
Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West?

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
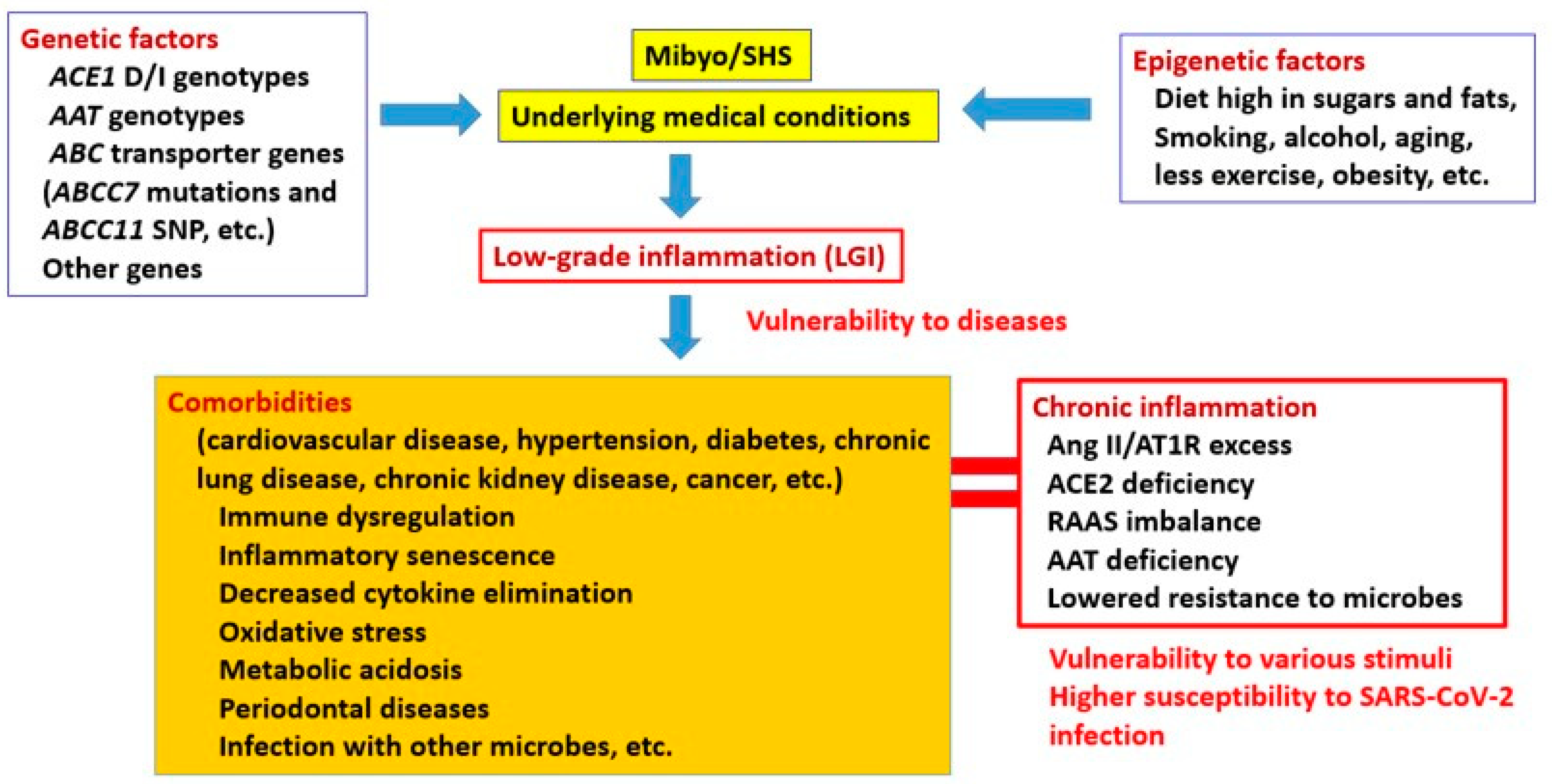
2. Pre-Existing Conditions and Inflammation
3. Association between Genetic Predispositions and Comorbidities
3.1. ACE1 D/I Genotype
3.2. AAT
3.3. Other Genes and Their Subtypes
3.3.1. FXIIIB and PV92
3.3.2. Neanderthal Haplotype
3.3.3. Human Leukocyte Antigen (HLA)
3.3.4. ATP-Binding Cassette (ABC) Transporter Genes
3.3.5. Epidermal Growth Factor Receptor (EGFR)
4. The Effect of the ACE1 DD Genotype Alone Seems to Be Modest
5. SARS-CoV-2 Infection and an Excessive Inflammatory Response
6. Viral Load and Disease Severity
7. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARDS | acute respiratory distress syndrome |
| ADAM17 | ADAM metallopeptidase domain 17 |
| AAT | alpha-1 anti-trypsin |
| Ang I | angiotensin I |
| Ang II | angiotensin II |
| ACE1 | angiotensin-converting enzyme 1 |
| ACE2 | angiotensin-converting enzyme 2 |
| ABC | ATP-binding cassette |
| COPD | chronic obstructive pulmonary disease |
| CRP | C-reactive protein |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| EGFR | epidermal growth factor receptor |
| E | envelopes |
| ERK | extracellular signal-regulated kinase |
| HDL | high-density lipoprotein |
| HLA | human leukocyte antigen |
| IL | interleukin |
| JAK | Janus kinase |
| STAT | signal transducer and activator of transcription |
| LGI | low-grade inflammation |
| MERS | Middle East respiratory syndrome |
| Nsps | non-structural proteins |
| NF-kB | nuclear factor kappa B |
| N | nucleocapsids |
| Orf | open reading frame |
| PRRs | pattern recognition receptors |
| ROS | reactive oxygen species |
| RAAS | renin–angiotensin–aldosterone system |
| RdRp | RNA-dependent RNA polymerase |
| SARS | severe acute respiratory syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| STAT | signal transducer and activator of |
| SHS | Suboptimal Health Status |
| TNF | tumor necrosis factor |
| TNFR | tumor necrosis factor receptor |
References
- Berlin, A.D.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Margallo, N.L.; Diaz, M.; Lim, P.P. 2019 Novel Coronavirus Pandemic: What Do We Know? S D Med. 2020, 73, 262–264. [Google Scholar] [PubMed]
- Colantuoni, A.; Martini, R.; Caprari, P.; Ballestri, M.; Capecchi, P.L.; Gnasso, A.; Lo Presti, R.; Marcoccia, A.; Rossi, M.; Caimi, G. COVID-19 Sepsis and Microcirculation Dysfunction. Front. Physiol. 2020, 11, 747. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Saab, B.Y.; Gard, P.R.; Overall, A.D.J. The geographic distribution of the ACE II genotype: A novel finding. Genet. Res. 2007, 89, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Ariumi, Y.; Nishida, N.; Yamamoto, R.; Bauer, G.; Gojobori, T.; Shimotohno, K.; Mizokami, M. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 2020, 758, 144944. [Google Scholar] [CrossRef]
- Yamamoto, N.; Bauer, G. Apparent difference in fatalities between Central Europe and East Asia due to SARS-COV-2 and COVID-19: Four hypotheses for possible explanation. Med. Hypotheses 2020, 144, 110160. [Google Scholar] [CrossRef]
