
Solvates of New Arylpiperazine Salicylamide Derivative-a Multi-Technique Approach to the Description of 5 HTR Ligand Structure and Interactions


Abstract
:1. Introduction
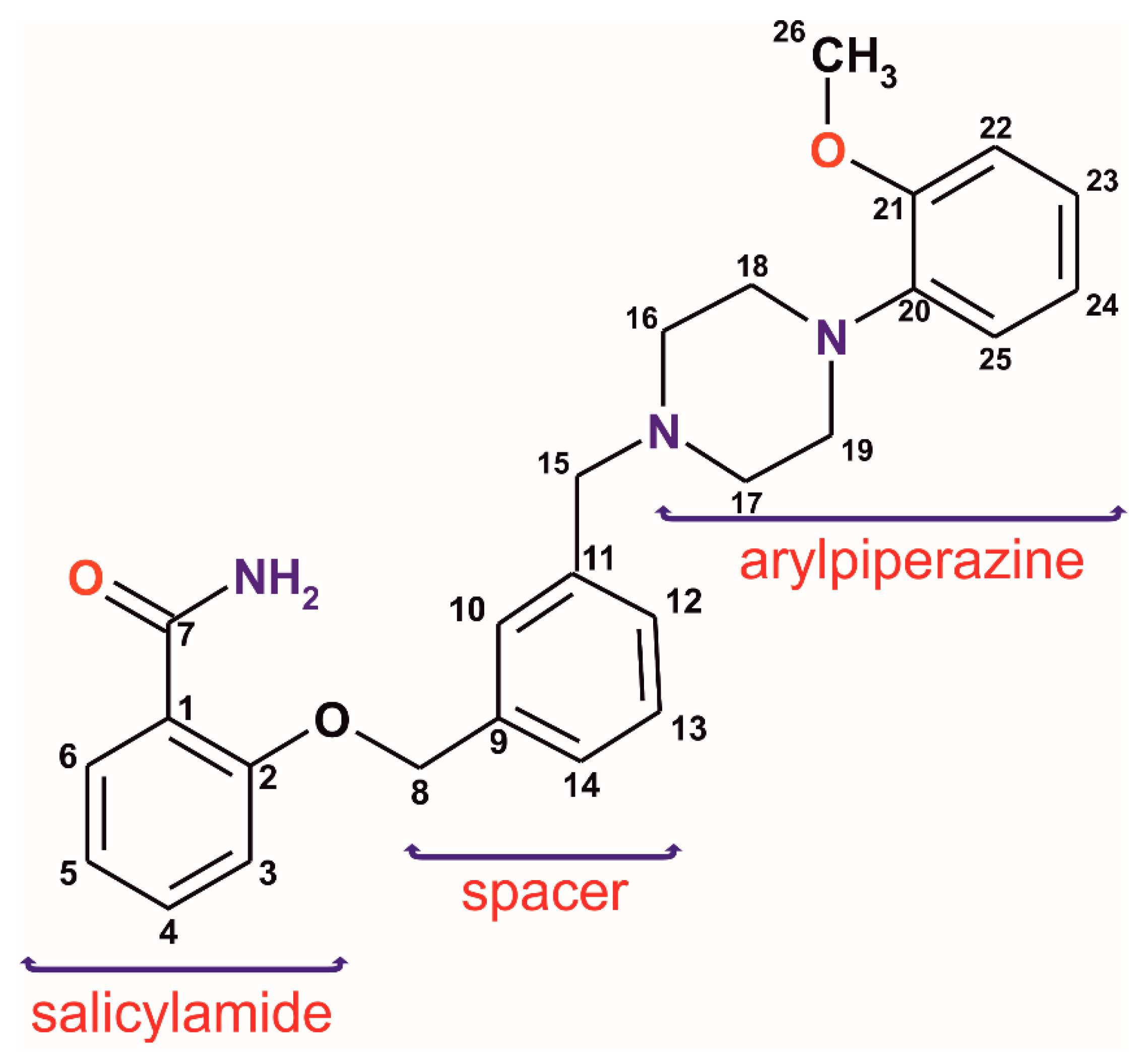
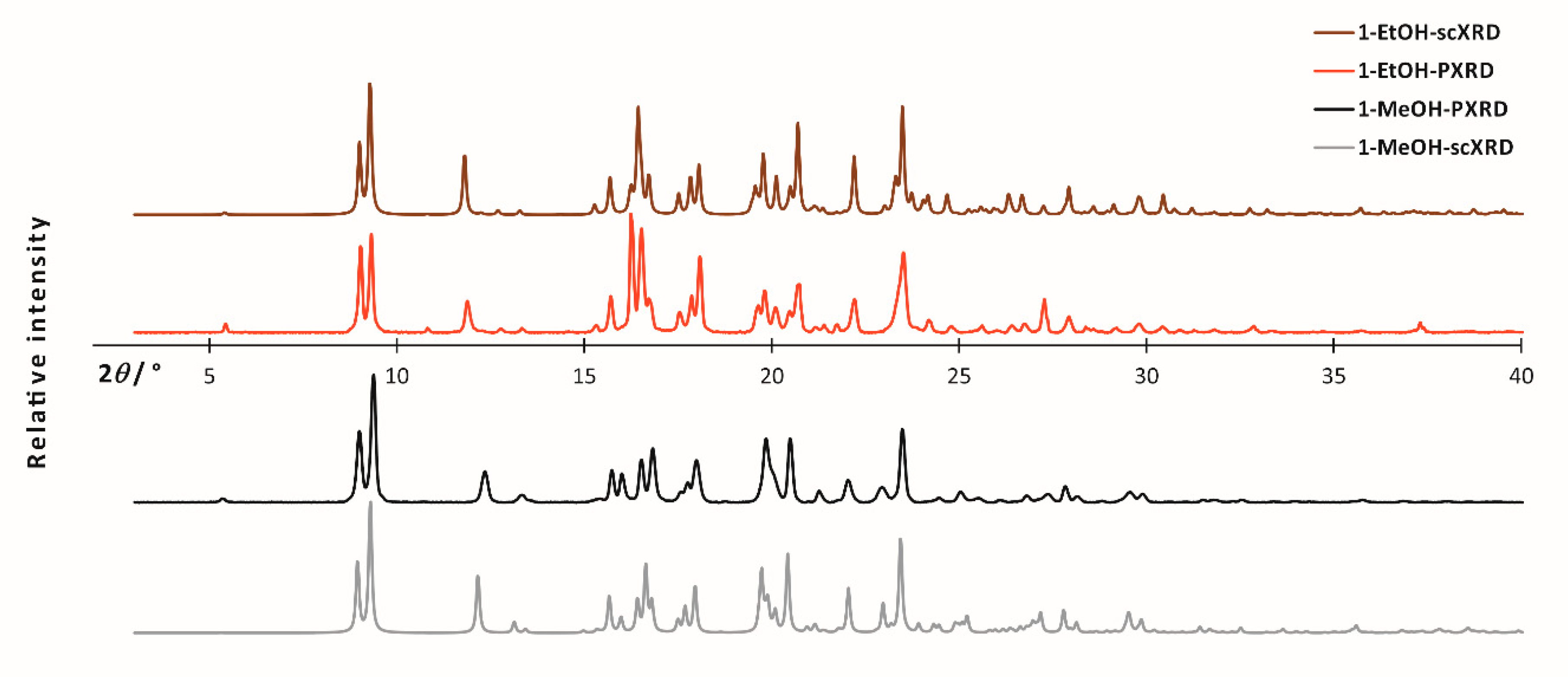
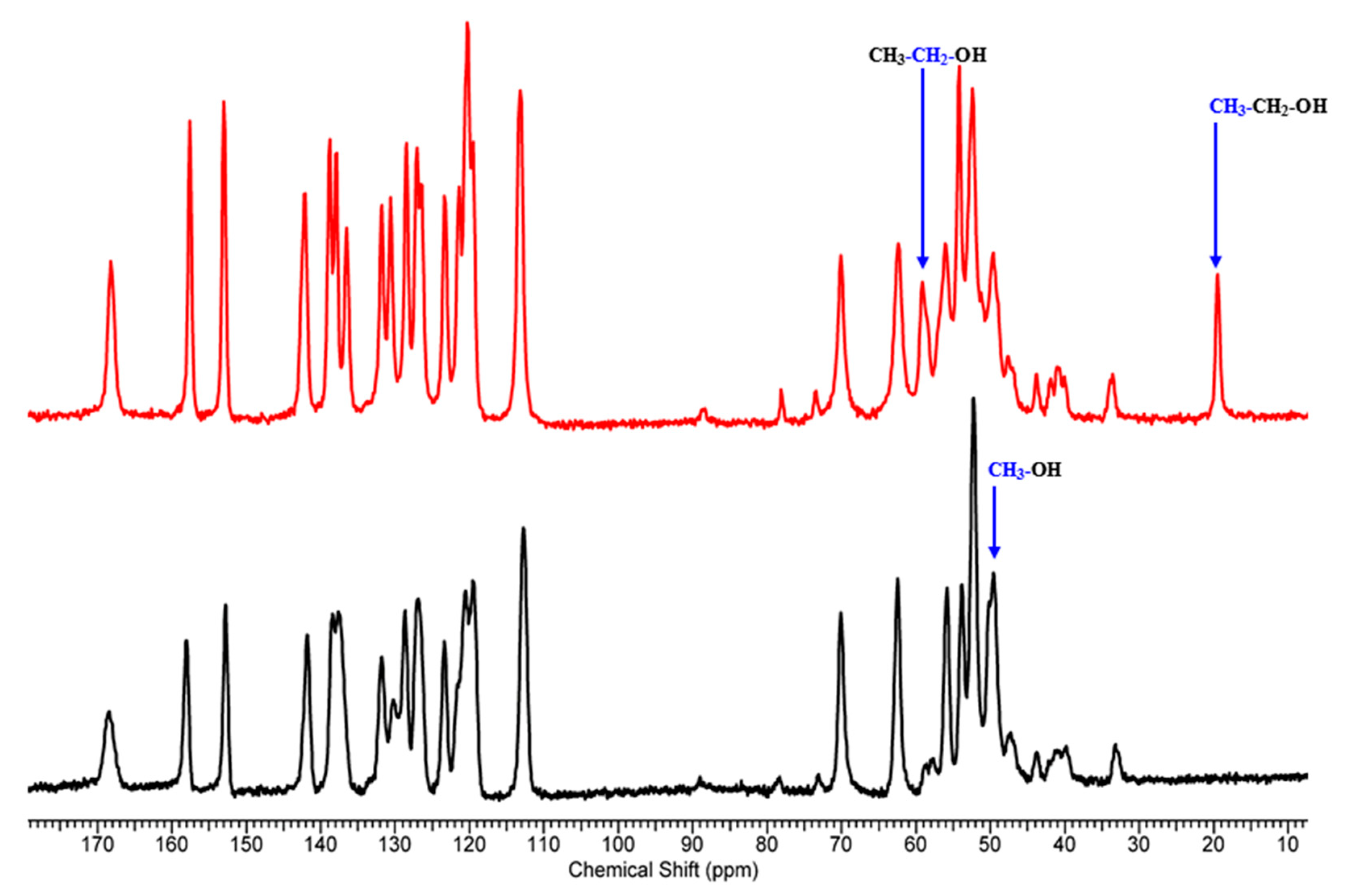
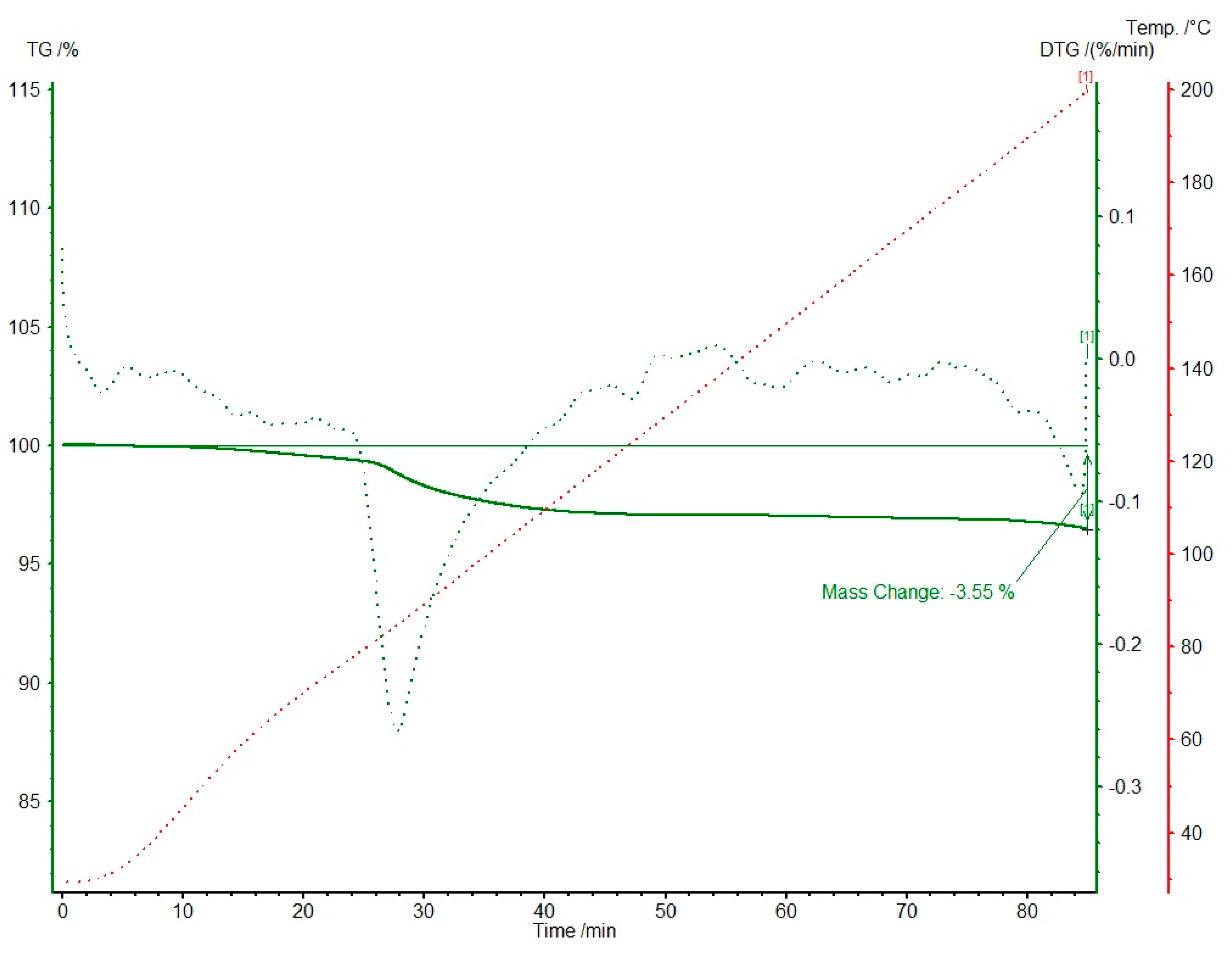
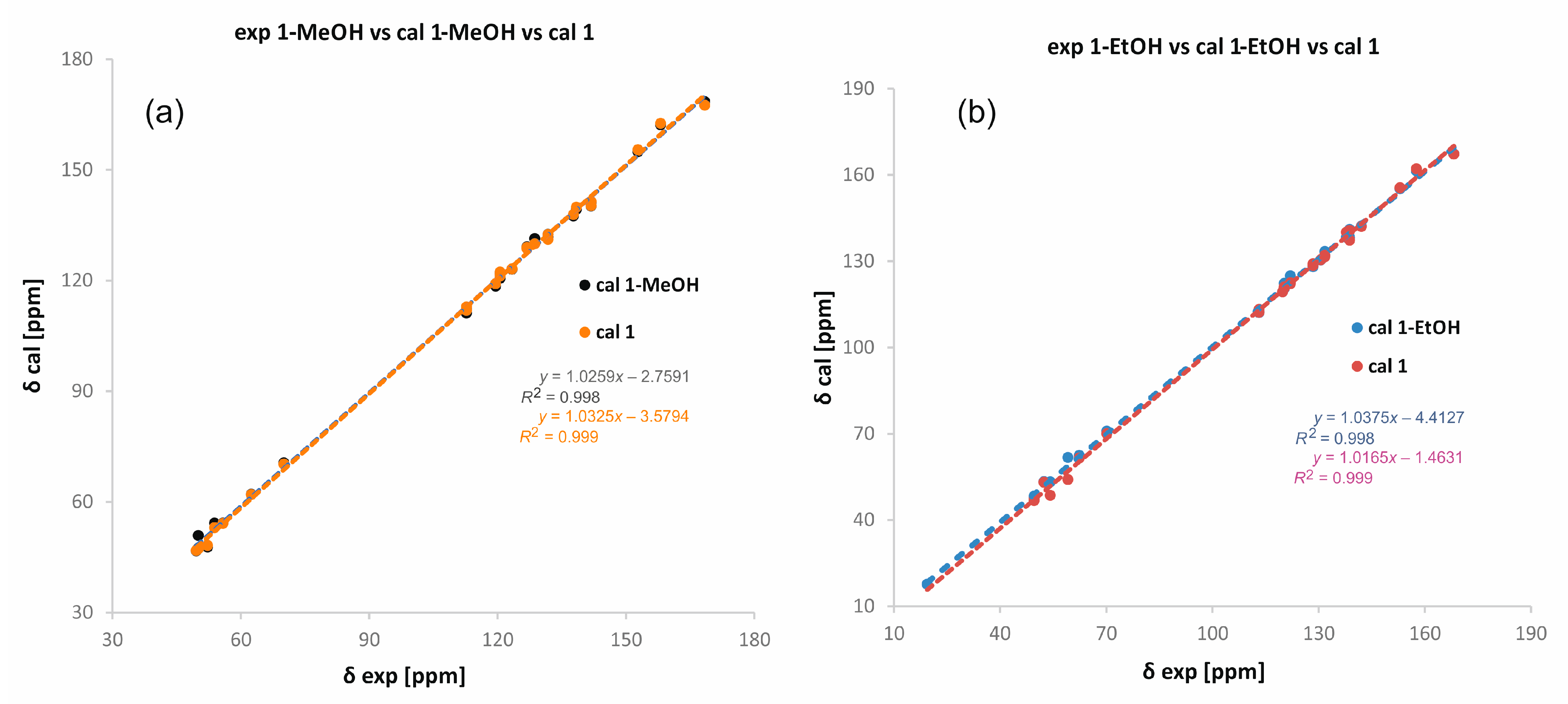
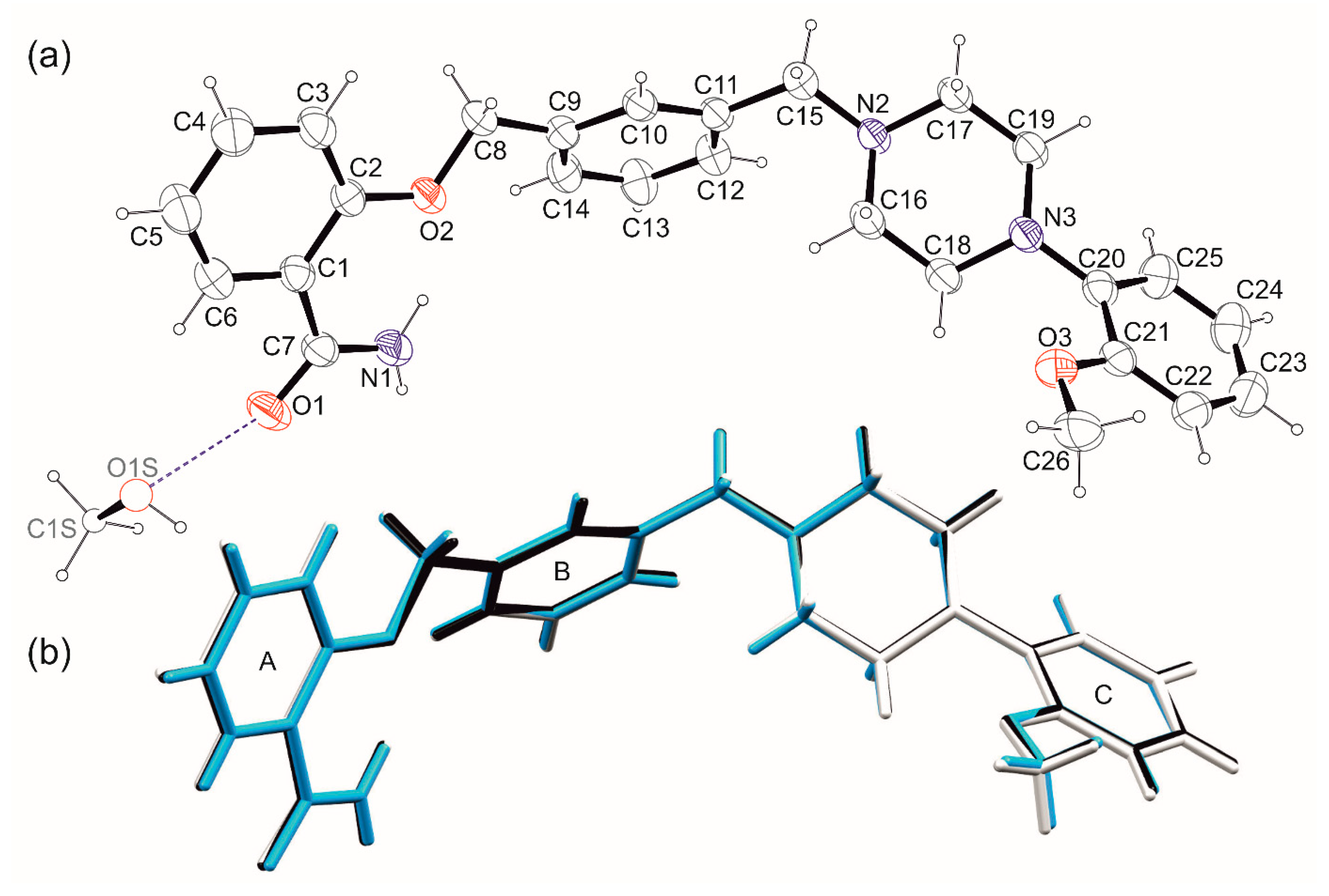
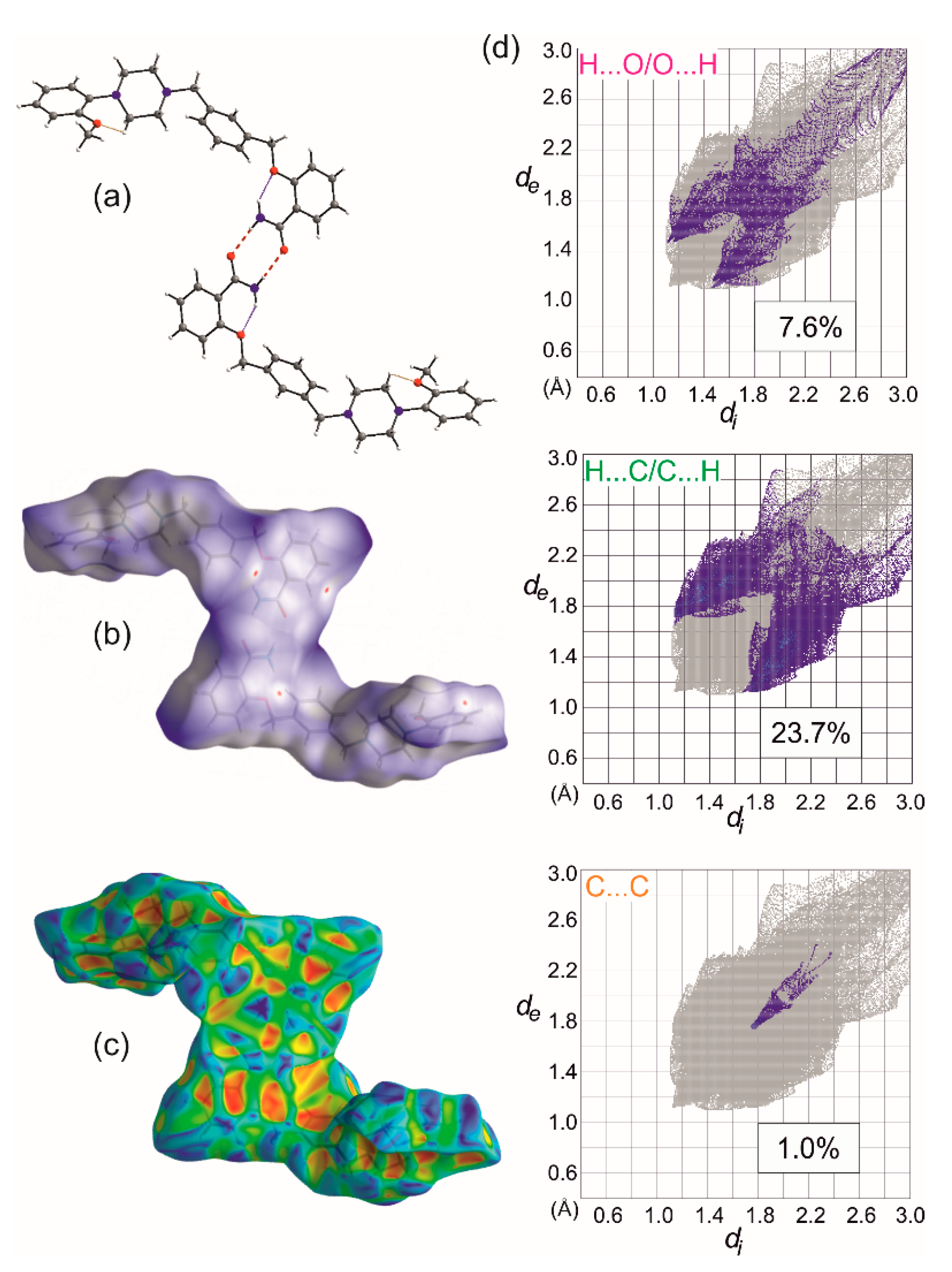
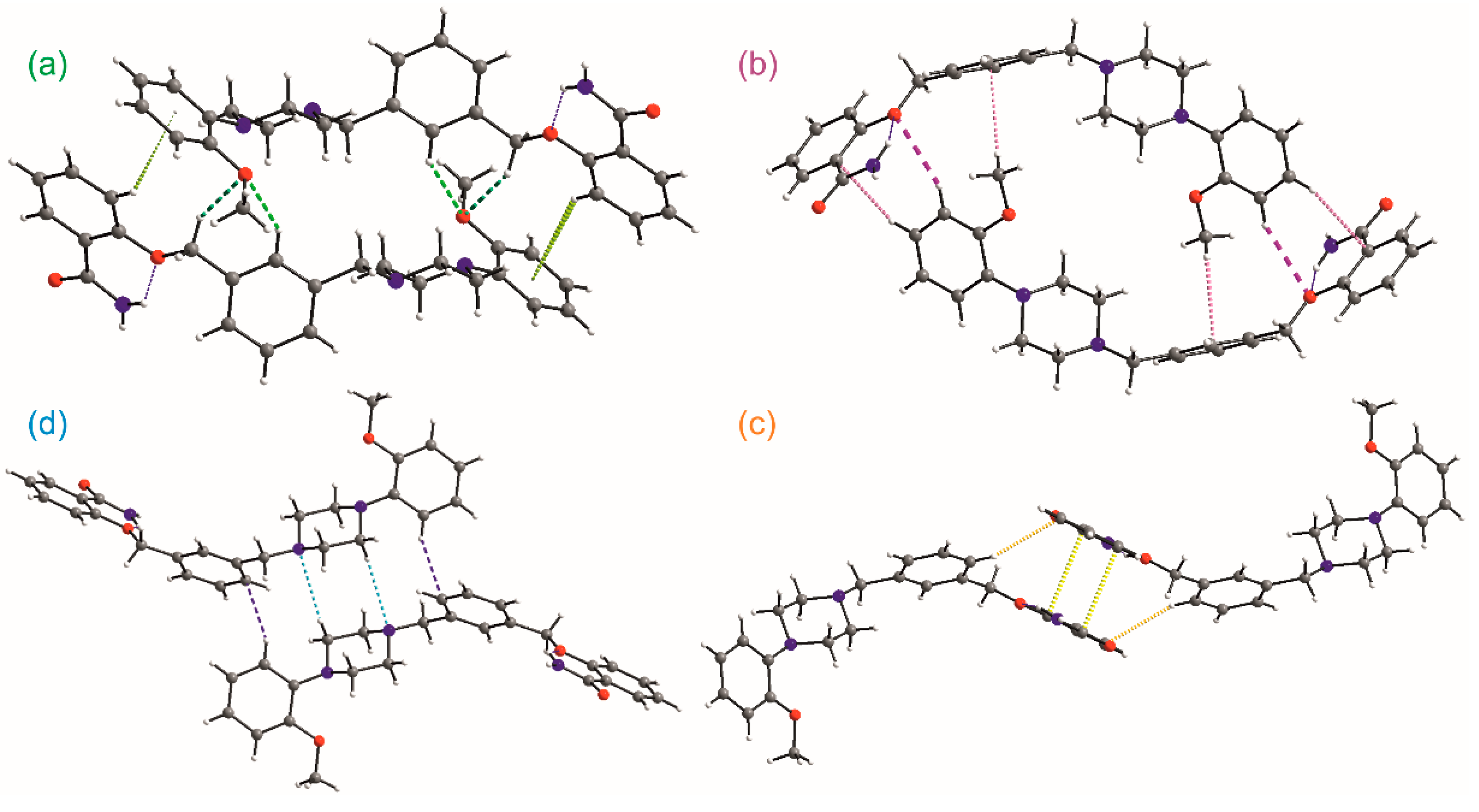
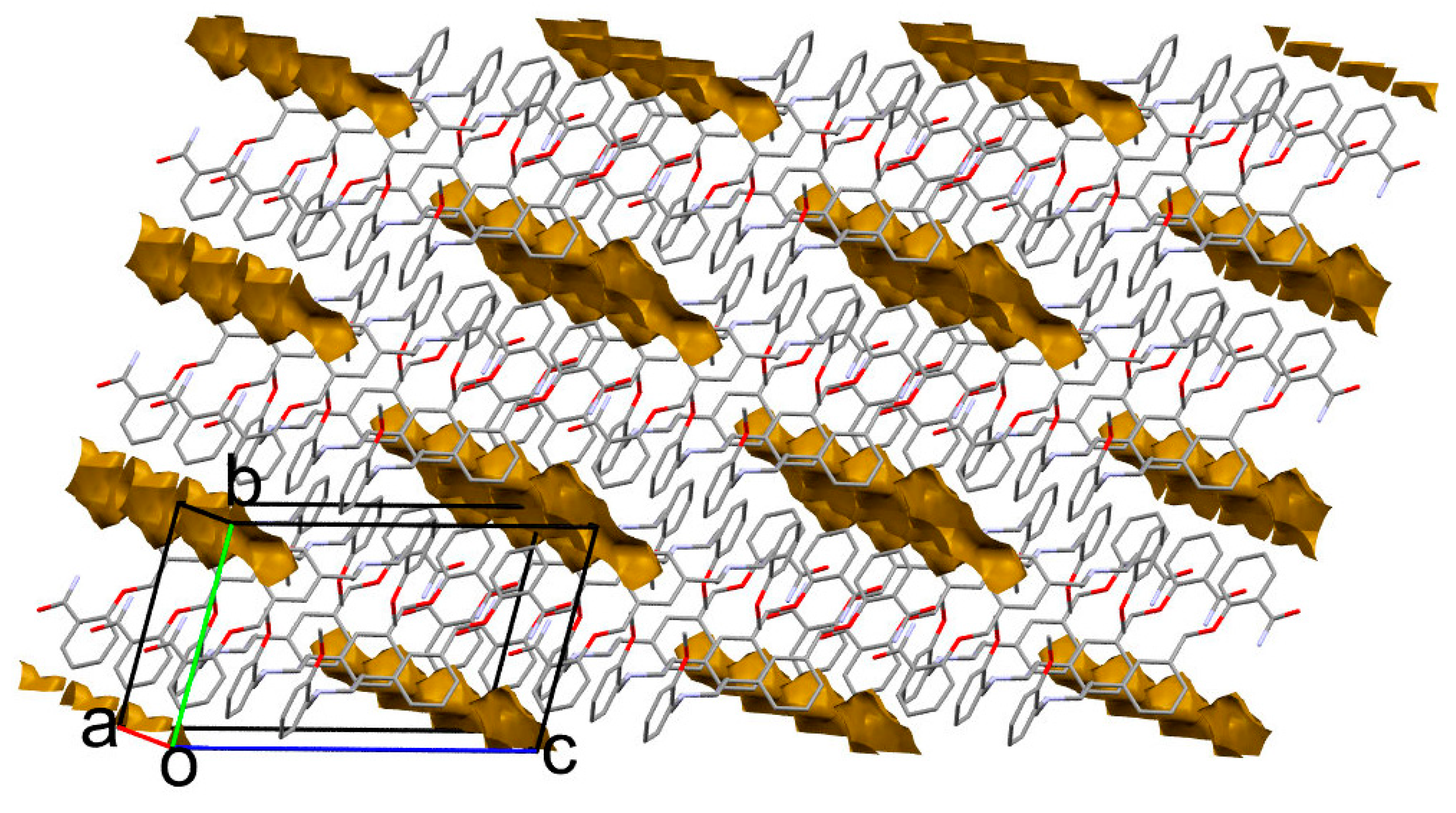
2. Results
3. Materials and Methods
3.1. Sample Preparation
3.2. Solid-State Nuclear Magnetic Resonance (ssNMR)
3.3. X-ray Diffraction
3.4. Thermal Analysis
3.5. GIPAW DFT Calculations
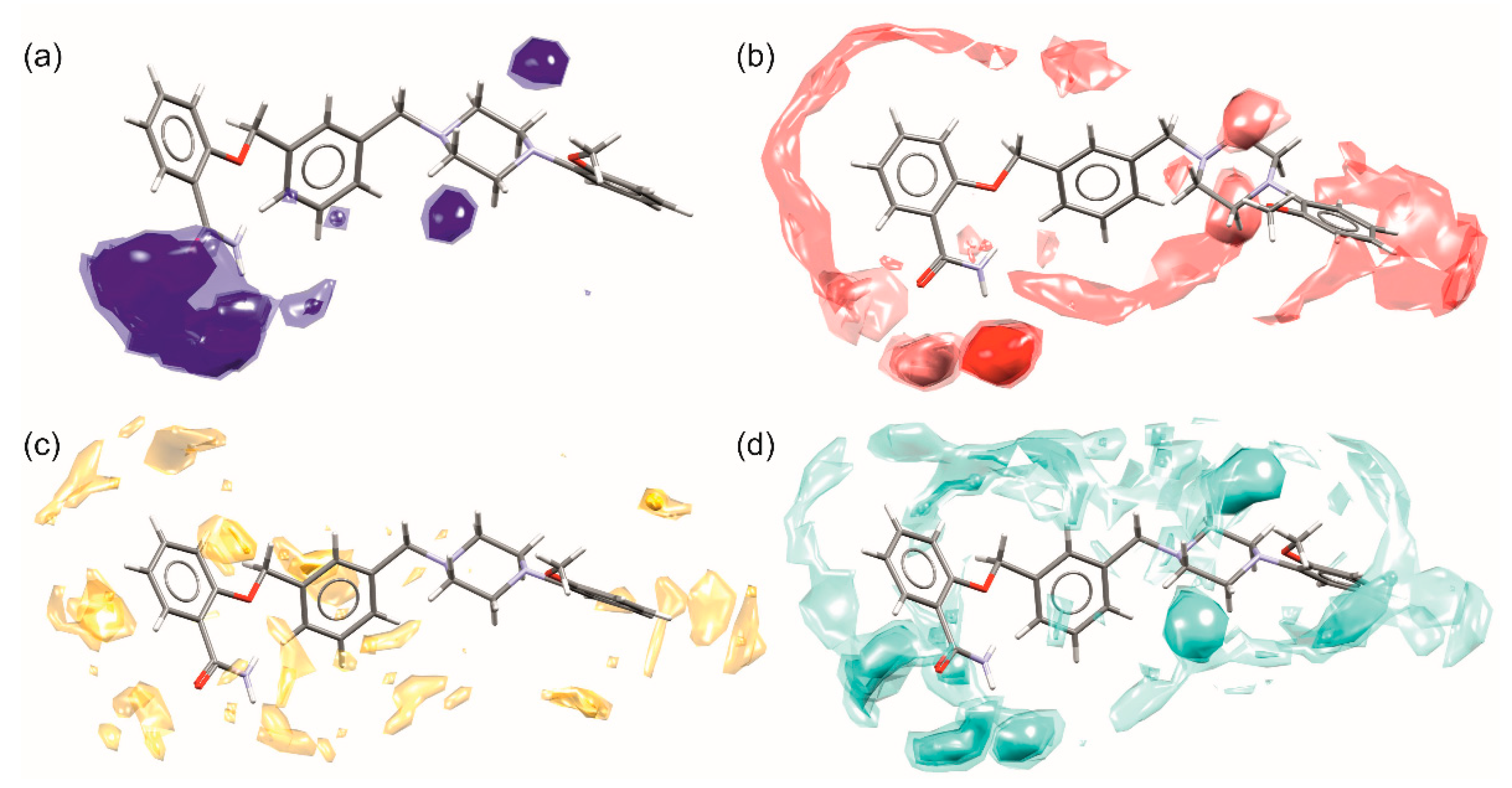
