
Identification of Novel Fusion Transcripts in High Grade Serous Ovarian Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Fusion Transcript Differences between Fallopian Tube and HGSC Samples
2.2. Prediction Model of HGSC Using Fusion Transcript Data
2.3. Association of Fusion Transcripts with Overall Survival
2.4. Association of Fusion Transcripts with Chemo-Response
2.5. Validation of Fusion Transcript Detection with FusionCatcher, DNA Sequencing
2.6. RT-PCR Validation
3. Discussion
4. Materials and Methods
4.1. Patient Inclusion Criteria
4.2. Clinical Data
4.3. RNA Purification and Whole Transcriptome Sequencing
4.4. DNA Extraction from Normal Fallopian Tubes
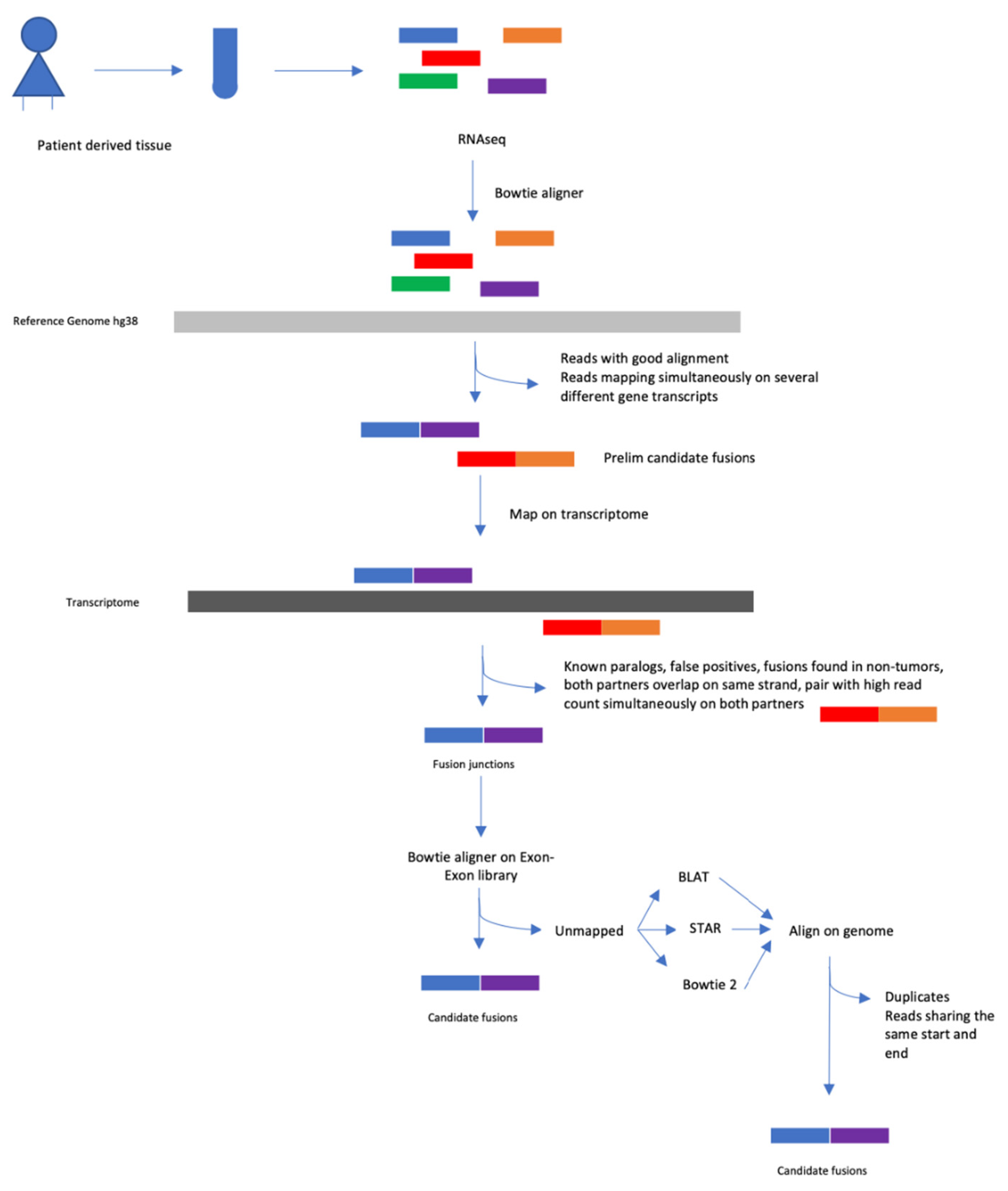
4.5. Fusion Transcript Detection
4.6. Statistical Analysis
4.7. Validation of Fusion Transcript Detection with FusionCatcher
4.8. RT-PCR Validation of Fusion Transcripts
4.8.1. RNA Purification
4.8.2. RT-PCR
| Fusion Transcript | PCR Primer Sequences | Tm * |
| CC2D1A-CPNE8 | For: ATGCACAAGAGGAAAGGAC Rev: GCAGGTGATGGCTTGATT | 59.3 °C 59.7 °C |
| FAM98B-FRMD5 | For: GTGCTGGACACACTGGAG Rev: TGCCGGGAAAGCAACAT | 61.5 °C 61.6 °C |
| AC004475.1-PRPF6 | For: GCAGCAGATGTACGACATGA Rev: CTTCAGGTTCTTCCAGCTCAA | 61.7 °C 61.7 °C |
| AUTS2-INO80C | For: CGGCAGAAGAGGACATCATT Rev: CAGGTTCTTCCCAGGTTCTGTT | 63.8 °C 61.5 °C |
4.8.3. Sequence Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fusion Transcript | Product | Predicted Effect |
|---|---|---|
| AC004475.1--PRPF6 | Protein coding | Inframe |
| Protein coding | ||
| AC007952.4--RN7SL2 | lncRNA | Unknown |
| SBP | ||
| AF235103.3--ZNF250 | lncRNA | Unknown |
| Protein coding | ||
| ARL17A--KANSL1 | Protein coding | Unknown |
| Protein coding | ||
| AUTS2--INO80C | Protein coding | Inframe |
| Protein coding | ||
| CC2D1A--CPNE8 | Protein coding | Inframe |
| Protein coding | ||
| FAM98B--FRMD5 | Protein coding | Frameshift |
| Protein coding | ||
| NRIP--AJ009632.2 | Protein coding | Unknown |
| lncRNA | ||
| Z68871.1--LINC00630 | lncRNA | Unknown |
| lncRNA | ||
| ZBTB8OS--AC090627.1 | Protein coding | Unknown |
| Novel transcript |
References
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; Depont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Network, T.C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Kangaspeska, S.; Hultsch, S.; Edgren, H.; Nicorici, D.; Murumägi, A.; Kallioniemi, O. Reanalysis of RNA-Sequencing Data Reveals Several Additional Fusion Genes with Multiple Isoforms. PLoS ONE 2012, 7, e48745. [Google Scholar] [CrossRef]