- Pati, A.; Mahto, H.; Padhi, S.; Panda, A.K. ACE deletion allele is associated with susceptibility to SARS-CoV-2 infection and mortality rate: An epidemiological study in the Asian population. Clin. Chim. Acta 2020, 510, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Hatami, N.; Ahi, S.; Sadeghinikoo, A.; Foroughian, M.; Javdani, F.; Kalani, N.; Fereydoni, M.; Keshavarz, P.; Hosseini, A. Worldwide ACE (I/D) polymorphism may affect COVID-19 recovery rate: An ecological meta-regression. Endocrine 2020, 68, 479–484. [Google Scholar] [CrossRef]
- Vianello, A.; Braccioni, F. Geographical Overlap between Alpha-1 Antitrypsin Deficiency and COVID-19 Infection in Italy: Casual or Causal? Arch. Bronconeumol. 2020, 56, 609–610. [Google Scholar] [CrossRef]
- De Loyola, M.B.; Dos Reis, T.T.A.; de Oliveira, G.X.L.M.; Palmeira, J.D.F.; Argañaraz, G.A.; Argañaraz, E.R. Alpha-1-antitrypsin: A possible host protective factor against Covid-19. Rev. Med. Virol. 2021, 31, e2157. [Google Scholar] [CrossRef]
- Yoshikura, H. Epidemiological correlation between COVID-19 epidemic and prevalence of α-1 antitrypsin deficiency in the world. Glob. Health Med. 2021, 3, 73–81. [Google Scholar] [CrossRef]
- Tsushima, M. Mibyo (preventive medicine) in aged society. Nihon. Ronen Igakkai Zasshi 2002, 39, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Russell, A.; Yan, Y. Global Health Epidemiology Reference Group (GHERG). Traditional Chinese medicine and new concepts of predictive, preventive and personalized medicine in diagnosis and treatment of suboptimal health. EPMA J. 2014, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.; Qiu, J.; Wang, H.; Liu, D.; Zhao, Z.; Song, M.; Song, Q.; Wang, X.; Zhou, Y.; et al. Association between Ideal Cardiovascular Health Metrics and Suboptimal Health Status in Chinese Population. Sci. Rep. 2017, 7, 14975. [Google Scholar] [CrossRef] [Green Version]
- Martin-Subero, M.; Anderson, G.; Kanchanatawan, B.; Berk, M.; Maes, M. Comorbidity between depression and inflammatory bowel disease explained by immune-inflammatory, oxidative, and nitrosative stress; tryptophan catabolite; and gut-brain pathways. Cns. Spectr. 2016, 21, 184–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korakas, E.; Ikonomidis, I.; Kousathana, F.; Balampanis, K.; Kountouri, A.; Raptis, A.; Palaiodimou, L.; Kokkinos, A.; Lambadiari, V. Obesity and COVID-19: Immune and metabolic derangement as a possible link to adverse clinical outcomes. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E105–E109. [Google Scholar] [CrossRef] [PubMed]
- Priyamvara, A.; Dey, A.K.; Bandyopadhyay, D.; Katikineni, V.; Zaghlol, R.; Basyal, B.; Barssoum, K.; Amarin, R.; Bhatt, D.L.; Lavie, C.J. Periodontal Inflammation and the Risk of Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 28. [Google Scholar] [CrossRef]
- Harambat, J.; Kunzmann, K.; Azukaitis, K.; Bayazit, A.K.; Canpolat, N.; Doyon, A.; Duzova, A.; Niemirska, A.; Sözeri, B.; Thurn-Valsassina, D.; et al. Metabolic acidosis is common and associates with disease progression in children with chronic kidney disease. Kidney Int. 2017, 92, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Tarantino, G. Gut microbiome, obesity-related comorbidities, and low-grade chronic inflammation. J. Clin. Endocrinol. Metab. 2014, 99, 2343–2346. [Google Scholar] [CrossRef] [Green Version]
