engagement to occur [153].
In view of the suggested prominent pathogenic involvement in ALS, complete understanding of the mechanism by which C5aR1 contributes to ALS may lead to optimal target selection for all forms of ALS, depending on the stages and severity of disease.
3.3. Huntington Disease
Huntington disease (HD) is a dominantly inherited autosomal disorder caused by expansion of three-base-pair (CAG) repeats. The CAG repeat is translated into an expanded polyglutamine tract in the ubiquitously expressed huntingtin protein within neurons and glial cells [155]. The inclusion of huntingtin leads to cognitive defects, motor dysfunction, psychiatric impairments, and subsequent neuronal loss [156]. The degenerative process is most severe in the striatum and cerebral cortex but is ultimately observed throughout the brain. However, how trinucleotide expansion in the huntingtin protein is linked to the observed disorders remains unclear. More recent studies implicate the immune system as being associated with HD pathology [157,158,159]. Because complement cascade activation is the best-known feature of the immune response, the detailed characterization of component functions in HD has become crucial due to the established strong association of the complement system with common diseases. One notable advance in this research came from the discovery of complement activation products in postmortem brain tissue from HD subjects. Furthermore, the expression of mRNA coding early components (C1q, C1r, C4, C3), complement regulators, and membrane cofactor proteins (CD46, CD55, and CD59) was upregulated in the striatum of HD patients [50]. The activation of the complement was further supported by findings of Hodges et al. [160], who observed increased expression of C4A, C4B, and C3 in the caudate nucleus and motor cortex. Additionally, some reports showed the elevation of certain components (C3, C4, C7, and C9) in the cerebrospinal fluid, serum, and plasma from HD patients. Importantly, detailed analysis of HD individuals revealed that the concentrations of these factors are positively correlated with disease severity [158,161]. It is worth noting that elevated levels of C4 and C7 were detected even before visible symptoms, while the level of C9 remained unchanged until an advanced stage of disease [158].
Taken together, the above data suggest that dysregulation of the complement system contributes to HD pathogenesis. This hypothesis was confirmed by using experimental models of HD. Significantly elevated levels of C3, C9, C5aR1, and C5aR2 were observed in the striatum of rats after administration of 3-nitropropionic acid (3-NP), which caused striatal degeneration [7]. Remarkable improvement in general knowledge relating to the role of the complement came from the beneficial effect of the receptor C5aR1 inhibitors PMX53 and PMX205. This approach reduced 3-NP-induced neuronal death and gliosis and ameliorated disease pathology and behavioral deficits in transgenic mice (R6/2 and C57BL/6). The relevance of C5a-C5aR1 to HD remains to be determined. Surprisingly, deletion of C3, the upstream component of the complement cascade, in a more relevant transgenic model of HD (R6/2) did not change disease progression and did not influence disease pathology [162]. Similarly, the lack of such an effect seems to be more general, as it was also observed in ALS [150] and PD models [127].
3.4. Multiple Sclerosis
Multiple sclerosis (MS) is a complex autoimmune demyelinating disease with a variable pathology and phenotypic presentation and an unknown disease course that cannot be predicted. The disease is characterized by recurrent episodes of inflammatory demyelination with a relapsing-remitting course, significant synapse loss, and CNS atrophy [163,164,165]. Although the exact molecular basis responsible for the pathogenesis of MS is still not clearly understood, increasing evidence suggests an autoimmune disorder that results in multiple inflammatory processes. Therefore, it seems logical to anticipate the participation of complement molecule activity in MS, similar to observations in other neurodegenerative diseases.
Growing clinical data show higher levels of various complement compounds in the plasma and CSF of MS patients than in healthy controls. Moreover, initial inspection of the database revealed that a large majority of increased component expression is closely related to the severity of disease [166,167,168]. Therefore, components C3 and C4a were significantly elevated in the serum of patients in the active relapsing-remitting (RRMS) phase compared to the stable stage of MS [166,167,169]. Similarly, enhanced expression of C3, C4b, and terminal complex C5b-9 was found in CSF in the matching phase of the disease [169,170]. Further research revealed that elevation of complement regulatory protein factor F can predict the transition from relapsing to progressive disease, since factor H increased progressively with disease advancement in patients transitioning from RRMs to secondary MS progression [167]. Another study detected an increase in soluble complement receptor 2 (sCR2) in patients with either RRMS or SPMS compared to controls [171].
It is worth noting that only a few studies were performed to clarify the involvement of the mannose binding lectin (MBL) pathway. However, despite the scarce data, a positive correlation between plasma levels and severity scores was demonstrated [172,173]. It is believed that full activation of the complement cascade may be restricted to patients with more advanced disease and is significantly correlated with the degree of neurological disability. Thus, according to existing knowledge, it is tempting to speculate that the presence of complement components and activation products in serum and spinal cortical fluid may serve as biomarkers of activity and probably help to differentiate the various MS subtypes [174].
The upregulation of complement components C1q, C3b, C4d, C3aR, and C5aR C5b-9 (MAC) was also evident in postmortem human tissue and in preclinical models of MS, suggesting the involvement of classical and alternative pathways [175,176,177]. The complement proteins were localized predominantly within plaques and adjacent white matter areas, in microglia, in synaptic elements, and in capillary endothelial cells. Information on all C activation products is consistent with possible direct involvement in myelin degradation in MS patients by lysis of oligodendrocytes and chemoattraction of macrophages to the inflammation sites [178]. For example, direct evidence for the role of MAC, the final complement product, in inflammatory disease progression in a mouse EAE model was documented by the effect of an inhibitor that prevents relapse after the first clinical attack. In addition, inhibition of the C5aR1 receptor reduced inflammatory gene expression [179]. This finding provides strong support for conclusions of earlier reports indicating that MAC exerts a direct impact on nervous tissue destruction in MS [175,177]. It should be pointed out that human myelin vulnerability to complement attack may be caused by the lack of complement inhibitors [180].
A component of MS pathology, in addition to demyelination and axon degeneration, is synapse loss. In this context, the compounds that attracted a great deal of attention are C1q and C3. The data reported by Michailidou et al. [179] in an EAE mouse model indicate that the main role in synapse loss is played by axis C1q-C3. Robust deposition of both complement components at synapses and colocalization with the receptor CR3 mediate signals that could strengthen synapse vulnerability to phagocytosis by microglia [181,182]. This statement is consistent with findings of earlier work showing that mice deficient in CR3 display an approximately 50% reduction in microglial synaptic engulfment in a model of AD [183,184]. Subsequent examination of synaptic changes in demyelinating disease was performed by Werneburg et al. [185]. The results obtained demonstrated the prominent role of C3 in synapse elimination. Moreover, they indicated that this process occurs through activation of the alternative complement pathway, contrary to the classical pathway engagement postulated by Michailidou et al. [182]. Thus, it is postulated that the elevated level of C1q may drive inflammatory reactive gliosis concomitant with the loss of synapses. The function of C3 was also confirmed by using the specific C3 inhibitor AAV-Crry. The presence of this agent leads to decreased microglial engulfment of synapses and preserved circuit function. This may indicate a strategy to prevent synapse loss. In addition, genetic deletion of C3, but not knockout of C1q, significantly reduced the EAE clinical score and preserved hippocampal synaptic density [186]. Another set of informative data regarding the participation of alternative pathways in MS came both from analysis of genetic deletion of factor B and the use of specific anti-B antibody. Both treatments showed promising results with delayed onset and reduced severity of EAE symptoms [187,188].
Although alternative pathway activation of the complement system in MS was documented, there remains a great deal of uncertainty regarding the roles of other pathways in this disease [189].
3.5. Epilepsy
Epilepsy is a chronic neurological disorder characterized by an enduring predisposition to recurrent spontaneous and unpredictable seizures. The clinical manifestation of epilepsy includes sudden and abnormal episodes of motor, sensory, autonomic, or psychological origin. Comprehensive studies revealed that epilepsies include a broad range of central nervous system disorders with different, not yet clarified, complex behavior at the molecular and cellular levels. A body of collected evidence demonstrated that inflammatory and immune processes are possible mechanisms controlling seizure recurrence and precipitation in both epileptic patients and animal models [190,191,192,193,194,195]. Noting the data indicating that the complement system is a major component of the innate immune system and participates in adaptive immunity, it seems logical to suggest its potential link to epileptic conditions. However, only a few studies showed the role of the complement system in the pathogenesis of epilepsy [196,197,198]. As a result, increased expression of genes involved in the complement pathway following seizures was reported and found to remain elevated during the chronic phase in a rat model of temporal lobe epilepsy (TLE) [199,200]. Furthermore, concentrations of multiple complement components, C1q, C3, C4, and the MAC consisting of the C5b-C9 complex, were documented to be higher than in healthy controls in surgically resected brain samples collected from patients and in experimental animal models of TLE epilepsy [190,199,201,202,203,204]. Epilepsy-induced increases in C1q signaling and the generation of C3a- and C3b-mediated activation of the C5a/band may contribute to the initiation and/or preservation of neuroinflammation in epilepsy [205]. This finding parallels altered expression of inflammatory cytokines that are widely associated with generation of seizures and epilepsy [192,206]. Moreover, the induction of both behavioral and electrophysiological seizures and neuronal death were also described after sequential infusion of individual MAC proteins (C5b, C7, C8, and C9) into the hippocampus [207].
Conversely, a follow-up study surprisingly revealed that complement component factor H (CFH), a specific inhibitor of the C3 to C3b transition, was downregulated in rat hippocampal tissue after epilepsy induced by electrical stimulation. The downregulation promoted acute seizures and upregulation of reduced seizure susceptibility. This suggests that CFH may contribute to epileptogenesis [208].
The contribution of complement components to epilepsy was supported strongly by experimentally blocking the C5aR1 receptor with a PMX53 antagonist. Such an approach diminished seizure power and protected hippocampal neurons from degeneration and epilepsy-associated mortality in two mouse models of epilepsy (induced by pilocarpine and intrahippocampal kainate) [209]. Moreover, C5aR1 deficiency was accompanied by reduced inflammation expressed by attenuation of TNF-alpha upregulation by microglia. This finding may represent an important opportunity for the prevention of epileptogenesis.