3.6. Hirshfeld Surface Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hilfiker, R. Polymorphism in the Pharmaceutical Industry; WILEY-VCH Verlag GmbH &, Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Generally Recognized as Safe (GRAS); U.S. FDA: College Park, MD, USA, 2021. Available online: https://www.fda.gov/food/food-ingredients-packaging/generally-recognized-safe-gras (accessed on 9 June 2019).
- Zhao, J.; Cao, Y.; Zhang, L. Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 2020, 18, 417–426. [Google Scholar] [CrossRef]
- Brink, A.; Helliwell, J.R. Why is interoperability between the two fields of chemical crystallography and protein crystallography so difficult? IUCrJ 2019, 6, 788–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wu, M.B.; Zhang, R.H.; Chen, Z.J.; Hua, C.; Lin, J.; Yang, L.R. Advances in Computational Structure-Based Drug Design and Application in Drug Discovery. Curr. Top. Med. Chem. 2016, 16, 901–916. [Google Scholar] [CrossRef]
- Kowalski, P.; Jaśkowska, J.; Bojarski, A.J.; Duszyńska, B. The synthesis of cyclic and acyclic long-chain arylpiperazine derivatives of salicylamide as serotonin receptor ligands. J. Heterocycl. Chem. 2008, 45, 209–214. [Google Scholar] [CrossRef]
- Kowalski, P.; Jaśkowska, J.; Bojarski, A.J.; Duszyńska, B.; Kołaczkowski, M. Evaluation of 1-arylpiperazine derivative of salicylamides as the 5-HT1A and 5-HT7 serotonin receptor ligands. J. Heterocycl. Chem. 2011, 48, 192–198. [Google Scholar] [CrossRef]
