
Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
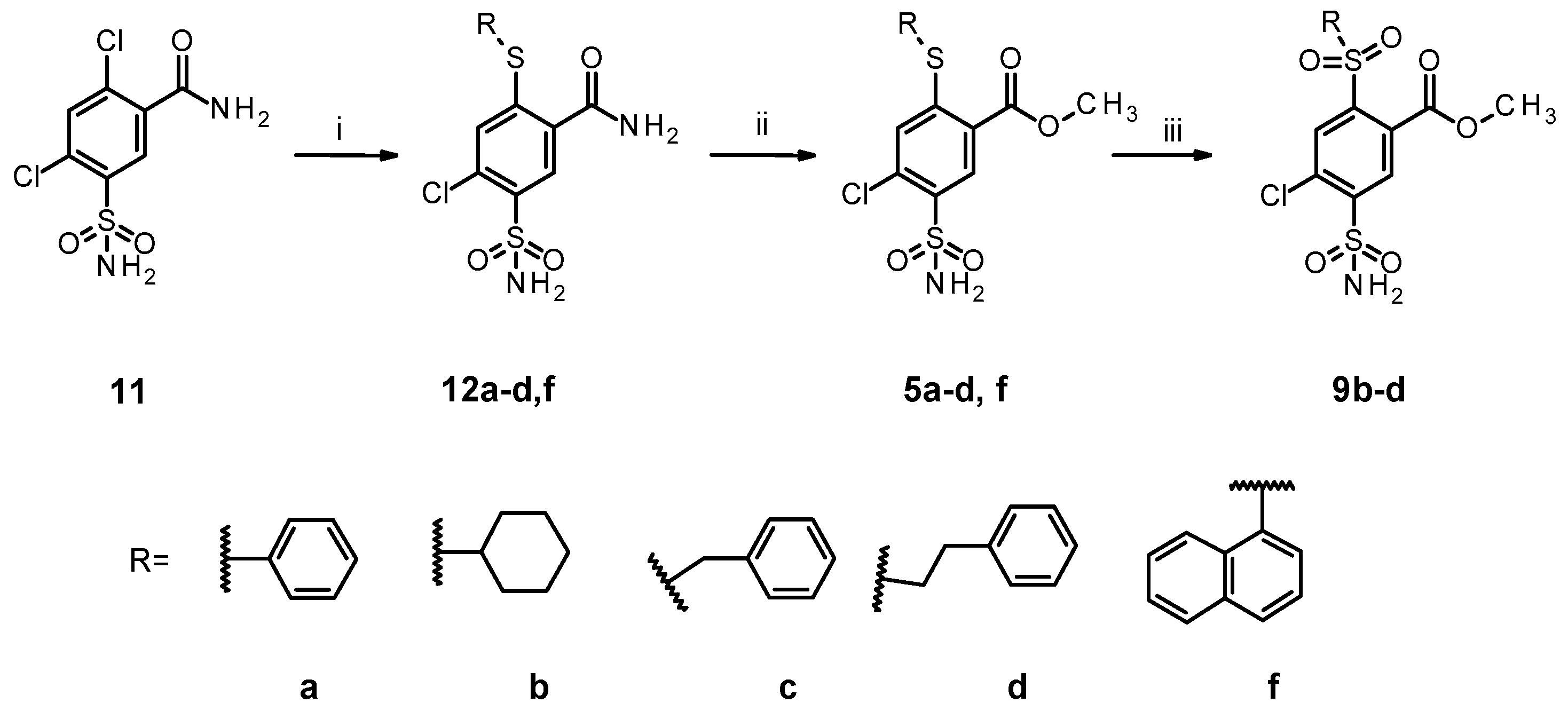
2.1. Organic Synthesis of Designed Compounds
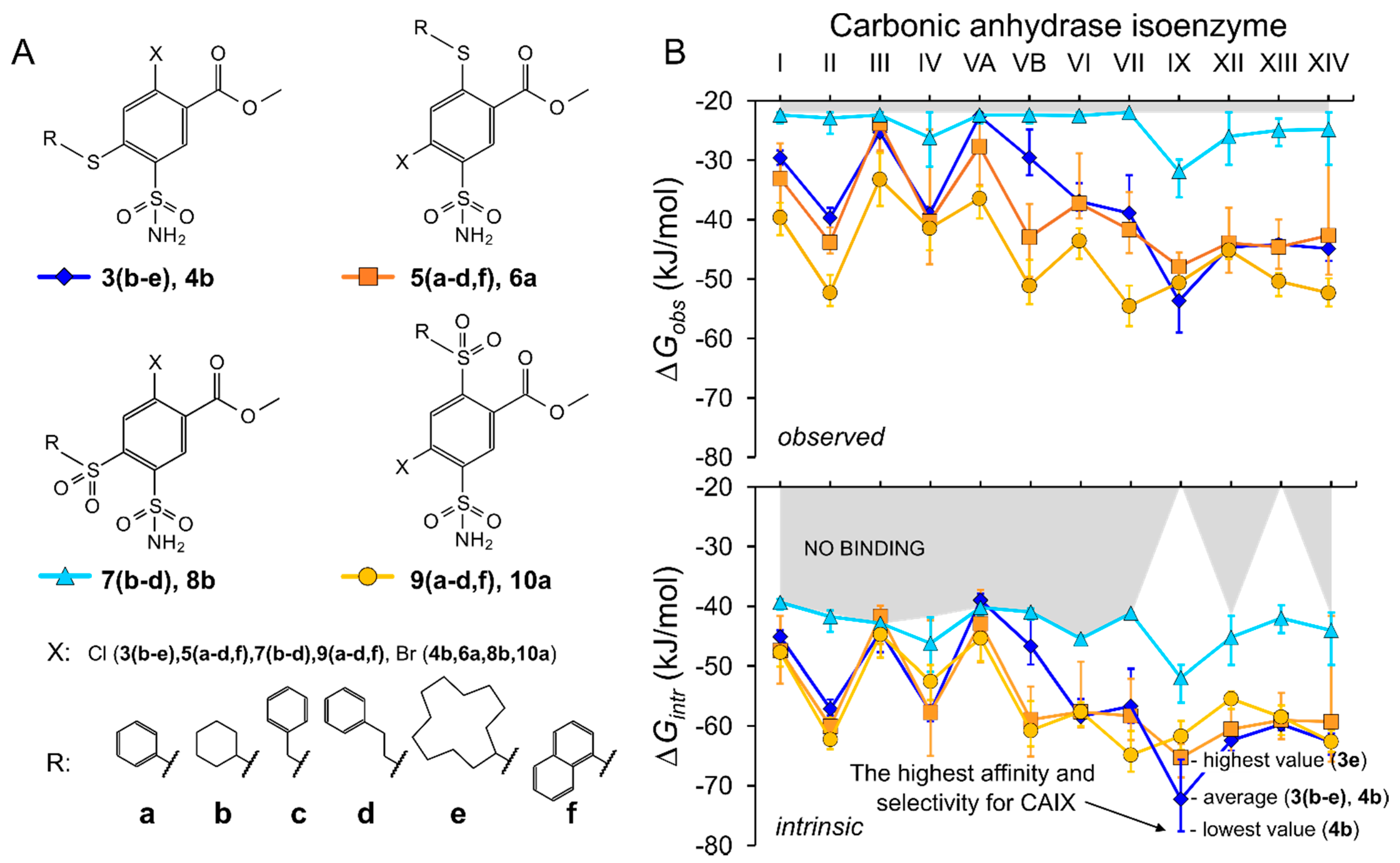