Furthermore, in another experimental setting, a C1q inhibitor blocked the classical and lectin pathways in rats following pilocarpine-induced epilepsy and showed at least some neuroprotective effects, including microgliosis promotion and accelerated weight gain, but had no effect on memory deficits [210]. It was postulated that the classical complement pathways C1q and C3b are involved in epileptogenic remodeling of the synaptic course and limit synaptic connectivity [204,211,212,213]. This suggestion is consistent with the reported removal of synapses during development of the visual system and the elimination of unnecessary structures of the synaptic hippocampus in models of neurodegeneration [64,184,214,215]. However, until now, there was a lack of direct data about the possible role of the complement in epilepsy processes. This subject will require additional investigation.
4. Psychiatric Disorders
Accumulating evidence suggests a dysregulated complement pathway involved in the pathogenic processes of psychiatric disorders such as schizophrenia (SZ) [216] and autism spectrum disorder (ASD) [217]. Abnormal complement signaling owing to genetic mutations or as a result of inflammatory insult during pre- and postnatal development may lead to changes in brain connectivity and may contribute to disease pathophysiology.
The present section summarizes the current data related to the role of the complement system in selected psychiatric disturbances—schizophrenia and autism spectrum disorder.
4.1. Schizophrenia
Schizophrenia (SZ) is a chronic and disabling mental disorder marked by symptoms including psychosis and deficits in cognition and social interactions. SZ commonly develops during late adolescence or early adulthood [218]. The principal pathologic findings in the brains of those affected with SZ include excessive loss of grey matter and fewer dendritic spines on neurons from the prefrontal cortex [219,220,221]. An early hypothesis stated that SZ may result from faulty synapse elimination in the postnatal period [222]. To date, the exact mechanism underlying schizophrenia is not clear. Studies in recent decades uncovered complex interactions among the immune system, systemic inflammation, and disturbed brain function demonstrated by changes in mood, cognition, and behavior [223,224]. Since then, elevated levels of inflammation markers (TNF-alpha, IL1-beta, IL6) in peripheral blood largely confirmed the participation of inflammation in SZ, at least in a subset of individuals with SZ [225,226]. Considering the established role of the complement as a mediator of innate and acquired immunity and its role in synapse elimination (as in neurodegenerative diseases), it is not surprising that alterations in complement genetics, or complement expression, might be linked to SZ. Regarding this matter, a series of studies reported changes in the hemolytic activity of classical pathway components, particularly C1q but also C2, C3, and C4, in SZ patients. The results obtained were inconsistent, and often failed to be replicated and have not provided significant insights into pathogenesis [227,228,229,230,231,232]. Nevertheless, these findings pointed towards abnormal activation of the classical complement pathway; however, the function of several components in SZ are not yet clarified. An example is C1q, whose increased level in the sera of schizophrenia patients, compared to those of controls, may be involved in linking autoimmune processes [223]. Additionally, elevated levels of C1q in blood cells were detected in mothers of infants who later developed schizophrenia. Therefore, it is postulated that C1q may be a contributing factor to the disease [233].
The availability of the large-scale genome-wide (GWAS) methodology provided clear views into the association between the complement system and schizophrenia. Through a collaborative effort of the Schizophrenia Working Group of the Psychiatric Genomics Consortium [234] and International Schizophrenia Consortium [235],, it was discovered that the locus most significantly associated with SZ lies within the extended human major histocompatibility complex (MHC), and the genomic region lies near the region encoding complement component C4. However, the precise determination of which genes force the SZ-MHC association is difficult due to extensive polymorphism. The subsequent research of the Schizophrenia Working Group of the Psychiatric Genomics Consortium [234] and Schizophrenia Psychiatric Genome-Wide Association Study Consortium [236] identified the connection of schizophrenia to CSMD1, which encodes a gene product that regulates the complement system by degrading C4 and C3. Deficiency in this factor is characteristic of SZ symptoms, such as diminished cognitive ability and memory function [237,238,239]. Thus, it was speculated that the association of SZ with CSMDI may indicate that C4 is a convincing genetic marker of schizophrenia [216].
The C4 component is encoded by two different genes, C4A and C4B, exhibiting distinct relationships with schizophrenia risk. The analysis revealed a strong correlation between schizophrenia risk alleles and copy number variation in the C4A gene. Additionally, postmortem analysis of brains from individuals with SZ showed higher expression of C4A than that in controls. It was therefore concluded that the presence of the C4A gene and high C4A expression predispose patients to SZ [216]. Subsequent brain imaging studies using magnetic resonance spectroscopy confirmed the association between high C4A gene copy numbers and neuropil contraction in different brain regions of patients with SZ [240].
The association of the C4A gene with schizophrenia and the presence of the C4 protein in synapses lead to the speculation that the pathogenic effect of this component may be mediated through dysfunction of synapse elimination and thus loss of important aspects of brain connectivity and sociability, consistent with the early hypothesis of Feniberg [222]. Recent work suggested that C4 might work with other components of the classical cascade to promote synaptic pruning by microglia. According to a report by Ishii et al. [241], the altered level of C5 may be associated with decreased cortical thickness [241,242].
It is worth noting that from the initial identification of C4A as a risk factor in SZ, several reviews on this subject were published [243,244]. Data from the literature point rather towards increased classical pathway involvement compared to controls. Data regarding alternative or lectin pathways [231] are too scarce to draw any conclusions. Similarly, the data related to the treatment of schizophrenia regarding the complement system are limited for the reasons presented in the elegant and detailed review by Woo et al. [244].
4.2. Autism Spectrum Disorder
Autism spectrum disorder (ASD) comprises a heterogeneous group of early-onset neurodevelopmental large-scale neuronal network diseases. Autism is characterized by impaired social communicative cognition, language impairment, and restricted and repetitive types of behavior [245,246]. Despite the considerable speculation about causes of ASD (such as neurotransmitters, genetics, environmental factors, and many others) [246,247,248], the exact pathophysiology of ASD remains unknown. However, the evidence presented thus far strongly suggests a potential role of immune activation/inflammation as a risk factor contributing to this disorder [249,250,251,252]. In the past decade, research emerged providing insight into the potential role of the complement system, the major effector of immunity, in ASD. One of the notable findings came early from Warren et al. [253], who described increased frequencies of C4B alleles in autistic patients and their mothers. Further breakthroughs in research on the participation of the complement system in ASD were presented in a series of publications reporting altered levels of complement molecules in the periphery of subjects with ASD. For example, increases in levels of C1q and C3 and C3 fragments were found in the plasma of children with this disorder [254,255]. In addition, analysis of postmortem brain tissue from ASD patients detected increased dendritic spine density, probably resulting from the reduction of synaptic pruning associated with the activation of the complement system [256,257].
Direct evidence on the association between complement components in the brain and ASD was reported by Fagan et al. [217]. Analysis of human postmortem samples from ASD patients showed elevated levels of mRNA coding for C2, C5, and MASP1 (MBL-associated serine protease 1) and decreased C1q, C3, and C4 mRNA levels in the middle frontal gyrus of ASD subjects. However, the availability of brain samples was not sufficient to make a confident conclusion. He further checked the effect of C3 knockdown in the prefrontal cortex (PFC) in rodents. The deletion of C3 resulted in social deficits and repetitive behavior in mice. This may indicate a possible role of C3 in the pathophysiology of ASD. Therefore, it is tempting to speculate that diminished complement-mediated synaptic pruning, among other mechanisms, may contribute to cortical hyperconnectivity and behavioral phenotypes in ASD. This is consistent with the previous statement that C3 deficiency may limit synaptic pruning processes [184].
Summarizing the available data, the evidence regarding the association between complement dysfunction and ASD is far weaker than the evidence of complement dysregulation in schizophrenia susceptibility. In addition, a recent genome-wide association did not identify common variants in complement genes significantly associated with ASD [258]. However, the altered complement expression in peripheral blood and in the brain from patients might suggest that ASD may somehow be attributed to aberrant activity.
5. Conclusions
Our review outlines recent discoveries that indicated complement system involvement in a wide range of brain disorders contributing to neuronal and synapse remodeling (summarized in Table 1). Over the last decade, understanding of the participation of complement components in brain pathology greatly advanced; however, the molecular mechanisms leading from alteration of the system to disturbances in brain functionality are not fully understood. The examination of the responses of different components to pathological conditions proved to be somewhat controversial, probably due to differing experimental paradigms. In addition, in many cases, the reported results are rather scarce and do not allow us to determine the roles of particular factors precisely. However, despite certain inconsistencies, dysregulation of the complement system pathways affords either beneficial or detrimental effects, depending on the stage of disease. It is hypothesized that modulation of specific members of the pathway could serve as an effective therapeutic approach for attenuating or ameliorating disease-specific signs. Therefore, future research should be focused on advancing the precise understanding of these processes.

Table 1.
The effects of dysregulated complement system in neurological disorders in humans and animal experimental models.