- Hunter, P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012, 13, 968–970. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.K.; Cathomas, F.; Russo, S.J. Central and Peripheral Inflammation Link Metabolic Syndrome and Major Depressive Disorder. Physiology 2019, 34, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, G.; Della Guardia, L.; Maurizi, A.; Poloni, A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J. Cell. Physiol. 2018, 233, 88–97. [Google Scholar] [CrossRef]
- Mustain, W.C.; Starr, M.E.; Valentino, J.D.; Cohen, N.A.; Okamura, D.; Wang, C.; Evers, B.M.; Saito, H. Inflammatory cytokine gene expression in mesenteric adipose tissue during acute experimental colitis. PLoS ONE 2013, 8, e83693. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Cortez, M.; Carmo, L.S.; Rogero, M.M.; Borelli, P.; Fock, R.A. A high-fat diet increases IL-1, IL-6, and TNF-alpha production by increasing NF-kappaB and attenuating PPAR-gamma expression in bone marrow mesenchymal stem cells. Inflammation 2013, 36, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Oliver, S.R.; Ganesan, G.; Zaldivar, F.P., Jr.; Radom-Aizik, S.; Galassetti, P.R. Fasting glucose level modulates cell surface expression of CD11b and CD66b in granulocytes and monocytes of patients with type 2 diabetes. J. Investig. Med. 2013, 61, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Lukic, L.; Lalic, N.M.; Rajkovic, N.; Jotic, A.; Lalic, K.; Milicic, T.; Seferovic, J.P.; Macesic, M.; Gajovic, J.S. Hypertension in obese type 2 diabetes patients is associated with increases in insulin resistance and IL-6 cytokine levels: Potential targets for an efficient preventive intervention. Int. J. Environ. Res. Public Health 2014, 11, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Lenart, N.; Brough, D.; Denes, A. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J. Cereb. Blood Flow Metab. 2016, 36, 1668–1685. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Zhang, S.; Xu, J.; Zhou, M.; Huang, Q.; Duan, L.; Lv, Z.; Xia, H.; Xiao, W.; Yin, Z.; et al. Comparison of clinical characteristics among younger and elderly deceased patients with COVID-19: A retrospective study. Aging 2020, 13, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.A.; McNamara, M.S.; Sinclair, A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981. [Google Scholar] [CrossRef]
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474. [Google Scholar] [CrossRef]
- Chanana, N.; Palmo, T.; Sharma, K.; Kumar, R.; Graham, B.B.; Pasha, Q. Sex-derived attributes contributing to SARS-CoV-2 mortality. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E562–E567. [Google Scholar] [CrossRef] [PubMed]
- Cambien, F.; Poirier, O.; Lecerf, L.; Evans, A.; Cambou, J.-P.; Arveiler, D.; Luc, G.; Bard, J.-M.; Bara, L.; Ricard, S.; et al. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 1992, 359, 641–644. [Google Scholar] [CrossRef]
- Butler, R. The DD-ACE genotype and cardiovascular disease. Pharmacogenomics 2000, 1, 153–167. [Google Scholar] [CrossRef]