2.2. Thermodynamics of Compound Binding to CA Isozymes
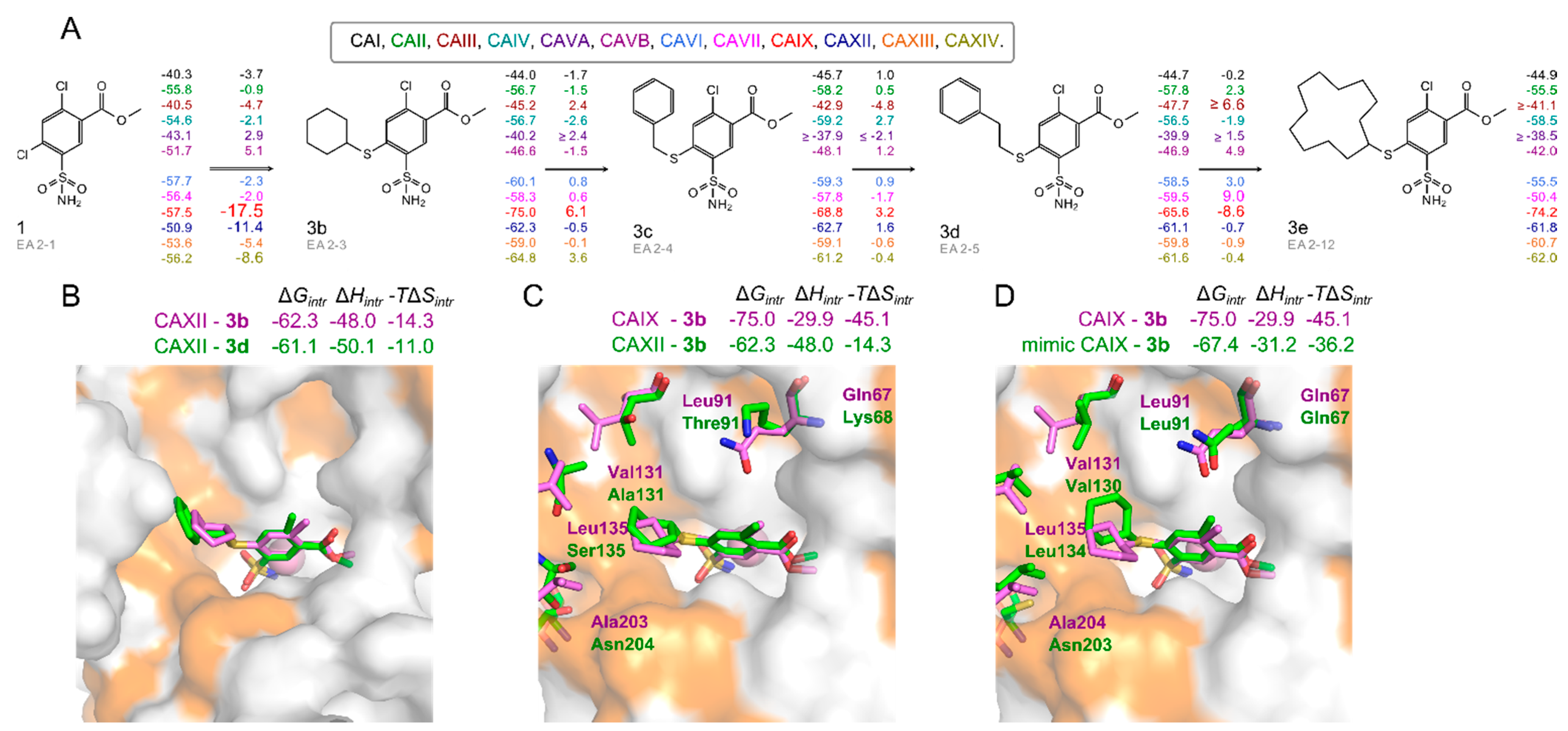
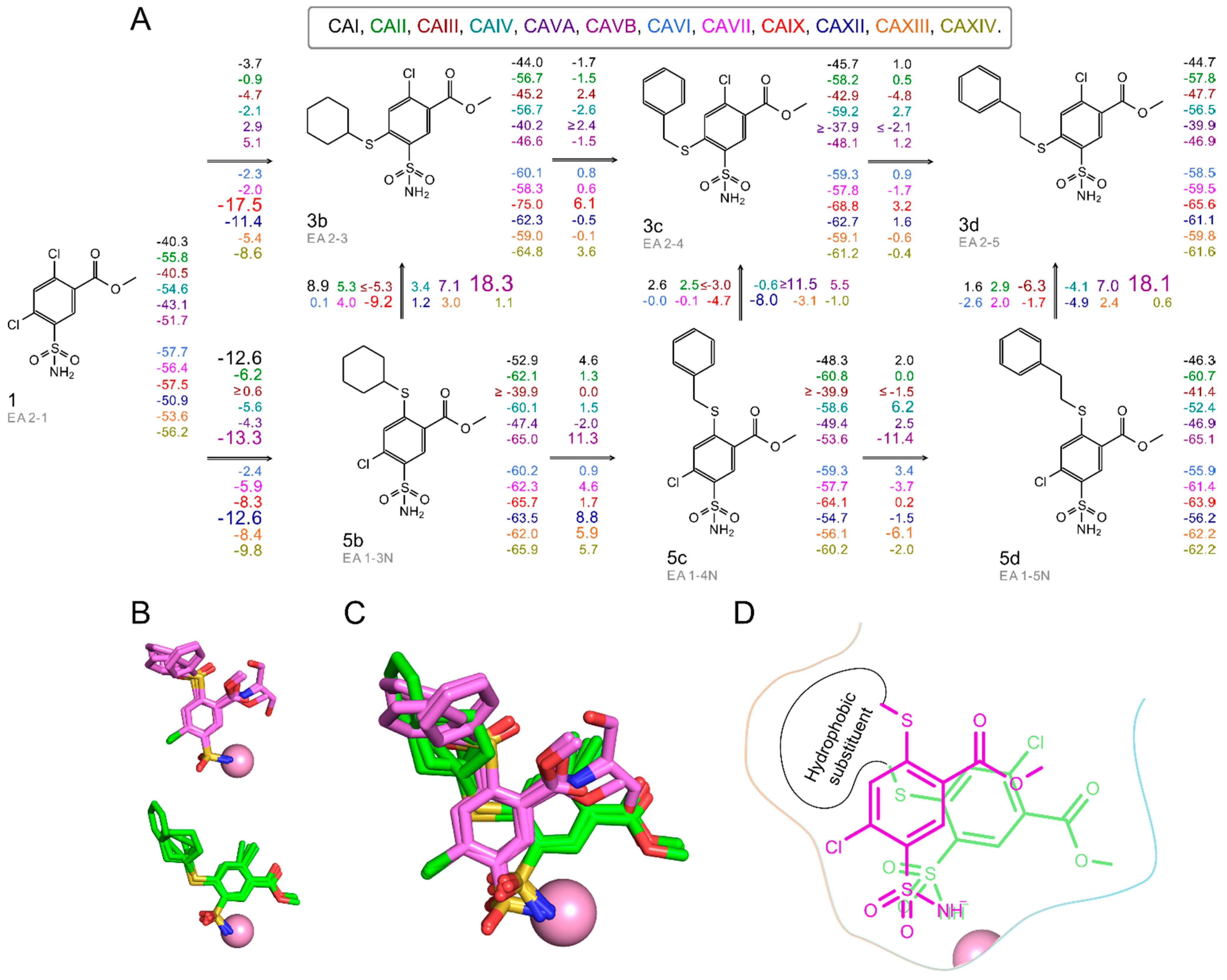
2.3. X-ray Crystal Structures and Correlations with Compound Binding Thermodynamics to CA Isozymes
- (i)
- methyl 2-halo-4-substituted-5-sulfamoyl-benzoates, abbreviated ortho- by substituent at the ortho- position relative to sulfonamide group;
- (ii)
- methyl 4-halo-2-substituted-5-sulfamoyl-benzoates, abbreviated as para-,
- (iii)
- the position of the substituent in benzenesulfonamide is compared in these two groups of compounds.