Author Contributions
T.Z. was the major contributor in collecting data and writing the manuscript. M.Z.-N., K.Z., P.P., and J.S. participated in writing the manuscript and prepared figures. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Science Centre, Poland grant no 2017/27/B/NZ3/00582. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflammation 2018, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.L.; Pouw, R.B.; Kavanagh, D.; Sun, R.; Ricklin, D. Developments in anti-complement therapy; from disease to clinical trial. Mol. Immunol. 2018, 102, 89–119. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Lee, M.; McGeer, E.G. A review of human diseases caused or exacerbated by aberrant complement activation. Neurobiol. Aging 2017, 52, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.-G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Barnum, S. Complement Biosynthesis in the Central Nervous System. Crit. Rev. Oral Biol. Med. 1995, 6, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Ager, R.R.; Tenner, A.J.; Noakes, P.G.; Taylor, S.M. The Role of the Complement System and the Activation Fragment C5a in the Central Nervous System. NeuroMolecular Med. 2009, 12, 179–192. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, D.; Fumagalli, S.; Orsini, F.; Neglia, L.; Perego, C.; Ortolano, F.; Zanier, E.R.; Picetti, E.; Locatelli, M.; Stocchetti, N.; et al. Human brain trauma severity is associated with lectin complement pathway activation. Br. J. Pharmacol. 2018, 39, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Hawksworth, O.A.; Woodruff, T.M. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci. 2018, 41, 373–384. [Google Scholar] [CrossRef]
- Lee, J.D.; Coulthard, L.G.; Woodruff, T.M. Complement dysregulation in the central nervous system during development and disease. Semin. Immunol. 2019, 45, 101340. [Google Scholar] [CrossRef]
- Alawieh, A.; Elvington, A.; Zhu, H.; Yu, J.; Kindy, M.S.; Atkinson, C.; Tomlinson, S. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J. Neuroinflammation 2015, 12, 1–15. [Google Scholar] [CrossRef]
- Ricklin, D.; Mastellos, D.C.; Lambris, J.D. Therapeutic targeting of the complement system. Nat. Rev. Drug Discov. 2019, 1. [Google Scholar] [CrossRef]
- Lindsberg, P.J.; Öhman, J.; Lehto, T.; Wuorimaa, T.; Meri, S.; Karjalainen-Lindsberg, M.-L.; Paetau, A.; Carpén, O.; Kaste, M. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann. Neurol. 1996, 40, 587–596. [Google Scholar] [CrossRef]
- Pedersen, M.B.; Zhou, X.; Larsen, E.K.U.; Sørensen, U.S.; Kjems, J.; Nygaard, J.V.; Nyengaard, J.R.; Meyer, R.L.; Boesen, T.; Vorup-Jensen, T. Curvature of Synthetic and Natural Surfaces Is an Important Target Feature in Classical Pathway Complement Activation. J. Immunol. 2010, 184, 1931–1945. [Google Scholar] [CrossRef]
- Perez-Alcazar, M.; Daborg, J.; Stokowska, A.; Wasling, P.; Bjorefeldt, A.; Kalm, M.; Zetterberg, H.; Carlström, K.E.; Blomgren, K.; Ekdahl, C.T.; et al. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp. Neurol. 2014, 253, 154–164. [Google Scholar] [CrossRef]
- Stokowska, A.; Olsson, S.; Holmegaard, L.; Jood, K.; Blomstrand, C.; Jern, C.; Pekna, M. Plasma C3 and C3a Levels in Cryptogenic and Large-Vessel Disease Stroke: Associations with Outcome. Cerebrovasc. Dis. 2011, 32, 114–122. [Google Scholar] [CrossRef]
- Zhang, Y.; Shao, D.; Ricklin, D.; Hilkin, B.M.; Nester, C.M.; Lambris, J.D.; Smith, R.J.H. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology 2015, 220, 993–998. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, Y.; Zhang, Z.; Yang, G.-Y. Significance of Complement System in Ischemic Stroke: A Comprehensive Review. Aging Dis. 2019, 10, 429–462. [Google Scholar] [CrossRef]
- Mocco, J.; Mack, W.J.; Ducruet, A.F.; Sosunov, S.A.; Sughrue, M.E.; Hassid, B.G.; Nair, M.N.; Laufer, I.; Komotar, R.J.; Claire, M.; et al. Complement Component C3 Mediates Inflammatory Injury Following Focal Cerebral Ischemia. Circ. Res. 2006, 99, 209–217. [Google Scholar] [CrossRef]
- Széplaki, G.; Szegedi, R.; Hirschberg, K.; Gombos, T.; Varga, L.; Karádi, I.; Entz, L.; Széplaki, Z.; Garred, P.; Prohászka, Z.; et al. Strong complement activation after acute ischemic stroke is associated with unfavorable outcomes. Atherosclerosis 2009, 204, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.; Bernaudin, M.; Petit, E.; Gasque, P.; Nouvelot, A.; MacKenzie, E.T.; Fontaine, M. Expression of Receptors for Complement Anaphylatoxins C3a and C5a Following Permanent Focal Cerebral Ischemia in the Mouse. Exp. Neurol. 2000, 161, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Huang, J. Neuronal Protection in Stroke by an sLex-Glycosylated Complement Inhibitory Protein. Science 1999, 285, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Vasthare, U.; Barone, F.; Sarau, H.; Rosenwasser, R.; DiMartino, M.; Young, W.F.; Tuma, R. Complement Depletion Improves Neurological Function in Cerebral Ischemia. Brain Res. Bull. 1998, 45, 413–419. [Google Scholar] [CrossRef]
- Cowell, R.M.; Plane, J.M.; Silverstein, F.S. Complement Activation Contributes to Hypoxic-Ischemic Brain Injury in Neonatal Rats. J. Neurosci. 2003, 23, 9459–9468. [Google Scholar] [CrossRef]
- Figueroa, E.; Gordon, L.E.; Feldhoff, P.W.; Lassiter, H.A. The administration of cobra venom factor reduces post-ischemic cerebral injury in adult and neonatal rats. Neurosci. Lett. 2005, 380, 48–53. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of Inflammation and Its Mediators in Acute Ischemic Stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef]
- Lew, S.M.; E Gross, C.; Bednar, M.M.; Russell, S.J.; Fuller, S.P.; Ellenberger, C.L.; Howard, D. Complement depletion does not reduce brain injury in a rabbit model of thromboembolic stroke. Brain Res. Bull. 1999, 48, 325–331. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Rossi, E.; Storini, C.; Pizzimenti, S.; Echart, C.; Bergamaschini, L. The Powerful Neuroprotective Action of C1-Inhibitor on Brain Ischemia-Reperfusion Injury Does Not Require C1q. Am. J. Pathol. 2004, 164, 1857–1863. [Google Scholar] [CrossRef]
- Schäfer, M.K.-H.; Schwaeble, W.J.; Post, C.; Salvati, P.; Calabresi, M.; Sim, R.B.; Petry, F.; Loos, M.; Weihe, E. Complement C1q Is Dramatically Up-Regulated in Brain Microglia in Response to Transient Global Cerebral Ischemia. J. Immunol. 2000, 164, 5446–5452. [Google Scholar] [CrossRef]
- Stenzel-Poore, M.P.; Stevens, S.L.; Xiong, Z.; Lessov, N.S.; A Harrington, C.; Mori, M.; Meller, R.; Rosenzweig, H.L.; Tobar, E.; E Shaw, T.; et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003, 362, 1028–1037. [Google Scholar] [CrossRef]
- Fraser, D.A.; Pisalyaput, K.; Tenner, A.J. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J. Neurochem. 2010, 112, 733–743. [Google Scholar] [CrossRef]
- Mack, W.J.; Sughrue, M.E.; Ducruet, A.F.; Mocco, J.; Sosunov, S.A.; Hassid, B.G.; Silverberg, J.Z.; Ten, V.S.; Pinsky, D.J.; Connolly, E.S.C. Temporal pattern of C1q deposition after transient focal cerebral ischemia. J. Neurosci. Res. 2006, 83, 883–889. [Google Scholar] [CrossRef]
- Satyam, A.; Kannan, L.; Matsumoto, N.; Geha, M.; Lapchak, P.H.; Bosse, R.; Shi, G.-P.; Lucca, J.J.D.; Tsokos, M.G.; Tsokos, G.C. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage during Mesenteric Ischemia. J. Immunol. 2016, 198, 788–797. [Google Scholar] [CrossRef]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4+ T Cells. Immunology 2008, 28, 425–435. [Google Scholar] [CrossRef]
- Yang, J.; Ahn, H.-N.; Chang, M.; Narasimhan, P.; Chan, P.H.; Song, Y.S. Complement component 3 inhibition by an antioxidant is neuroprotective after cerebral ischemia and reperfusion in mice. J. Neurochem. 2013, 124, 523–535. [Google Scholar] [CrossRef]
- Kildsgaard, J.; Hollmann, T.J.; Matthews, K.W.; Bian, K.; Murad, F.; Wetsel, R.A. Cutting edge: Targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J. Immunol. 2000, 165, 5406–5409. [Google Scholar] [CrossRef]
- Barnum, S.R.; Ames, R.S.; Maycox, P.R.; Hadingham, S.J.; Meakin, J.; Harrison, D.; Parsons, A.A. Expression of the complement C3a and C5a receptors after permanent focal ischemia: An alternative interpretation. Glia 2002, 38, 169–173. [Google Scholar] [CrossRef]
- Ducruet, A.F.; Hassid, B.G.; Mack, W.J.; Sosunov, S.A.; Otten, M.L.; Fusco, D.J.; Hickman, Z.L.; Kim, G.H.; Komotar, R.J.; Mocco, J.; et al. C3a Receptor Modulation of Granulocyte Infiltration after Murine Focal Cerebral Ischemia is Reperfusion Dependent. Br. J. Pharmacol. 2008, 28, 1048–1058. [Google Scholar] [CrossRef]