- Lindpaintner, K.; Pfeffer, M.A.; Kreutz, R.; Stampfer, M.J.; Grodstein, F.; LaMotte, F.; Buring, J.; Hennekens, C.H. A prospective evaluation of an angiotensin-converting-enzyme gene polymorphism and the risk of ischemic heart disease. N. Engl. J. Med. 1995, 332, 706–711. [Google Scholar] [CrossRef]
- Arbustini, E.; Grasso, M.; Fasani, R.; Klersy, C.; Diegoli, M.; Porcu, E.; Banchieri, N.; Fortina, P.; Danesino, C.; Specchia, G. Angiotensin converting enzyme gene deletion allele is independently and strongly associated with coronary atherosclerosis and myocardial infarction. Br. Heart J. 1995, 74, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Gregg, P.L.; Hedayati, S.S. Management of Traditional Cardiovascular Risk Factors in CKD: What Are the Data? Am. J. Kidney Dis. 2018, 72, 728–744. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; McKarns, S.; DeMarco, V.G.; Jia, G.; Sowers, J.R. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism 2013, 62, 1543–1552. [Google Scholar] [CrossRef] [Green Version]
- Boureau, A.S.; De Decker, L.; Berrut, G.; Hanon, O. COVID-19 and cardiovascular disease: Characteristic features in older patients. Geriatr. Psychol. Neuropsychiatr. Vieil. 2020, 18, 141–148. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharm. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Boguski, S.M.; McCormick, F. Proteins regulating Ras and its relatives. Nature 1993, 366, 643–654. [Google Scholar] [CrossRef]
- Ramalingam, L.; Menikdiwela, K.; LeMieux, M.; Dufour, J.M.; Kaur, G.; Kalupahana, N.; Moustaid-Moussa, N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, H.; Brull, D.; Humphries, E.S. Analysis of gene-environment interactions by “stressing-the-genotype” studies: The angiotensin converting enzyme and exercise-induced left ventricular hypertrophy as an example. Ital. Heart J. 2002, 3, 10–14. [Google Scholar] [PubMed]
- Morris, B.J. Hypothesis: An angiotensin converting enzyme genotype, present in one in three caucasians, is associated with increased mortality rate. Clin. Exp. Pharm. Physiol. 1996, 23, 1–10. [Google Scholar] [CrossRef]
- Staessen, J.A.; Wang, J.G.; Ginocchio, G.; Petrov, V.; Saavedra, A.P.; Soubrier, F.; Vlietinck, R.; Fagard, R. The deletion/insertion polymorphism of the angiotensin converting enzyme gene and cardiovascular-renal risk. J. Hypertens. 1997, 15 Pt 2, 1579–1592. [Google Scholar] [CrossRef]
- Pabst, S.; Theis, B.; Gillissen, A.; Lennarz, M.; Tuleta, I.; Nickenig, G.; Skowasch, D.; Grohé, C. Angiotensin-converting enzyme I/D polymorphism in chronic obstructive pulmonary disease. Eur. J. Med. Res. 2009, 14 (Suppl. 4), 177–181. [Google Scholar] [CrossRef] [Green Version]
- Margaglione, M.; Grandone, E.; Vecchione, G.; Cappucci, G.; Giuliani, N.; Colaizzo, D.; Celentano, E.; Panico, S.; Di Minno, G. Plasminogen activator inhibitor-1 (PAI-1) antigen plasma levels in subjects attending a metabolic ward: Relation to polymorphisms of PAI-1 and angiontensin converting enzyme (ACE) genes. Arter. Thromb. Vasc. Biol. 1997, 17, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Mizgerd, J.P. Inflammation and Pneumonia: Why Are Some More Susceptible than Others? Clin. Chest Med. 2018, 39, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell COVID-19 Research Consortium; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; et al. Presenting Characteristics, Comorbidities, and Outcomes among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- De Serres, F.J.; Blanco, I.; Fernandez-Bustillo, E. Genetic epidemiology of alpha-1 antitrypsin deficiency in southern Europe: France, Italy, Portugal and Spain. Clin. Genet. 