- Pindelska, E.; Madura, I.D.; Szeleszczuk, Ł.; Żeszko, A.; Jaśkowska, J.; Marek, P.H.; Kolodziejski, W. Alkyl Spacer Length and Protonation Induced Changes in Crystalline Psychoactive Arylpiperazine Derivatives: Single-Crystal X-ray, Solid-State NMR, and Computational Studies. Cryst. Growth Des. 2016, 16, 63716380. [Google Scholar] [CrossRef]
- Kowalski, P.; Mitka, K.; Jaśkowska, J.; Bojarski, A.J.; Duszyńska, B. New arypiperazines with flexible vs partly constrained linker as serotonin 5-HT1A/5-HT7 receptor ligands. Arch. Pharm. 2013, 346, 339–348. [Google Scholar] [CrossRef]
- Jaśkowska, J.; Kowalski, P.; Bucki, A.; Kołaczkowski, M. Synthesis and evaluation of novel flexible and partly constrained long-chain arylpiperazine derivatives of salicylamide as serotonin receptors ligands. In Proceedings of the 4th Congress of the European Association for Chemical and Molecular Sciences, Prague, Czech Republic, 26–30 October 2012. [Google Scholar]
- Nowak, M.; Kołaczkowski, M.; Pawłowski, M.; Bojarski, A.J. Homology modeling of the serotonin 5-HT1A receptor using automated docking of bioactive compounds with defined geometry. J. Med. Chem. 2006, 49, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.C.; Salum, L.B.; Honório, K.M.; Andricopulo, A.D.; da Silva, A.B. Pharmacophore-based 3D QSAR studies on a series of high affinity 5-HT1A receptor ligands. Eur. J. Med. Chem. 2010, 45, 1508–1514. [Google Scholar] [CrossRef]