- Soriano, S.G.; Coxon, A.; Wang, Y.F.; Frosch, M.P.; Lipton, S.A.; Hickey, P.R.; Mayadas, T.N. Mice Deficient in Mac-1 (CD11b/CD18) Are Less Susceptible to Cerebral Ischemia/Reperfusion Injury. Stroke 1999, 30, 134–139. [Google Scholar] [CrossRef]
- Su, E.J.; Cao, C.; Fredriksson, L.; Nilsson, I.; Stefanitsch, C.; Stevenson, T.K.; Zhao, J.; Ragsdale, M.; Sun, Y.-Y.; Yepes, M.; et al. Microglial-mediated PDGF-CC activation increases cerebrovascular permeability during ischemic stroke. Acta Neuropathol. 2017, 134, 585–604. [Google Scholar] [CrossRef]
- Ducruet, A.F.; Zacharia, B.E.; Sosunov, S.A.; Gigante, P.R.; Yeh, M.L.; Gorski, J.W.; Otten, M.L.; Hwang, R.Y.; DeRosa, P.A.; Hickman, Z.L.; et al. Complement Inhibition Promotes Endogenous Neurogenesis and Sustained Anti-Inflammatory Neuroprotection following Reperfused Stroke. PLoS ONE 2012, 7, e38664. [Google Scholar] [CrossRef]
- Rahpeymai, Y.; Hietala, M.A.; Wilhelmsson, U.; Fotheringham, A.; Davies, I.; Nilsson, A.-K.; Zwirner, J.; A Wetsel, R.; Gerard, C.; Pekny, M.; et al. Complement: A novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006, 25, 1364–1374. [Google Scholar] [CrossRef]
- Shinjyo, N.; Ståhlberg, A.; Dragunow, M.; Pekny, M.; Pekna, M. Complement-Derived Anaphylatoxin C3a Regulates In Vitro Differentiation and Migration of Neural Progenitor Cells. Stem Cells 2009, 27, 2824–2832. [Google Scholar] [CrossRef]
- Stokowska, A.; Atkins, A.L.; Morán, J.; Pekny, T.; Bulmer, L.; Pascoe, M.C.; Barnum, S.R.; Wetsel, R.A.; Nilsson, J.A.; Dragunow, M.; et al. Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain 2016, 140, 353–369. [Google Scholar] [CrossRef]
- Hasegawa, M.; Yada, S.; Liu, M.Z.; Kamada, N.; Muñoz-Planillo, R.; Do, N.; Núñez, G.; Inohara, N. Interleukin-22 Regulates the Complement System to Promote Resistance against Pathobionts after Pathogen-Induced Intestinal Damage. Immunology 2014, 41, 620–632. [Google Scholar] [CrossRef]
- Gasque, P.; Chan, P.; Mauger, C.; Schouft, M.T.; Singhrao, S.; Dierich, M.P.; Morgan, B.P.; Fontaine, M. Identification and characterization of complement C3 receptors on human astrocytes. J. Immunol. 1996, 156, 2247–2255. [Google Scholar]
- Molina, H.; Kinoshita, T.; Inoue, K.; Carel, J.C.; Holers, V.M. A molecular and immunochemical characterization of mouse CREvidence for a single gene model of mouse complement receptors 1 and 2. J. Immunol. 1990, 145, 2974–2983. [Google Scholar]
- Molina, H.; Holers, V.M.; Li, B.; Fung, Y.; Mariathasan, S.; Goellner, J.; Strauss-Schoenberger, J.; Karr, R.W.; Chaplin, D.D. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA 1996, 93, 3357–3361. [Google Scholar] [CrossRef] [PubMed]
- Singhrao, S.; Neal, J.; Morgan, B.; Gasque, P. Increased Complement Biosynthesis by Microglia and Complement Activation on Neurons in Huntington’s Disease. Exp. Neurol. 1999, 159, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Yates, D. Microglial cell-mediated depression. Nat. Rev. Neurosci. 2014, 15, 281. [Google Scholar] [CrossRef]
- Arumugam, T.; Woodruff, T.; Lathia, J.; Selvaraj, P.; Mattson, M.; Taylor, S. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience 2009, 158, 1074–1089. [Google Scholar] [CrossRef]
- Wagner, E.; Frank, M.M. Therapeutic potential of complement modulation. Nat. Rev. Drug Discov. 2009, 9, 43–56. [Google Scholar] [CrossRef]
- Arumugam, T.V.; Tang, S.-C.; Lathia, J.D.; Cheng, A.; Mughal, M.R.; Chigurupati, S.; Magnus, T.; Chan, S.L.; Jo, D.-G.; Ouyang, X.; et al. Intravenous Immunoglobulin (IVIG) Protects the Brain against Experimental Stroke by Preventing Com-plement-Mediated Neuronal Cell Death. Proc. Natl. Acad. Sci. USA 2007, 104, 14104–14109. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Thomas, S.; Pasinetti, G.M. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 In Vitro and In Vivo. J. Neuroinflammation 2008, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Pekna, M.; Hansson, E.; Rönnbäck, L. The complement-derived anaphylatoxin C5a increases microglial GLT-1 expression and glutamate uptake in a TNF-α-independent manner. Eur. J. Neurosci. 2009, 29, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Harhausen, D.; Khojasteh, U.; Stahel, P.F.; Morgan, B.P.; Nietfeld, W.; Dirnagl, U.; Trendelenburg, G. Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. J. Neuroinflammation 2010, 7, 15. [Google Scholar] [CrossRef]
- Cervera, A.; Planas, A.M.; Justicia, C.; Urra, X.; Jensenius, J.C.; Torres, F.; Lozano, F.; Chamorro, A. Genetically-Defined Deficiency of Mannose-Binding Lectin Is Associated with Protection after Experimental Stroke in Mice and Outcome in Human Stroke. PLoS ONE 2010, 5, e8433. [Google Scholar] [CrossRef]
- De la Rosa, X.; Cervera, A.; Kristoffersen, A.K.; Valdés, C.P.; Varma, H.M.; Justicia, C.; Durduran, T.; Chamorro, Á; Planas, A.M. Mannose-Binding Lectin Promotes Local Microvascular Thrombosis After Transient Brain Ischemia in Mice. Stroke 2014, 45, 1453–1459. [Google Scholar] [CrossRef]
- Orsini, F.; Fumagalli, S.; Császár, E.; Tóth, K.; de Blasio, D.; Zangari, R.; Lénárt, N.; Dénes, Á.; De Simoni, M.-G. Mannose-Binding Lectin Drives Platelet Inflammatory Phenotype and Vascular Damage After Cerebral Ischemia in Mice via IL (Interleukin)-1α. Arter. Thromb. Vasc. Biol. 2018, 38, 2678–2690. [Google Scholar] [CrossRef]
- Morrison, H.; Frye, J.; Davis-Gorman, G.; Funk, J.; McDonagh, P.; Stahl, G.; Ritter, L. The Contribution of Mannose Binding Lectin to Reperfusion Injury after Ischemic Stroke. Curr. Neurovascular Res. 2011, 8, 52–63. [Google Scholar] [CrossRef]
- Ducruet, A.F.; Sosunov, S.A.; Zacharia, B.E.; Gorski, J.; Yeh, M.L.; DeRosa, P.; Cohen, G.; Gigante, P.R.; Connolly, E.S. The Neuroprotective Effect of Genetic Mannose-Binding Lectin Deficiency Is Not Sustained in the Subacute Phase of Stroke. Transl. Stroke Res. 2011, 2, 588–599. [Google Scholar] [CrossRef]
- Elvington, A.; Atkinson, C.; Zhu, H.; Yu, J.; Takahashi, K.; Stahl, G.L.; Kindy, M.S.; Tomlinson, S. The Alternative Complement Pathway Propagates Inflammation and Injury in Murine Ischemic Stroke. J. Immunol. 2012, 189, 4640–4647. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Jakubs, K.; Bonde, S.; Iosif, R.E.; Ekdahl, C.T.; Kokaia, Z.; Kokaia, M.; Lindvall, O. Inflammation Regulates Functional Integration of Neurons Born in Adult Brain. J. Neurosci. 2008, 28, 12477–12488. [Google Scholar] [CrossRef]
- Grether, J.K.; Nelson, K.B.; Dambrosia, J.M.; Phillips, T.M. Interferons and cerebral palsy. J. Pediatr. 1999, 134, 324–332. [Google Scholar] [CrossRef]
- Sonntag, J.; Dähnert, I.; Stiller, B.; Hetzer, R.; Lange, P.E. Complement and Contact Activation during Cardiovascular Operations in Infants. Ann. Thorac. Surg. 1998, 65, 525–531. [Google Scholar] [CrossRef]
- Aly, H.; Khashaba, M.T.; Nada, A.; Hasanen, B.M.; McCarter, R.; Schultz, S.J.; Gordon, L.; Feldhoff, P.W.; Lassiter, H.A. The Role of Complement in Neurodevelopmental Impairment following Neonatal Hypoxic-Ischemic Encephalopathy. Am. J. Perinatol. 2009, 26, 659–665. [Google Scholar] [CrossRef]
- Maj, H.; Mallard, C.; Hagberg, H. Inflammatory Gene Profiling in the Developing Mouse Brain after Hypoxia-Ischemia. Br. J. Pharmacol. 2004, 24, 1333–1351. [Google Scholar] [CrossRef]
- Mallard, C.; Hagberg, H. Inflammation-induced preconditioning in the immature brain. Semin. Fetal Neonatal Med. 2007, 12, 280–286. [Google Scholar] [CrossRef]
- Shah, T.A.; Mauriello, C.T.; Hair, P.S.; Sandhu, A.; Stolz, M.P.; Bass, W.T.; Krishna, N.K.; Cunnion, K.M. Clinical hypothermia temperatures increase complement activation and cell destruction via the classical pathway. J. Transl. Med. 2014, 12, 181. [Google Scholar] [CrossRef]
- Shah, T.A.; Nejad, J.E.; Pallera, H.K.; Lattanzio, F.A.; Farhat, R.; Kumar, P.S.; Hair, P.S.; Bass, W.T.; Krishna, N.K. Therapeutic hypothermia modulates complement factor C3a and C5a levels in a rat model of hypoxic ischemic encephalopathy. Pediatr. Res. 2016, 81, 654–662. [Google Scholar] [CrossRef]
- Ten, V.S.; Sosunov, S.A.; Mazer, S.P.; Stark, R.I.; Caspersen, C.; Sughrue, M.E.; Botto, M.; Connolly, E.S.; Pinsky, D.J. C1q-Deficiency Is Neuroprotective Against Hypoxic-Ischemic Brain Injury in Neonatal Mice. Stroke 2005, 36, 2244–2250. [Google Scholar] [CrossRef]
- Järlestedt, K.; Rousset, C.I.; Ståhlberg, A.; Sourkova, H.; Atkins, A.L.; Thornton, C.; Barnum, S.R.; Wetsel, R.A.; Dragunow, M.; Pekny, M.; et al. Receptor for complement peptide C3a: A therapeutic target for neonatal hypoxic-ischemic brain injury. FASEB J. 2013, 27, 3797–3804. [Google Scholar] [CrossRef]