2003, 63, 490–509. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Welte, T. Well-Known and Less Well-Known Functions of Alpha-1 Antitrypsin. Its Role in Chronic Obstructive Pulmonary Disease and Other Disease Developments. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S4), S280–S288. [Google Scholar] [CrossRef]
- De Serres, F.; Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Bueno, P.; Diego, I.; Pérez-Holanda, S.; Lara, B.; Casas-Maldonado, F.; Esquinas, C.; Miravitlles, M. Alpha-1 antitrypsin Pi * SZ genotype: Estimated prevalence and number of SZ subjects worldwide. Int. J. Chron. Obs. Pulmon. Dis. 2017, 12, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- De Serres, F.J. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ. Health Perspect 2003, 111, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Guttman, O.; Baranovski, B.M.; Schuster, R.; Kaner, Z.; Freixo-Lima, G.S.; Bahar, N.; Kalay, N.; Mizrahi, M.I.; Brami, I.; Ochayon, D.E.; et al. Acute-phase protein alpha1-anti-trypsin: Diverting injurious innate and adaptive immune responses from non-authentic threats. Clin. Exp. Immunol. 2015, 179, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Stone, H.; Pye, A.; Stockley, R.A. Disease associations in alpha-1-antitrypsin deficiency. Respir. Med. 2014, 108, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott, G.B.; Chan, E.D.; Dinarello, C.A.; Shapiro, L. Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J. Leukoc. Biol. 2009, 85, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. Alpha1-antitrypsin Deficiency: A Misfolded Secretory Protein Variant with Unique Effects on the Endoplasmic Reticulum. Endoplasmic Reticulum Stress Dis. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.C. Expanding the clinical indications for alpha(1)-antitrypsin therapy. Mol. Med. 2012, 18, 957–970. [Google Scholar] [CrossRef]
- Gialeraki, A.; Politou, M.; Rallidis, L.; Merkouri, E.; Markatos, C.; Kremastinos, D.; Travlou, A. Prevalence of prothrombotic polymorphisms in Greece. Genet. Test. 2008, 12, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Giraldo, Y.; Rodriguez-Duenas, M.; Forero, D.A. Development of Novel High-Resolution Melting-Based Assays for Genotyping Two Alu Insertion Polymorphisms (FXIIIB and PV92). Mol. Biotechnol. 2016, 58, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Paabo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Ueda, K. ABC proteins protect the human body and maintain optimal health. Biosci. Biotechnol. Biochem. 2011, 75, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Yoshiura, K.; Kinoshita, A.; Ishida, T.; Ninokata, A.; Ishikawa, T.; Kaname, T.; Bannai, M.; Tokunaga, K.; Sonoda, S.; Komaki, R.; et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet. 2006, 38, 324–330. [Google Scholar] [CrossRef]
- Klann, K.; Bojkova, D.; Tascher, G.; Ciesek, S.; Münch, C.; Cinatl, J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 2020, 80, 164–174.e4. [Google Scholar] [CrossRef]