- Kołaczkowski, M.; Nowak, M.; Pawłowski, M.; Bojarski, A.J. Receptor-based pharmacophores for serotonin 5-HT7R antagonists-implications to selectivity. J. Med. Chem. 2006, 49, 6732–6741. [Google Scholar] [CrossRef] [PubMed]
- Rague, A.; Tidgewell, K. Pharmacophore Comparison and Development of Recently Discovered Long Chain Arylpiperazine and Sulfonamide Based 5-HT7 Ligands. Mini Rev. Med. Chem. 2018, 18, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Day, G.M. Combining forces: Complementary techniques brought together to determine tricky crystal structures. Acta Cryst. 2020, 76, 294–295. [Google Scholar] [CrossRef]
- Dudek, M.K.; Paluch, P.; Pindelska, E. Crystal structures of two furazidin polymorphs revealed by a joint effort of crystal structure prediction and NMR crystallography. Acta Cryst. 2020, 76, 322–335. [Google Scholar] [CrossRef]
- Gobetto, R.; Nervi, C.; Chierotti, M.R.; Braga, D.; Maini, L.; Grepioni, F.; Harris, R.K.; Hodgkinson, P. Hydrogen Bonding and Dynamic Behaviour in Crystals and Polymorphs of Dicarboxylic–Diamine Adducts: A Comparison between NMR Parameters and X-ray Diffraction Studies. Chem. A Eur. J. 2005, 11, 7461–7471. [Google Scholar] [CrossRef]
- Dudek, M.K.; Wielgus, E.; Paluch, P.; Sniechowska, J.; Kostrzewa, M.; Day, G.M.; Bujacz, G.D.; Potrzebowski, M.J. Understanding the formation of apremilast cocrystals. Acta Cryst. 2019, 75, 803–814. [Google Scholar] [CrossRef]
- Available online: https://xray.chem.wisc.edu/idealized-molecular-geometry-library/ (accessed on 18 March 2010).