- Morán, J.; Stokowska, A.; Walker, F.R.; Mallard, C.; Hagberg, H.; Pekna, M. Intranasal C3a treatment ameliorates cognitive impairment in a mouse model of neonatal hypoxic–ischemic brain injury. Exp. Neurol. 2017, 290, 74–84. [Google Scholar] [CrossRef]
- Schultz, S.J.; Aly, H.; Hasanen, B.M.; Khashaba, M.T.; Lear, S.C.; Bendon, R.W.; Gordon, L.E.; Feldhoff, P.W.; Lassiter, H.A. Complement component 9 activation, consumption, and neuronal deposition in the post-hypoxic–ischemic central nervous system of human newborn infants. Neurosci. Lett. 2005, 378, 1–6. [Google Scholar] [CrossRef]
- Imm, M.D.; Feldhoff, P.W.; Feldhoff, R.C.; Lassiter, H.A. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci. Lett. 2002, 325, 175–178. [Google Scholar] [CrossRef]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation After Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef]
- Bao, W.; He, F.; Yu, L.; Gao, J.; Meng, F.; Ding, Y.; Zou, H.; Luo, B. Complement cascade on severe traumatic brain injury patients at the chronic unconscious stage: Implication for pathogenesis. Expert Rev. Mol. Diagn. 2018, 18, 761–766. [Google Scholar] [CrossRef]
- Bellander, B.-M.; Olafsson, I.H.; Ghatan, P.H.; Skejo, H.P.B.; Hansson, L.-O.; Wanecek, M.; Svensson, M.A. Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochir. 2011, 153, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, T.; Stahel, P.F.; Morganti-Kossmann, M.C.; Jones, J.L.; Barnum, S.R. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 1997, 73, 63–69. [Google Scholar] [CrossRef]
- Manek, R.; Moghieb, A.; Yang, Z.; Kumar, D.; Kobessiy, F.; Sarkis, G.A.; Raghavan, V.; Wang, K.K. Protein Biomarkers and Neuroproteomics Characterization of Microvesicles/Exosomes from Human Cerebrospinal Fluid Following Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 6112–6128. [Google Scholar] [CrossRef]
- Stahel, P.F.; Morganti-Kossmann, M.C.; Perez, D.; Redaelli, C.; Gloor, B.; Trentz, O.; Kossmann, T. Intrathecal Levels of Complement-Derived Soluble Membrane Attack Complex (sC5b-9) Correlate with Blood-Brain Barrier Dysfunction in Patients with Traumatic Brain Injury. J. Neurotrauma 2001, 18, 773–781. [Google Scholar] [CrossRef]
- Kaczorowski, S.L.; Schiding, J.K.; Toth, C.A.; Kochanek, P.M. Effect of Soluble Complement Receptor-1 on Neutrophil Accumulation after Traumatic Brain Injury in Rats. Br. J. Pharmacol. 1995, 15, 860–864. [Google Scholar] [CrossRef]
- Leinhase, I.; Holers, V.M.; Thurman, J.M.; Harhausen, D.; I Schmidt, O.; Pietzcker, M.; E Taha, M.; Rittirsch, D.; Huber-Lang, M.; Smith, W.R.; et al. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. 2006, 7, 55. [Google Scholar] [CrossRef]
- Leinhase, I.; Rozanski, M.; Harhausen, D.; Thurman, J.M.; Schmidt, O.I.; Hossini, A.M.; Taha, M.E.; Rittirsch, D.; Ward, P.A.; Holers, V.M.; et al. Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: A randomized placebo-controlled study in mice. J. Neuroinflammation 2007, 4, 13. [Google Scholar] [CrossRef]
- Alawieh, X.A.; Langley, E.F.; Weber, X.S.; Adkins, X.D.; Tomlinson, X.S. Identifying the Role of Complement in Triggering Neuroinflammation after Traumatic Brain Injury. J. Neurosci. 2018, 14, 2519–2532. [Google Scholar] [CrossRef]
- Longhi, L.; Perego, C.; Ortolano, F.; Zanier, E.R.; Bianchi, P.; Stocchetti, N.; McIntosh, T.K.; De Simoni, M.G. C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit. Care Med. 2009, 37, 659–665. [Google Scholar] [CrossRef]
- Rancan, M.; Morganti-Kossmann, M.C.; Barnum, S.R.; Saft, S.; Schmidt, O.I.; Ertel, W.; Stahel, P.F. Central Nervous System-Targeted Complement Inhibition Mediates Neuroprotection after Closed Head Injury in Transgenic Mice. Br. J. Pharmacol. 2003, 23, 1070–1074. [Google Scholar] [CrossRef]
- Rich, M.C.; Keene, C.N.; Neher, M.D.; Johnson, K.; Yu, Z.-X.; Ganivet, A.; Holers, V.M.; Stahel, P.F. Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci. Lett. 2016, 617, 188–194. [Google Scholar] [CrossRef]
- Sewell, D.L.; Nacewicz, B.; Liu, F.; Macvilay, S.; Erdei, A.; Lambris, J.D.; Sandor, M.; Fabry, Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: Blocking effects on neutrophil extravasation by C5a receptor antagonist. J. Neuroimmunol. 2004, 155, 55–63. [Google Scholar] [CrossRef]
- Yang, S.; Nakamura, T.; Hua, Y.; Keep, R.F.; Younger, J.G.; He, Y.; Hoff, J.T.; Xi, G. The Role of Complement C3 in Intracerebral Hemorrhage-Induced Brain Injury. Br. J. Pharmacol. 2006, 26, 1490–1495. [Google Scholar] [CrossRef]
- Stahel, P.F.; Flierl, M.A.; Morgan, B.P.; Persigehl, I.; Stoll, C.; Conrad, C.; Touban, B.M.; Smith, W.R.; Beauchamp, K.; Schmidt, O.I.; et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J. Neuroinflammation 2009, 6, 2. [Google Scholar] [CrossRef]
- Fluiter, K.; Opperhuizen, A.L.; Morgan, B.P.; Baas, F.; Ramaglia, V. Inhibition of the Membrane Attack Complex of the Complement System Reduces Secondary Neuroaxonal Loss and Promotes Neurologic Recovery after Traumatic Brain Injury in Mice. J. Immunol. 2014, 192, 2339–2348. [Google Scholar] [CrossRef]
- Ruseva, M.M.; Ramaglia, V.; Morgan, B.P.; Harris, C.L. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 14319–14324. [Google Scholar] [CrossRef]
- De Blasio, D.; Fumagalli, S.; Longhi, L.; Orsini, F.; Palmioli, A.; Stravalaci, M.; Vegliante, G.; Zanier, E.R.; Bernardi, A.; Gobbi, M.; et al. Pharmacological inhibition of mannose-binding lectin ameliorates neurobehavioral dysfunction following experimental traumatic brain injury. Br. J. Pharmacol. 2016, 37, 938–950. [Google Scholar] [CrossRef]
- Longhi, L.; Orsini, F.; de Blasio, D.; Fumagalli, S.; Ortolano, F.; Locatelli, M.; Stocchetti, N.; de Simoni, M.-G. Mannose-Binding Lectin Is Expressed after Clinical and Experimental Traumatic Brain Injury and Its Deletion Is Protective. Crit. Care Med. 2014, 42, 1910–1918. [Google Scholar] [CrossRef]
- Yager, P.H.; You, Z.; Qin, T.; Kim, H.-H.; Takahashi, K.; Ezekowitz, A.B.; Stahl, G.L.; Carroll, M.C.; Whalen, M.J. Mannose Binding Lectin Gene Deficiency Increases Susceptibility to Traumatic Brain Injury in Mice. Br. J. Pharmacol. 2008, 28, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Osthoff, M.; Walder, B.; Delhumeau, C.; Trendelenburg, M.; Turck, N. Association of Lectin Pathway Protein Levels and Genetic Variants Early after Injury with Outcomes after Severe Traumatic Brain Injury: A Prospective Cohort Study. J. Neurotrauma 2017, 34, 2560–2566. [Google Scholar] [CrossRef]
- Kwon, B.K. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004, 4, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.H.; Lee, J.D.; Ruitenberg, M.J.; Woodruff, T.M. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin. Immunol. 2016, 28, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Rebhun, J.; Botvin, J. Complement elevation in spinal cord injury. Ann. Allergy 1980, 44, 287–288. [Google Scholar] [PubMed]
- Rebhun, J.; Madorsky, J.G.; Glovsky, M.M. Proteins of the complement system and acute phase reactants in sera of patients with spinal cord injury. Ann. Allergy 1991, 66, 335–338. [Google Scholar]
- Anderson, A.J.; Robert, S.; Huang, W.; Young, W.; Cotman, C.W. Activation of Complement Pathways after Contusion-Induced Spinal Cord Injury. J. Neurotrauma 2004, 21, 1831–1846. [Google Scholar] [CrossRef]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. From the Cover: A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Galvan, M.D.; Anderson, A.J. Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes In Vitro and In Vivo after spinal cord injury. J. Neuroinflammation 2008, 5, 26. [Google Scholar] [CrossRef]
- Törnqvist, E.; Liu, L.; Aldskogius, H.; Von Holst, H.; Svensson, M. Complement and clusterin in the injured nervous system. Neurobiol. Aging 1996, 17, 695–705. [Google Scholar] [CrossRef]
- Galvan, M.D.; Luchetti, S.; Burgos, A.M.; Nguyen, H.X.; Hooshmand, M.J.; Hamers, F.P.T.; Anderson, A.J. Deficiency in Complement C1q Improves Histological and Functional Locomotor Outcome after Spinal Cord Injury. J. Neurosci. 2008, 28, 13876–13888. [Google Scholar] [CrossRef]
- Guo, Q.; Li, S.; Liang, Y.; Zhang, Y.; Zhang, J.; Wen, C.; Lin, S.; Wang, H.; Su, B. Effects of C3 deficiency on inflammation and regeneration following spinal cord injury in mice. Neurosci. Lett. 2010, 485, 32–36. [Google Scholar] [CrossRef]
- Qiao, F.; Atkinson, C.; Song, H.; Pannu, R.; Singh, I.; Tomlinson, S. Complement Plays an Important Role in Spinal Cord Injury and Represents a Therapeutic Target for Improving Recovery following Trauma. Am. J. Pathol. 2006, 169, 1039–1047. [Google Scholar] [CrossRef][Green Version]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation 2020, 17, 1–25. [Google Scholar] [CrossRef]