- Shafiee, S.M.; Firoozrai, M.; Salimi, S.; Zand, H.; Hesabi, B.; Mohebbi, A. Angiotensin converting enzyme DD genotype not associated with increased risk of coronary artery disease in the Iranian population. Pathophysiology 2010, 17, 163–167. [Google Scholar] [CrossRef]
- Wuyts, B.; Delanghe, J.; de Buyzere, M. Angiotensin I-converting enzyme insertion/deletion polymorphism: Clinical implications. Acta Clin. Belg. 1997, 52, 338–349. [Google Scholar] [CrossRef]
- Gilkes, A.; Ashworth, M.; Schofield, P.; Harries, T.H.; Durbaba, S.; Weston, C.; White, P. Does COPD risk vary by ethnicity? A retrospective cross-sectional study. Int. J. Chron. Obs. Pulmon. Dis. 2016, 11, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Lan, F.; Yan, F.; Shen, H. Angiotensin-converting enzyme I/D polymorphism is associated with COPD risk in Asian population: Evidence from a meta-analysis. COPD 2013, 10, 35–39. [Google Scholar] [CrossRef]
- Staessen, J.A.; Ginocchio, G.; Wang, J.G.; Saavedra, A.P.; Soubrier, F.; Vlietinck, R.; Fagard, R. Genetic variability in the renin-angiotensin system: Prevalence of alleles and genotypes. J. Cardiovasc. Risk 1997, 4, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Garatachea, N.; Marin, P.J.; Lucia, A. The ACE DD genotype and D-allele are associated with exceptional longevity: A meta-analysis. Ageing Res. Rev. 2013, 12, 1079–1087. [Google Scholar] [CrossRef]
- Phillips, I.M.; Kagiyama, S. Angiotensin II as a pro-inflammatory mediator. Curr. Opin. Investig. Drugs 2002, 3, 569–577. [Google Scholar]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, T.; Kawai, T.; Forrester, S.J.; Obama, T.; Tsuji, T.; Fukuda, Y.; Elliott, K.J.; Tilley, D.G.; Davisson, R.L.; Park, J.Y.; et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 2015, 65, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.T.; Lakkappa, N.; Lazartigues, E. ADAM17-Mediated Shedding of Inflammatory Cytokines in Hypertension. Front. Pharm. 2020, 11, 1154. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Mukerjee, S.; Silva-Alves, C.R.; Carvalho-Galvão, A.; Cruz, J.C.; Balarini, C.M.; Braga, V.A.; Lazartigues, E.; França-Silva, M.S. A Disintegrin and Metalloprotease 17 in the Cardiovascular and Central Nervous Systems. Front. Physiol. 2016, 7, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor alpha to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chapman, K.R.; Wong, A.; Liu, M. alpha1-Antitrypsin deficiency and the risk of COVID-19: An urgent call to action. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Lockett, A.D.; Van Demark, M.; Gu, Y.; Schweitzer, K.S.; Sigua, N.; Kamocki, K.; Fijalkowska, I.; Garrison, J.; Fisher, A.J.; Serban, K.; et al. Effect of cigarette smoke exposure and structural modifications on the alpha-1 Antitrypsin interaction with caspases. Mol. Med. 2012, 18, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ní Choileáin, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Maiese, A.; Manetti, A.C.; La Russa, R.; Di Paolo, M.; Turillazzi, E.; Frati, P.; Fineschi, V. Autopsy findings in COVID-19-related deaths: A literature review. Forensic Sci. Med. Pathol. 2020. [Google Scholar] [CrossRef]