- Soller, M.J.; Isaksson, M.; Elfving, P.; Soller, W.; Lundgren, R.; Panagopoulos, I. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer 2006, 45, 717–719. [Google Scholar] [CrossRef]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes (Basel) 2020, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Mano, H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Smebye, M.L.; Agostini, A.; Johannessen, B.; Thorsen, J.; Davidson, B.; Tropé, C.G.; Heim, S.; Skotheim, R.I.; Micci, F. Involvement of DPP9 in gene fusions in serous ovarian carcinoma. BMC Cancer 2017, 17, 642. [Google Scholar] [CrossRef] [PubMed]
- Jonna, S.; Feldman, R.A.; Swensen, J.; Gatalica, Z.; Korn, W.M.; Borghaei, H.; Ma, P.C.; Nieva, J.J.; Spira, A.I.; Vanderwalde, A.M.; et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin. Cancer Res. 2019, 25, 4966–4972. [Google Scholar] [CrossRef] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earp, M.A.; Raghavan, R.; Li, Q.; Dai, J.; Winham, S.J.; Cunningham, J.M.; Natanzon, Y.; Kalli, K.R.; Hou, X.; Weroha, S.J.; et al. Characterization of fusion genes in common and rare epithelial ovarian cancer histologic subtypes. Oncotarget 2017, 8, 46891–46899. [Google Scholar] [CrossRef]
- Yin, L.; Li, W.; Xu, A.; Shi, H.; Wang, K.; Yang, H.; Wang, R.; Peng, B. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. EBioMedicine 2020, 51, 102596. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-D.; Deng, L.; Hu, S.-Y.; Zhang, F.-L.; Li, D.-Q. SH3BGRL2 exerts a dual function in breast cancer growth and metastasis and is regulated by TGF-β1. Am. J. Cancer Res. 2020, 10, 1238–1254. [Google Scholar]
- Popow, J.; Jurkin, J.; Schleiffer, A.; Martinez, J. Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature 2014, 511, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2019, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- López-Nieva, P.; Fernández-Navarro, P.; Graña-Castro, O.; Andrés-León, E.; Santos, J.; Villa-Morales, M.; Cobos-Fernández, M.Á.; González-Sánchez, L.; Malumbres, M.; Salazar-Roa, M.; et al. Detection of novel fusion-transcripts by RNA-Seq in T-cell lymphoblastic lymphoma. Sci. Rep. 2019, 9, 5179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.X.; Yang, X.; Ning, S.; Wang, L.; Wang, K.; Zhang, Y.; Yuan, F.; Li, F.; Zhuo, D.D.; Tang, L.; et al. Identification of KANSARL as the first cancer predisposition fusion gene specific to the population of European ancestry origin. Oncotarget 2017, 8, 50594–50607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhard, D.S.; Wagner, L.; Feingold, E.A.; Shenmen, C.M.; Grouse, L.H.; Schuler, G.; Klein, S.L.; Old, S.; Rasooly, R.; Good, P.; et al. The Status, Quality, and Expansion of the NIH Full-Length cDNA Project: The Mammalian Gene Collection (MGC). Genome Res. 2004, 14, 2121–2127. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Cayrou, C.; Huang, R.; Lane, W.S.; Cote, J.; Lucchesi, J.C. A Human Protein Complex Homologous to the Drosophila MSL Complex Is Responsible for the Majority of Histone H4 Acetylation at Lysine 16. Mol. Cell. Biol. 2005, 25, 9175–9188. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.31–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Akter, K.A.; Mansour, M.A.; Hyodo, T.; Senga, T. FAM98A associates with DDX1-C14orf166-FAM98B in a novel complex involved in colorectal cancer progression. Int. J. Biochem. Cell Biol. 2017, 84, 1–13. [Google Scholar] [CrossRef]
- Hu, J.; Niu, M.; Li, X.; Lu, D.; Cui, J.; Xu, W.; Li, G.; Zhan, J.; Zhang, H. FERM domain-containing protein FRMD5 regulates cell motility via binding to integrin β5 subunit and ROCK1. FEBS Lett. 2014, 588, 4348–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Pei, X.; Zhan, J.; Hu, J.; Yu, Y.; Zhang, H. FERM-containing protein FRMD5 is a p120-catenin interacting protein that regulates tumor progression. FEBS Lett. 2012, 586, 3044–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashkin, S.R.; Graff, R.E.; Kachuri, L.; Thai, K.K.; Alexeeff, S.E.; Blatchins, M.A.; Cavazos, T.B.; Corley, D.A.; Emami, N.C.; Hoffman, J.D.; et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 2020, 11, 4423. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xia, J.; Jia, P.; Pao, W.; Zhao, Z. Application of next generation sequencing to human gene fusion detection: Computational tools, features and perspectives. Brief. Bioinform. 2013, 14, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Salzman, J.; Marinelli, R.J.; Wang, P.L.; Green, A.E.; Nielsen, J.S.; Nelson, B.H.; Drescher, C.W.; Brown, P.O. ESRRA-C11orf20 is a recurrent gene fusion in serous ovarian carcinoma. PLoS Biol. 2011, 9, e1001156. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Coarfa, C.; Rajapakshe, K.; Hawkins, S.M.; Matzuk, M.M.; Milosavljevic, A.; Yen, L. CDKN2D-WDFY2 is a cancer-specific fusion gene recurrent in high-grade serous ovarian carcinoma. PLoS Genet. 2014, 10, e1004216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newtson, A.M.; Devor, E.J.; Gonzalez Bosquet, J. Prediction of Epithelial Ovarian Cancer Outcomes With Integration of Genomic Data. Clin. Obstet. Gynecol. 2020, 63, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 15, 11.14.11–11.14.19. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Bosquet, J.; Newtson, A.M.; Chung, R.K.; Thiel, K.W.; Ginader, T.; Goodheart, M.J.; Leslie, K.K.; Smith, B.J. Prediction of chemo-response in serous ovarian cancer. Mol. Cancer 2016, 15, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005, 23, 7332–7341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Olsen, T.K.; Panagopoulos, I.; Gorunova, L.; Micci, F.; Andersen, K.; Kilen Andersen, H.; Meling, T.R.; Due-Tonnessen, B.; Scheie, D.; Heim, S.; et al. Novel fusion genes and chimeric transcripts in ependymal tumors. Genes Chromosomes Cancer 2016, 55, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tsai, W.H.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.; Kim, S.; Ma, T.; Chang, T.Y.; Priedigkeit, N.M.; et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016, 44, e47. [Google Scholar] [CrossRef]





| FusionCatcher | AUC | Specificity | Accuracy | NPV | PPV |
|---|---|---|---|---|---|
| AP3D1--ARHGDIA | 1 | 1 | 0.998 | 0.998 | 1 |
| ARHGAP1--CKAP5 | 1 | 1 | 0.998 | 0.998 | 1 |
| ARL17A--KANSL1 | 0.55 | 0.22 | 0.321 | 0.844 | 0.175 |
| BTBD10--TEAD1 | 1 | 1 | 0.998 | 0.998 | 1 |
| CC2D1A--CPNE8 | 1 | 1 | 0.998 | 0.998 | 1 |
| CHTOP--PCAT1 | 1 | 1 | 0.998 | 0.998 | 1 |
| DOT1L--GCGR | 1 | 1 | 0.998 | 0.998 | 1 |
| FAM20C--AC093627.4 | 1 | 1 | 0.998 | 0.998 | 1 |
| FAM98B--FRMD5 | 1 | 1 | 0.998 | 0.998 | 1 |
| FBXO34--SORCS3 | 1 | 1 | 0.998 | 0.998 | 1 |
| GRIN2A--C16ORF72 | 1 | 1 | 0.998 | 0.998 | 1 |
| INPP5B--PLEKHO1 | 1 | 1 | 0.998 | 0.998 | 1 |
| LUC7L--AXIN1 | 1 | 1 | 0.998 | 0.998 | 1 |
| MAGED2--ZFAT | 1 | 1 | 0.998 | 0.998 | 1 |
| MECOM--AC116337.3 | 1 | 1 | 0.998 | 0.998 | 1 |
| NFE2L1--PNPO | 1 | 1 | 0.998 | 0.998 | 1 |
| NFKBIB--TEAD1 | 1 | 1 | 0.998 | 0.998 | 1 |
| NRIP1--AJ009632.2 | 1 | 1 | 0.998 | 0.998 | 1 |
| PACS1--HAUS3 | 1 | 1 | 0.998 | 0.998 | 1 |
| PCAT1--C1ORF210 | 1 | 1 | 0.998 | 0.998 | 1 |
| PGM2L1--POLD3 | 1 | 1 | 0.998 | 0.998 | 1 |
| PSPC1--ZMYM5 | 0.50 | 0.20 | 0.231 | 0.948 | 0.052 |
| RB1CC1--LINC02091 | 1 | 1 | 0.998 | 0.998 | 1 |
| SMARCA4--ZNF700 | 1 | 1 | 0.998 | 0.998 | 1 |
| TMCC1--CD96 | 1 | 1 | 0.998 | 0.998 | 1 |
| TOGARAM1--FANCM | 1 | 1 | 0.998 | 0.998 | 1 |
| TRAPPC3--MAP7D1 | 1 | 1 | 0.998 | 0.998 | 1 |
| TRMT1--CPA4 | 1 | 1 | 0.998 | 0.998 | 1 |
| UBA2--RAD51B | 1 | 1 | 0.998 | 0.998 | 1 |
| UBE2F--LRRFIP1 | 1 | 1 | 0.998 | 0.998 | 1 |
| ZNF609--SNX1 | 1 | 1 | 0.998 | 0.998 | 1 |
| HGSC Patients | HR | 95% CI | p-Value | |||
|---|---|---|---|---|---|---|
| N = 103 | ||||||
| Age | (mean) | 59.8 | 1.01 | 0.99, 1.03 | 0.164 | |
| BMI | (mean) | 27.2 | 1.00 | 0.97, 1.03 | 0.764 | |