- Tenner, A.J.; Stevens, B.; Woodruff, T.M. New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018, 102, 3–13. [Google Scholar] [CrossRef]
- Gao, L.-L.; Wu, T. The study of brain functional connectivity in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 1–7. [Google Scholar] [CrossRef]
- Sharma, S.; Deshmukh, R. Vinpocetine attenuates MPTP-induced motor deficit and biochemical abnormalities in Wistar rats. Neuroscience 2015, 286, 393–403. [Google Scholar] [CrossRef]
- McDonald, C.; Gordon, G.; Hand, A.; Walker, R.W.; Fisher, J.M. 200 Years of Parkinson’s disease: What have we learnt from James Parkinson? Age Ageing 2018, 47, 209–214. [Google Scholar] [CrossRef]
- Moore, J.W.; Schneider, S.A.; Schwingenschuh, P.; Moretto, G.; Bhatia, K.P.; Haggard, P. Dopaminergic medication boosts action-effect binding in Parkinson’s disease. Neuropsychologia 2010, 48, 1125–1132. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Chemistry of Mood and Emotion. Annu. Rev. Psychol. 1980, 31, 273–307. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and neurodegeneration in Parkinson’s disease. Park. Relat. Disord. 2004, 10, S3–S7. [Google Scholar] [CrossRef]
- Depboylu, C.; Schäfer, M.K.-H.; Arias-Carrión, O.; Oertel, W.H.; Weihe, E.; Höglinger, G.U. Possible Involvement of Complement Factor C1q in the Clearance of Extracellular Neuromelanin From the Substantia Nigra in Parkinson Disease. J. Neuropathol. Exp. Neurol. 2011, 70, 125–132. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Camp, D.M.; Conant, S.B. Complement activation in the Parkinson’s disease substantia nigra: An immunocytochemical study. J. Neuroinflammation 2006, 3, 29. [Google Scholar] [CrossRef]
- Yamada, M.; Iwatsubo, T.; Mizuno, Y.; Mochizuki, H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J. Neurochem. 2004, 91, 451–461. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Sheta, E.A.; Bryson, J.; Folsom, B.; Wilson, C.; Duty, J.; Yen, A.A.; Appel, S.H. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 342, 1034–1039. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, E.G.; McGeer, P.L. Therapeutic approaches to inflammation in neurodegenerative disease. Curr. Opin. Neurol. 2007, 20, 351–357. [Google Scholar] [CrossRef]
- Wang, X.-J.; Yan, Z.-Q.; Lu, G.-Q.; Stuart, S.; Chen, S.-D. Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: Role of microglia. Neurochem. Int. 2007, 50, 39–50. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Guo, Q.; Zhang, Y.; Wen, C.; Zou, Q.; Su, B. Complement 3-deficient mice are not protected against MPTP-induced dopaminergic neurotoxicity. Brain Res. 2007, 1178, 132–140. [Google Scholar] [CrossRef]
- Hou, L.; Wang, K.; Zhang, C.; Sun, F.; Che, Y.; Zhao, X.; Zhang, D.; Li, H.; Wang, Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 2018, 14, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms In Vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Tovar-Y-Romo, L.B.; Tapia, R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin. Ther. Targets 2007, 11, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Kjældgaard, A.-L.; Pilely, K.; Olsen, K.S.; Pedersen, S.W.; Lauritsen, A. Øberg; Møller, K.; Garred, P. Amyotrophic lateral sclerosis: The complement and inflammatory hypothesis. Mol. Immunol. 2018, 102, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Bahia, P.K.; Parks, T.A.; Stanford, K.R.; Mitchell, D.A.; Varma, S.; Stevens, S.M., Jr.; Taylor-Clark, T.E. The exceptionally high reactivity of Cys 621 is critical for electrophilic activation of the sensory nerve ion channel TRPA1. J. Gen. Physiol. 2016, 147, 451–465. [Google Scholar] [CrossRef]
- Donnenfeld, H.; Kascsak, R.; Bartfeld, H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J. Neuroimmunol. 1984, 6, 51–57. [Google Scholar] [CrossRef]
- Sta, M.; Sylva-Steenland, R.; Casula, M.; de Jong, J.; Troost, D.; Aronica, E.; Baas, F. Innate and adaptive immunity in amyotrophic lateral sclerosis: Evidence of complement activation. Neurobiol. Dis. 2011, 42, 211–220. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.; Baimbridge, K.; McGeer, E. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990, 526, 303–307. [Google Scholar] [CrossRef]
- Annunziata, P.; Volpi, N. High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 2009, 72, 61–64. [Google Scholar] [CrossRef]
- Mantovani, S.; Gordon, R.; Macmaw, J.; Pfluger, C.; Henderson, R.; Noakes, P.; McCombe, P.; Woodruff, T. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J. Neuroimmunol. 2014, 276, 213–218. [Google Scholar] [CrossRef]
- Yu, J.; Wang, N.; Qi, F.; Wang, X.; Zhu, Q.; Lu, Y.; Zhang, H.; Che, F.; Li, W. Serum ferritin is a candidate biomarker of disease aggravation in amyotrophic lateral sclerosis. Biomed. Rep. 2018, 9, 333–338. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Heath, P.R.; Holden, H.; Kasher, P.; Kirby, J.; Shaw, P.J. Microarray Analysis of the Cellular Pathways Involved in the Adaptation to and Progression of Motor Neuron Injury in the SOD1 G93A Mouse Model of Familial ALS. J. Neurosci. 2007, 27, 9201–9219. [Google Scholar] [CrossRef]
- Fukada, Y.; Yasui, K.; Kitayama, M.; Doi, K.; Nakano, T.; Watanabe, Y.; Nakashima, K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: Studies of the Leu126delTT mutation in SOD1. Brain Res. 2007, 1160, 1–10. [Google Scholar] [CrossRef]
- Heurich, B.; El Idrissi, N.B.; Donev, R.M.; Petri, S.; Claus, P.; Neal, J.; Morgan, B.P.; Ramaglia, V. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J. Neuroimmunol. 2011, 235, 104–109. [Google Scholar] [CrossRef]
- Lee, J.D.; Levin, S.C.; Willis, E.F.; Li, R.; Woodruff, T.M.; Noakes, P.G. Complement components are upregulated and correlate with disease progression in the TDP-43Q331K mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 2018, 15, 171. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, J.H.; Gil Hwang, S.; Gwag, B.J.; McKee, A.C.; Kowall, N.W.; Ryu, H.; Lim, D.-S.; Choi, E.-J. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2013, 110, 12066–12071. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Boillée, S.; Cleveland, D.W. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7319–7326. [Google Scholar] [CrossRef]
- Wang, H.A.; Lee, J.D.; Lee, K.M.; Woodruff, T.M.; Noakes, P.G. Complement C5a-C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Skelet. Muscle 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Costantini, K.J.; Crane, J.W.; Atkin, J.D.; Monk, P.N.; Taylor, S.M.; Noakes, P.G. The Complement Factor C5a Contributes to Pathology in a Rat Model of Amyotrophic Lateral Sclerosis. J. Immunol. 2008, 181, 8727–8734. [Google Scholar] [CrossRef]
- Chiu, I.M.; Phatnani, H.; Kuligowski, M.; Tapia, J.C.; Carrasco, M.A.; Zhang, M.; Maniatis, T.; Carroll, M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20960–20965. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Boillee, S.; Pozniak, C.; Khan, A.M.; McAlonis-Downes, M.; Lewcock, J.W.; Cleveland, D.W. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc. Natl. Acad. Sci. USA 2013, 110, E4385–E4392. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Hawksworth, O.A.; Li, R.; Balachandran, A.; Lee, J.D.; Sepehrband, F.; Kurniawan, N.; Jeanes, A.; Simmons, D.G.; Wolvetang, E.; et al. Complement C5aR1 Signaling Promotes Polarization and Proliferation of Embryonic Neural Progenitor Cells through PKCζ. J. Neurosci. 2017, 37, 5395–5407. [Google Scholar] [CrossRef]
- Lee, J.D.; Kumar, V.; Fung, J.N.T.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Lee, J.D.; Noakes, P.G. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc. Natl. Acad. Sci. USA 2013, 111, E3–E4. [Google Scholar] [CrossRef]
- Humayun, S.; Gohar, M.; Volkening, K.; Moisse, K.; Leystra-Lantz, C.; Mepham, J.; McLean, J.; Strong, M.J. The complement factor C5a receptor is upregulated in NFL−/− mouse motor neurons. J. Neuroimmunol. 2009, 210, 52–62. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Björkqvist, M.; Petersén, Å.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic Profiling of Plasma in Huntington’s Disease Reveals Neuroinflammatory Activation and Biomarker Candidates. J. Proteome Res. 2007, 6, 2833–2840. [Google Scholar] [CrossRef]
- Nayak, A.; Ansar, R.; Verma, S.K.; Bonifati, D.M.; Kishore, U. Huntington’s Disease: An Immune Perspective. Available online: https://www.hindawi.com/journals/nri/2011/563784/ (accessed on 8 February 2021).