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0–New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Gavezzotti, A. Are Crystal Structures Predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Filippini, G. Geometry of the Intermolecular X-H…Y (X, Y = N, O) Hydrogen Bond and the Calibration of Empirical Hydrogen-Bond Potentials. J. Phys. Chem. 1994, 98, 4831–4837. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory. In International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK, 2009; Volume 23, p. 336. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Branko, G.; Shephard, G.C. Tilings and Patterns, 2nd ed.; Dover Publications Inc.: Mineola, NY, USA, 2016; pp. 473–481. [Google Scholar]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer (Version 17.5). University of Western Australia. 2017. Available online: https://crystalexplorer.scb.uwa.edu.au/ (accessed on 5 April 2017).
- Ganguly, P.; Desiraju, G.R. Long-range synthon Aufbau modules (LSAM) in crystal structures: Systematic changes in C6H6-nFn (0 ≤ n ≤ 6) fluorobenzenes. CrystEngComm 2010, 12, 817–833. [Google Scholar] [CrossRef]
- Kitajgorodskij, A. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. CrystEngComm 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Jaśkowska, J. Method for Obtaining O-arylpiperazinyalkyl Salicylamides. Patent PL236884 (B1), 22 February 2021. [Google Scholar]
- CrysAlisPro 1.171.38.41; Rigaku Oxford Diffraction; Rigaku Corporation: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND: Visual Crystal Structure Information System; Version 2.1d; Crystal Impact GbR: Bonn, Germany, 2004. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. PLATON: A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2020. [Google Scholar]
- CASTEP Material.Studio v. 6.1; Accelrys Software Inc.: San Diego, CA, USA, 2013.
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B-Condens. Matter Mater. Phys. 2001, 63, 245101–2451013. [Google Scholar] [CrossRef] [Green Version]
- Liégeois, J.F.; Lespagnard, M.; Meneses Salas, E.; Mangin, F.; Scuvée-Moreau, J.; Dilly, S. Enhancing a CH−π Interaction to Increase the Affinity for 5-HT1A Receptors. ACS Med. Chem. Lett. 2014, 5, 358–362. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1-MeOH | 1-MeOH Low | 1-EtOH | Calc 1-MeOH * | Calc 1-EtOH * | Calc 1-H2O * | Calc 1 | |
|---|---|---|---|---|---|---|---|
| a/Å | 7.2792(4) | 7.2165(4) | 7.5240(3) | 7.1704 | 7.3751 | 7.1916 | 7.1932 |
| b/Å | 10.1555(5) | 9.9585(6) | 10.1584(4) | 10.1861 | 10.7564 | 9.7899 | 9.9247 |
| c/Å | 17.1001(7) | 17.030(1) | 16.9167(6) | 16.9461 | 16.9272 | 17.0122 | 16.9496 |
| α/° | 76.527(4) | 76.185(6) | 75.404(3) | 74.8319 | 69.7323 | 74.9742 | 75.1011 |
| β/° | 87.792(4) | 87.712(5) | 85.663(3) | 88.3336 | 84.9575 | 87.4625 | 87.5922 |
| γ/° | 87.810(4) | 87.901(5) | 85.721(4) | 89.8875 | 84.3890 | 88.7825 | 87.8934 |
| V/Å3 | 1227.9(1) | 1187.1(1) | 1245.59(8) | 1194.1 | 1251.5 | 1155.6 | 1167.9 |
| Solvent accessible voids/Å3 ** | 34.5 | 25.8 | 42.1 | 24.5 | 43.1 | 17.4 | 19.7 |
| Voids per unit cell /% | 2.8 | 2.2 | 3.4 | 2.1 | 3.4 | 1.5 | 1.7 |
| No. of electrons per void | 19.3 | 16.9 | 29.0 | – | – | – | – |
| Packing index | 62.3 | 65.1 | 61.4 | 73.3 | 72.5 | 72.5 | 68.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pindelska, E.; Marczewska-Rak, A.; Jaśkowska, J.; Madura, I.D. Solvates of New Arylpiperazine Salicylamide Derivative-a Multi-Technique Approach to the Description of 5 HTR Ligand Structure and Interactions. Int. J. Mol. Sci. 2021, 22, 4992. https://doi.org/10.3390/ijms22094992
Pindelska E, Marczewska-Rak A, Jaśkowska J, Madura ID. Solvates of New Arylpiperazine Salicylamide Derivative-a Multi-Technique Approach to the Description of 5 HTR Ligand Structure and Interactions. International Journal of Molecular Sciences. 2021; 22(9):4992. https://doi.org/10.3390/ijms22094992
Chicago/Turabian StylePindelska, Edyta, Anna Marczewska-Rak, Jolanta Jaśkowska, and Izabela D. Madura. 2021. "Solvates of New Arylpiperazine Salicylamide Derivative-a Multi-Technique Approach to the Description of 5 HTR Ligand Structure and Interactions" International Journal of Molecular Sciences 22, no. 9: 4992. https://doi.org/10.3390/ijms22094992