- Stephen-Victor, E.; Das, M.; Karnam, A.; Pitard, B.; Gautier, J.F.; Bayry, J. Potential of regulatory T-cell-based therapies in the management of severe COVID-19. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Netea, M.G.; Giamarellos-Bourboulis, E.J.; Domínguez-Andrés, J.; Curtis, N.; van Crevel, R.; van de Veerdonk, F.L.; Bonten, M. Trained Immunity: A Tool for Reducing Susceptibility to and the Severity of SARS-CoV-2 Infection. Cell 2020, 181, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Zhu, G.; Zhu, C.; Zhu, Y.; Sun, F. Minireview of progress in the structural study of SARS-CoV-2 proteins. Curr. Res. Microb. Sci. 2020, 1, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.; Cheng, V.C.; Wu, A.K.; Tang, B.S.; Chan, K.H.; Chu, C.M.; Wong, M.M.; Hui, W.T.; Poon, L.L.; Tse, D.M.; et al. Viral loads in clinical specimens and SARS manifestations. Emerg. Infect. Dis. 2004, 10, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Hagman, K.; Hedenstierna, M.; Gille-Johnson, P.; Hammas, B.; Grabbe, M.; Dillner, J.; Ursing, J. SARS-CoV-2 RNA in serum as predictor of severe outcome in COVID-19: A retrospective cohort study. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- To, K.K.; Tsang, O.T.; Leung, W.S.; Tam, A.R.; Wu, T.C.; Lung, D.C.; Yip, C.C.; Cai, J.P.; Chan, J.M.; Chik, T.S.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated With Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Shi, P.Y. Antagonism of Type I Interferon by Severe Acute Respiratory Syndrome Coronavirus 2. J. Interferon Cytokine Res. 2020, 40, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398. [Google Scholar] [CrossRef] [PubMed]
- BBC Bews, New Coronavirus Variant: What Do We Know? 20 December 2020. Available online: https://www.bbc.com/news/health-55388846 (accessed on 10 February 2021).
- Feinstein, R.A.; Schimpff, C.R.; Hull, E.W. A reappraisal of staging and therapy for patients with cancer of the rectum. I. Development of two new systems of staging. Arch. Intern. Med. 1975, 135, 1441–1453. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, N.; Yamamoto, R.; Ariumi, Y.; Mizokami, M.; Shimotohno, K.; Yoshikura, H. Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West? Int. J. Mol. Sci. 2021, 22, 5000. https://doi.org/10.3390/ijms22095000
Yamamoto N, Yamamoto R, Ariumi Y, Mizokami M, Shimotohno K, Yoshikura H. Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West? International Journal of Molecular Sciences. 2021; 22(9):5000. https://doi.org/10.3390/ijms22095000
Chicago/Turabian StyleYamamoto, Naoki, Rain Yamamoto, Yasuo Ariumi, Masashi Mizokami, Kunitada Shimotohno, and Hiroshi Yoshikura. 2021. "Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West?" International Journal of Molecular Sciences 22, no. 9: 5000. https://doi.org/10.3390/ijms22095000
APA StyleYamamoto, N., Yamamoto, R., Ariumi, Y., Mizokami, M., Shimotohno, K., & Yoshikura, H. (2021). Does Genetic Predisposition Contribute to the Exacerbation of COVID-19 Symptoms in Individuals with Comorbidities and Explain the Huge Mortality Disparity between the East and the West? International Journal of Molecular Sciences, 22(9), 5000. https://doi.org/10.3390/ijms22095000