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Leblhuber, F.; Walli, J.; Jellinger, K.; Tilz, G.P.; Widner, B.; Laccone, F.; Fuchs, D. Activated Immune System in Patients with Huntington’s Disease. Clin. Chem. Lab. Med. 1998, 36, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Larkin, P.B.; Muchowski, P.J. Genetic Deficiency of Complement Component 3 does not Alter Disease Progression in a Mouse Model of Huntington’s Disease. J. Huntington’s Dis. 2012, 1, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.P.; Portaccio, E.; Goretti, B.; Zipoli, V.; Hakiki, B.; Giannini, M.; Pastò, L.; Razzolini, L. Cognitive impairment in early stages of multiple sclerosis. Neurol. Sci. 2010, 31, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Reich, D.S.; Zackowski, K.M.; Gordon-Lipkin, E.M.; Smith, S.A.; Chodkowski, B.A.; Cutter, G.R.; Calabresi, P.A. Corticospinal Tract Abnormalities Are Associated with Weakness in Multiple Sclerosis. Am. J. Neuroradiol. 2008, 29, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Aeinehband, S.; Lindblom, R.P.F.; Al Nimer, F.; Vijayaraghavan, S.; Sandholm, K.; Khademi, M.; Olsson, T.; Nilsson, B.; Ekdahl, K.N.; Darreh-Shori, T.; et al. Complement Component C3 and Butyrylcholinesterase Activity Are Associated with Neurodegeneration and Clinical Disability in Multiple Sclerosis. PLoS ONE 2015, 10, e0122048. [Google Scholar] [CrossRef]
- Ingram, G.; Hakobyan, S.; Hirst, C.L.; Harris, C.L.; Loveless, S.; Mitchell, J.P.; Pickersgill, T.P.; Robertson, N.P.; Morgan, B.P. Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult. Scler. J. 2012, 18, 1401–1411. [Google Scholar] [CrossRef]
- Watkins, L.M.; Neal, J.W.; Loveless, S.; Michailidou, I.; Ramaglia, V.; Rees, M.I.; Reynolds, R.; Robertson, N.P.; Morgan, B.P.; Howell, O.W. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J. Neuroinflammation 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Long, Y.; Lu, Z.; Hu, X. Increased memory Th17 cells in patients with neuromyelitis optica and multiple sclerosis. J. Neuroimmunol. 2011, 234, 155–160. [Google Scholar] [CrossRef]
- Sanders, V.J.; Felisan, S.; Waddell, A.; Tourtellotte, W.W. Detection of Herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J. Neurovirology 1996, 2, 249–258. [Google Scholar] [CrossRef]
- Lindblom, R.P.; Aeinehband, S.; Ström, M.; Al Nimer, F.; Sandholm, K.; Khademi, M.; Nilsson, B.; Piehl, F.; Ekdahl, K.N. Complement Receptor 2 is increased in cerebrospinal fluid of multiple sclerosis patients and regulates C3 function. Clin. Immunol. 2016, 166–167, 89–95. [Google Scholar] [CrossRef]
- Farrokhi, M.; Dabirzadeh, M.; Dastravan, N.; Etemadifar, M.; Ghadimi, K.; Saadatpour, Z.; Rezaei, A. Mannose-binding Lectin Mediated Complement Pathway in Autoimmune Neurological Disorders. Iran. J. Allergy Asthma Immunol. 2016, 15, 251–256. [Google Scholar]
- Kwok, J.Y.; Vaida, F.; Augst, R.M.; Yu, D.Y.; Singh, K.K. Mannose binding lectin mediated complement pathway in multiple sclerosis. J. Neuroimmunol. 2011, 239, 98–100. [Google Scholar] [CrossRef][Green Version]
- Tatomir, A.; Talpos-Caia, A.; Anselmo, F.; Kruszewski, A.M.; Boodhoo, D.; Rus, V.; Rus, H. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol. Res. 2017, 65, 1103–1109. [Google Scholar] [CrossRef]
- Compston, D.A.S.; Morgan, B.P.; Campbell, A.K.; Wilkins, P.; Cole, G.; Thomas, N.D.; Jasani, B. Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol. Appl. Neurobiol. 1989, 15, 307–316. [Google Scholar] [CrossRef]
- Ingram, G.; Loveless, S.; Howell, O.W.; Hakobyan, S.; Dancey, B.; Harris, C.L.; Robertson, N.P.; Neal, J.W.; Morgan, B.P. Complement activation in multiple sclerosis plaques: An immunohistochemical analysis. Acta Neuropathol. Commun. 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Storch, M.K.; Piddlesden, S.; Haltia, M.; Iivanainen, M.; Morgan, P.; Lassmann, H. Multiple sclerosis: In Situ evidence for antibody and complement-mediated demyelination. Ann. Neurol. 1998, 43, 465–471. [Google Scholar] [CrossRef]
- Schwab, C.; McGeer, P.L. Complement Activated C4d Immunoreactive Oligodendrocytes Delineate Small Cortical Plaques in Multiple Sclerosis. Exp. Neurol. 2002, 174, 81–88. [Google Scholar] [CrossRef]
- Michailidou, I.; Jongejan, A.; Vreijling, J.P.; Georgakopoulou, T.; de Wissel, M.B.; Wolterman, R.A.; Ruizendaal, P.; Klar-Mohamad, N.; Grootemaat, A.E.; Picavet, D.I.; et al. Systemic inhibition of the membrane attack complex impedes neuroinflammation in chronic relapsing experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Rus, H.; Cudrici, C.; David, S.; Niculescu, F. The complement system in central nervous system diseases. Autoimmunity 2006, 39, 395–402. [Google Scholar] [CrossRef]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef]
- Michailidou, I.; Willems, J.G.P.; Kooi, E.-J.; van Eden, C.; Gold, S.M.; Geurts, J.J.G.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.-K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020, 52, 167–182.e7. [Google Scholar] [CrossRef]
- Hammond, J.W.; Bellizzi, M.J.; Ware, C.; Qiu, W.Q.; Saminathan, P.; Li, H.; Luo, S.; Ma, S.A.; Li, Y.; Gelbard, H.A. Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain Behav. Immun. 2020, 87, 739–750. [Google Scholar] [CrossRef]
- Nataf, S.; Carroll, S.L.; Wetsel, R.A.; Szalai, A.J.; Barnum, S.R. Attenuation of Experimental Autoimmune Demyelination in Complement-Deficient Mice. J. Immunol. 2000, 165, 5867–5873. [Google Scholar] [CrossRef]
- Xu, W.; Li, R.; Dai, Y.; Wu, A.; Wang, H.; Cheng, C.; Qiu, W.; Lu, Z.; Zhong, X.; Shu, Y.; et al. IL-22 secreting CD4+ T cells in the patients with neuromyelitis optica and multiple sclerosis. J. Neuroimmunol. 2013, 261, 87–91. [Google Scholar] [CrossRef]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Complement in multiple sclerosis: Its role in disease and potential as a biomarker. Clin. Exp. Immunol. 2009, 155, 128–139. [Google Scholar] [CrossRef]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Löscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58, 27–38. [Google Scholar] [CrossRef]
- Cerri, C.; Caleo, M.; Bozzi, Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy. Epilepsy Res. 2017, 136, 77–83. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; de Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Murugan, M.; Wu, L.-J. Microglia-Neuron Communication in Epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q.J. Epilepsy and brain inflammation. Exp. Neurol. 2013, 244, 11–21. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Kooij, G.; Frenkel, D.; Georgopoulos, S.; Monsonego, A.; Janigro, D. Inflammatory events at blood-brain barrier in neuroinflammatory and neurodegenerative disorders: Implications for clinical disease. Epilepsia 2012, 53, 45–52. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular Intercommunication between the Complement and Coagulation Systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef]
- Chen, M.; Edwards, S.R.; Reutens, D.C. Complement in the Development of Post-Traumatic Epilepsy: Prospects for Drug Repurposing. J. Neurotrauma 2020, 37, 692–705. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Aronica, E.; Boer, K.; van Vliet, E.A.; Redeker, S.; Baayen, J.C.; Spliet, W.G.M.; van Rijen, P.C.; Troost, D.; da Silva, F.H.L.; Wadman, W.J.; et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiol. Dis. 2007, 26, 497–511. [Google Scholar] [CrossRef]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E.; Breit, T.; Rauwerda, H.; da Silva, F.H.L.; Wadman, W.J. Potential New Antiepileptogenic Targets Indicated by Microarray Analysis in a Rat Model for Temporal Lobe Epilepsy. J. Neurosci. 2006, 26, 11083–11110. [Google Scholar] [CrossRef]
- Jamali, S.; Salzmann, A.; Perroud, N.; Ponsole-Lenfant, M.; Cillario, J.; Roll, P.; Roeckel-Trevisiol, N.; Crespel, A.; Balzar, J.; Schlachter, K.; et al. Functional Variant in Complement C3 Gene Promoter and Genetic Susceptibility to Temporal Lobe Epilepsy and Febrile Seizures. PLoS ONE 2010, 5, e12740. [Google Scholar] [CrossRef]
- Kharatishvili, I.; Shan, Z.Y.; She, D.T.; Foong, S.; Kurniawan, N.D.; Reutens, D.C. MRI changes and complement activation correlate with epileptogenicity in a mouse model of temporal lobe epilepsy. Brain Struct. Funct. 2014, 219, 683–706. [Google Scholar] [CrossRef]
- Liguori, C.; Romigi, A.; Izzi, F.; Placidi, F.; Nuccetelli, M.; Cordella, A.; Bernardini, S.; Biagio, M.N. Complement system dysregulation in patients affected by Idiopathic Generalized Epilepsy and the effect of antiepileptic treatment. Epilepsy Res. 2017, 137, 107–111. [Google Scholar] [CrossRef]
- Wyatt, S.K.; Witt, T.; Barbaro, N.M.; Cohen-Gadol, A.A.; Brewster, A.L. Enhanced classical complement pathway activation and altered phagocytosis signaling molecules in human epilepsy. Exp. Neurol. 2017, 295, 184–193. [Google Scholar] [CrossRef]
- Schartz, N.D.; Wyatt-Johnson, S.K.; Price, L.R.; Colin, S.A.; Brewster, A.L. Status epilepticus triggers long-lasting activation of complement C1q-C3 signaling in the hippocampus that correlates with seizure frequency in experimental epilepsy. Neurobiol. Dis. 2018, 109, 163–173. [Google Scholar] [CrossRef]
- Choi, J.; Koh, S. Role of Brain Inflammation in Epileptogenesis. Yonsei Med. J. 2008, 49, 1–18. [Google Scholar] [CrossRef]