| Preop CA-125 | (mean) | 2413.6 | 1.00 | 0.99, 1.00 | 0.488 | |
| Charlson Comorbidity Index | 1–3 | 17 | 1.14 | 1.01, 1.31 | 0.044 * | |
| 4–6 | 64 | |||||
| >6 | 18 | |||||
| FIGO Stage | 2 | 3 | 2.72 | 0, N/A | 0.995 | |
| 3 | 68 | |||||
| 4 | 25 | |||||
| Disease in Upper abdomen (Other than Omentum) by Imaging | Yes | Large Bowel (N = 4) | 63 | 1.60 | 1.02, 2.50 | 0.039 * |
| Porta—Hepatis (N = 4) | ||||||
| Mesenteric Mets (N = 4) | ||||||
| Other (N = 26) | ||||||
| No | 40 | |||||
| Disease in the Chest by Imaging | Yes | Chest (N = 5) | 7 | 1.11 | 0.44, 2.79 | 0.813 |
| Pleural effusion (N = 5) | ||||||
| No | 96 | |||||
| Grade | 2 | 21 | 1.30 | 0.82, 2.07 | 0.270 | |
| 3 | 67 | |||||
| Residual disease after surgery | Microscopic | 20 | 0.59 | 0.32, 1.09 | 0.093 | |
| Macroscopic | 82 | |||||
| Optimal (<1 cm) | 66 | 1.11 | 0.71, 1.73 | 0.639 | ||
| Suboptimal (>1 cm) | 36 | |||||
| Removal of Pelvic LN | Yes | 17 | 1.83 | 0.27, 1.09 | 0.088 | |
| No | 86 | |||||
| Removal of Para-Aortic LN | Yes | 10 | 0.41 | 0.15, 1.11 | 0.080 | |
| No | 93 | |||||
| Surgery of large bowel | Yes | 29 | 1.43 | 0.91, 2.26 | 0.123 | |
| No | 74 | |||||
| Surgical complexity score ** | Low | 52 | 1.58 | 0.56, 4.43 | 0.381 | |
| Intermediate | 47 | |||||
| High | 4 | |||||
| Neoadjuvant Chemotherapy | Yes | 13 | 2.11 | 1.16, 3.83 | 0.015 * | |
| No | 88 | |||||
| Number of Cycles delivered | < 6 | 15 | 0.96 | 0.87, 1.07 | 0.476 | |
| ≥6 | 87 | |||||
| Dose Dense Chemotherapy | Yes | 3 | 0.60 | 0.15, 2.46 | 0.480 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newtson, A.; Reyes, H.; Devor, E.J.; Goodheart, M.J.; Bosquet, J.G. Identification of Novel Fusion Transcripts in High Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 4791. https://doi.org/10.3390/ijms22094791
Newtson A, Reyes H, Devor EJ, Goodheart MJ, Bosquet JG. Identification of Novel Fusion Transcripts in High Grade Serous Ovarian Cancer. International Journal of Molecular Sciences. 2021; 22(9):4791. https://doi.org/10.3390/ijms22094791
Chicago/Turabian StyleNewtson, Andreea, Henry Reyes, Eric J. Devor, Michael J. Goodheart, and Jesus Gonzalez Bosquet. 2021. "Identification of Novel Fusion Transcripts in High Grade Serous Ovarian Cancer" International Journal of Molecular Sciences 22, no. 9: 4791. https://doi.org/10.3390/ijms22094791
APA StyleNewtson, A., Reyes, H., Devor, E. J., Goodheart, M. J., & Bosquet, J. G. (2021). Identification of Novel Fusion Transcripts in High Grade Serous Ovarian Cancer. International Journal of Molecular Sciences, 22(9), 4791. https://doi.org/10.3390/ijms22094791