- Xiong, Z.-Q.; Qian, W.; Suzuki, K.; McNamara, J.O. Formation of Complement Membrane Attack Complex in Mammalian Cerebral Cortex Evokes Seizures and Neurodegeneration. J. Neurosci. 2003, 23, 955–960. [Google Scholar] [CrossRef]
- He, F.; Liu, B.; Meng, Q.; Sun, Y.; Wang, W.; Wang, C. Modulation of miR-146a/complement factor H-mediated inflammatory responses in a rat model of temporal lobe epilepsy. Biosci. Rep. 2016, 36, e00433. [Google Scholar] [CrossRef]
- Benson, M.J.; Thomas, N.K.; Talwar, S.; Hodson, M.P.; Lynch, J.W.; Woodruff, T.M.; Borges, K. A novel anticonvulsant mechanism via inhibition of complement receptor C5ar1 in murine epilepsy models. Neurobiol. Dis. 2015, 76, 87–97. [Google Scholar] [CrossRef]
- Schartz, N.D.; Sommer, A.L.; Colin, S.A.; Mendez, L.B.; Brewster, A.L. Early treatment with C1 esterase inhibitor improves weight but not memory deficits in a rat model of status epilepticus. Physiol. Behav. 2019, 212, 112705. [Google Scholar] [CrossRef]
- Brewster, A.L.; Lugo, J.N.; Patil, V.V.; Lee, W.L.; Qian, Y.; Vanegas, F.; Anderson, A.E. Rapamycin Reverses Status Epilepticus-Induced Memory Deficits and Dendritic Damage. PLoS ONE 2013, 8, e57808. [Google Scholar] [CrossRef]
- Dachet, F.; Bagla, S.; Keren-Aviram, G.; Morton, A.; Balan, K.; Saadat, L.; Valyi-Nagy, T.; Kupsky, W.; Song, F.; Dratz, E.; et al. Predicting novel histopathological microlesions in human epileptic brain through transcriptional clustering. Brain 2014, 138, 356–370. [Google Scholar] [CrossRef]
- Schartz, N.D.; Herr, S.A.; Madsen, L.; Butts, S.J.; Torres, C.; Mendez, L.B.; Brewster, A.L. Spatiotemporal profile of Map2 and microglial changes in the hippocampal CA1 region following pilocarpine-induced status epilepticus. Sci. Rep. 2016, 6, 24988. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Sekar, A.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; et al. Schizophrenia risk from complex variation of complement component 4. Nat. Cell Biol. 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Fagan, K.; Crider, A.; Ahmed, A.O.; Pillai, A. Complement C3 Expression Is Decreased in Autism Spectrum Disorder Subjects and Contributes to Behavioral Deficits in Rodents. Mol. Neuropsychiatry 2017, 3, 19–27. [Google Scholar] [CrossRef]
- Howes, O.D.; Murray, R.M. Schizophrenia: An integrated sociodevelopmental-cognitive model. Lancet 2014, 383, 1677–1687. [Google Scholar] [CrossRef]
- Cannon, M.; Jones, P.B.; Murray, R.M. Obstetric Complications and Schizophrenia: Historical and Meta-Analytic Review. Am. J. Psychiatry 2002, 159, 1080–1092. [Google Scholar] [CrossRef]
- Cannon, T.D.; Chung, Y.; He, G.; Sun, D.; Jacobson, A.; van Erp, T.G.; McEwen, S.; Addington, J.; Bearden, C.E.; Cadenhead, K.; et al. Progressive Reduction in Cortical Thickness as Psychosis Develops: A Multisite Longitudinal Neuroimaging Study of Youth at Elevated Clinical Risk. Biol. Psychiatry 2015, 77, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Glantz, L.A.; Lewis, D.A. Decreased Dendritic Spine Density on Prefrontal Cortical Pyramidal Neurons in Schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Howes, O.D.; McCutcheon, R.; Owen, M.J.; Murray, R.M. The Role of Genes, Stress, and Dopamine in the Development of Schizophrenia. Biol. Psychiatry 2017, 81, 9–20. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-Analysis of Cytokine Alterations in Schizophrenia: Clinical Status and Antipsychotic Effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Upthegrove, R.; Manzanares-Teson, N.; Barnes, N.M. Cytokine function in medication-naive first episode psychosis: A systematic review and meta-analysis. Schizophr. Res. 2014, 155, 101–108. [Google Scholar] [CrossRef]
- Arakelyan, A.; Zakharyan, R.; Khoyetsyan, A.; Poghosyan, D.; Aroutiounian, R.; Mrazek, F.; Petrek, M.; Boyajyan, A. Functional characterization of the complement receptor type 1 and its circulating ligands in patients with schizophrenia. BMC Clin. Pathol. 2011, 11, 10. [Google Scholar] [CrossRef]
- Hakobyan, S.; Boyajyan, A.; Sim, R.B. Classical pathway complement activity in schizophrenia. Neurosci. Lett. 2005, 374, 35–37. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Q.; Li, N.; Wang, F.; Xiang, H.; Zhang, Z.; Su, Y.; Huang, Y.; Zhang, S.; Zhao, G.; et al. Plasma levels of Th17-related cytokines and complement C3 correlated with aggressive behavior in patients with schizophrenia. Psychiatry Res. 2016, 246, 700–706. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Kenis, G.; de Jong, R.; Smith, R.S.; Meltzer, H.Y. In vivo immunomodulatory effects of clozapine in schizophrenia. Schizophr. Res. 1997, 26, 221–225. [Google Scholar] [CrossRef]
- Mayilyan, K.R.; Weinberger, D.R.; Sim, R.B. The complement system in schizophrenia. Drug News Perspect. 2008, 21, 200–210. [Google Scholar] [CrossRef]
- Spivak, B.; Radwan, M.; Brandon, J.; Baruch, Y.; Stawski, M.; Tyano, S.; Weizman, A. Reduced total complement haemolytic activity in schizophrenic patients. Psychol. Med. 1993, 23, 315–318. [Google Scholar] [CrossRef]
- Severance, E.G.; Gressitt, K.L.; Buka, S.L.; Cannon, T.D.; Yolken, R.H. Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophr. Res. 2014, 159, 14–19. [Google Scholar] [CrossRef]
- Hon-Cheong Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nat. Cell Biol. 2014, 511, 421–427. [CrossRef]
- The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nat. Cell Biol. 2009, 460, 748–752. [Google Scholar] [CrossRef]
- The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium; Ripke, S.; Sanders, A.R.; Kendler, K.S.; Levinson, D.F.; Sklar, P.; Holmans, P.A.; Lin, D.-Y.; Duan, J.; Ophoff, R.A.; et al. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef]
- Donohoe, G.; Walters, J.; Hargreaves, A.; Rose, E.; Morris, D.; Fahey, C.; Bellini, S.; Cummins, E.; Giegling, I.; Hartmann, A.; et al. Neuropsychological effects of theCSMD1genome-wide associated schizophrenia risk variant rs10503253. Genes Brain Behav. 2013, 12, 203–209. [Google Scholar] [CrossRef]
- Escudero-Esparza, A.; Kalchishkova, N.; Kurbasic, E.; Jiang, W.G.; Blom, A.M. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 2013, 27, 5083–5093. [Google Scholar] [CrossRef]
- Koiliari, E.; Roussos, P.; Pasparakis, E.; Lencz, T.; Malhotra, A.; Siever, L.J.; Giakoumaki, S.G.; Bitsios, P. The CSMD1 genome-wide associated schizophrenia risk variant rs10503253 affects general cognitive ability and executive function in healthy males. Schizophr. Res. 2014, 154, 42–47. [Google Scholar] [CrossRef]
- Prasad, K.M.; Chowdari, K.V.; D’Aiuto, L.A.; Iyengar, S.; Stanley, J.A.; Nimgaonkar, V.L. Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl. Psychiatry 2018, 8, 134. [Google Scholar] [CrossRef]
- Ishii, T.; Hattori, K.; Miyakawa, T.; Watanabe, K.; Hidese, S.; Sasayama, D.; Ota, M.; Teraishi, T.; Hori, H.; Yoshida, S.; et al. Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem. Biophys. Res. Commun. 2018, 497, 683–688. [Google Scholar] [CrossRef]
- Allswede, D.M.; Zheutlin, A.B.; Chung, Y.; Anderson, K.; Hultman, C.M.; Ingvar, M.; Cannon, T.D. Complement Gene Expression Correlates with Superior Frontal Cortical Thickness in Humans. Neuropsychopharmacology 2017, 43, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Nimgaonkar, V.L.; Prasad, K.M.; Chowdari, K.V.; Severance, E.G.; Yolken, R.H. The complement system: A gateway to gene–environment interactions in schizophrenia pathogenesis. Mol. Psychiatry 2017, 22, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.J.; Pouget, J.G.; Zai, C.C.; Kennedy, J.L. The complement system in schizophrenia: Where are we now and what’s next? Mol. Psychiatry 2020, 25, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016, 113, E7049–E7058. [Google Scholar] [CrossRef]
- Ashwood, P.; van de Water, J. Is autism an autoimmune disease? Autoimmun. Rev. 2004, 3, 557–562. [Google Scholar] [CrossRef]
- Crider, A.; Thakkar, R.; Ahmed, A.O.; Pillai, A. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol. Autism 2014, 5, 46. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Sertié, A.L. Autism spectrum disorders: An updated guide for genetic counseling. Einstein (São Paulo) 2017, 15, 233–238. [Google Scholar] [CrossRef]
- Meltzer, A.; Van De Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Immune Dysfunction: Epidemiological Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144. [Google Scholar] [CrossRef]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef]
- Warren, R.P.; Singh, V.K.; Cole, P.; Odell, J.D.; Pingree, C.B.; Warren, W.L.; White, E. Increased frequency of the null allele at the complement C4b locus in autism. Clin. Exp. Immunol. 1991, 83, 438–440. [Google Scholar] [CrossRef]
- Corbett, B.A.; Kantor, A.B.; Schulman, H.; Walker, W.L.; Lit, L.; Ashwood, P.; Rocke, D.M.; Sharp, F.R. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol. Psychiatry 2006, 12, 292–306. [Google Scholar] [CrossRef]
- Momeni, N.; Brudin, L.; Behnia, F.; Nordström, B.; Yosefi-Oudarji, A.; Sivberg, B.; Joghataei, M.T.; Persson, B.L. High Complement Factor I Activity in the Plasma of Children with Autism Spectrum Disorders. Autism Res. Treat. 2011, 2012, 1–6. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).