2.3.1. Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates Binding to CA Isozymes
2.3.2. Methyl 4-Halo-2-Substituted-5-Sulfamoyl-Benzoates Binding CA Isozymes
2.3.3. Comparison of Methyl Halo 2- and 4-Substituted-5-Sulfamoyl-Benzoates Binding to CA Isozymes
3. Conclusions
4. Materials and Methods
4.1. Organic Synthesis
4.2. General Procedure for the Syntheses of 1, 2
4.3. General Procedure for the Syntheses of 3b, 3e, and 4b
4.4. General Procedure for the Syntheses of 3c and 3d
4.5. General Procedure for the Syntheses of 5a, 5f, and 6a
4.6. General Procedure for the Syntheses of 7b–d, 8b, 9a–d, 9f, 10a
4.7. General Procedure for the Syntheses of 12a–d,f
4.8. General Procedure for the Syntheses of 5a–d,f
4.9. Protein Preparation
4.10. Determination of Binding Parameters
4.10.1. Fluorescent Thermal Shift Assay (FTSA)
4.10.2. Isothermal Titration Calorimetry (ITC)
4.10.3. Calculation of Intrinsic Binding Parameters
4.11. Determination of Protonation Parameters
4.11.1. Determination of pKa Values
4.11.2. Determination of Protonation Enthalpy
4.12. X-ray Crystallography: Crystallization, Data Collection, and Structure Determination
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CA | carbonic anhydrase |
| FTSA | fluorescent thermal shift assay (or differential scanning fluorimetry, DSF) |
| intr | intrinsic |
| ITC | isothermal titration calorimetry |
| obs | observed |
| VARIABLES | |
| ΔGintr | change of the intrinsic standard Gibbs energy upon binding |
| ΔGobs | change of the observed standard Gibbs energy upon binding |
| ΔHintr | change of the intrinsic standard enthalpy upon binding |
| ΔHobs | change of the observed standard enthalpy upon binding |
| Kd_intr | the intrinsic equilibrium dissociation constant |
| Kd_obs | the observed equilibrium dissociation constant |
| Tm | melting temperature (midpoint of the unfolding transition) |
| TΔSintr | change of the intrinsic standard entropy upon binding multiplied by absolute temperature |
References
- Dodgson, S.J.; Tashian, R.E.; Gros, G.; Carter, N.D. (Eds.) The Carbonic Anhydrases; Springer: Boston, MA, USA, 1991. [Google Scholar]
- Chegwidden, W.R.; Carter, N.D.; Edwards, Y.H. (Eds.) The Carbonic Anhydrases; Birkhäuser: Basel, Switzerland, 2000. [Google Scholar]
- Oosterwijk, E.; Ruiter, D.J.; Hoedemaeker, P.J.; Pauwels, E.K.; Jonas, U.; Zwartendijk, J.; Warnaar, S.O. Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int. J. Cancer 1986, 38, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.P.; Zelník, V.; Opavský, R.; Zat’ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994, 9, 2877–2888. [Google Scholar]
- Saarnio, J.; Parkkila, S.; Parkkila, A.-K.; Haukipuro, K.; Pastoreková, S.; Pastorek, J.; Kairaluoma, M.I.; Karttunen, T.J. Immunohistochemical Study of Colorectal Tumors for Expression of a Novel Transmembrane Carbonic Anhydrase, MN/CA IX, with Potential Value as a Marker of Cell Proliferation. Am. J. Pathol. 1998, 153, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein−Ligand Binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [CrossRef] [Green Version]
- Lomelino, C.L.; Andring, J.T.; McKenna, R. Crystallography and Its Impact on Carbonic Anhydrase Research. Int. J. Med. Chem. 2018, 2018, 9419521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakšauskas, A.; Čapkauskaitė, E.; Jezepčikas, L.; Linkuvienė, V.; Kišonaitė, M.; Smirnov, A.; Manakova, E.; Gražulis, S.; Matulis, D. Design of two-tail compounds with rotationally fixed benzenesulfonamide ring as inhibitors of carbonic anhydrases. Eur. J. Med. Chem. 2018, 156, 61–78. [Google Scholar] [CrossRef]
- Zakšauskas, A.; Čapkauskaitė, E.; Jezepčikas, L.; Linkuvienė, V.; Paketurytė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Manakova, E.; et al. Halogenated and di-substituted benzenesulfonamides as selective inhibitors of carbonic anhydrase isoforms. Eur. J. Med. Chem. 2020, 185, 111825. [Google Scholar] [CrossRef]
- Dudutienė, V.; Zubrienė, A.; Kairys, V.; Smirnov, A.; Smirnovienė, J.; Leitans, J.; Kazaks, A.; Tars, K.; Manakova, L.; Gražulis, S.; et al. Isoform-Selective Enzyme Inhibitors by Exploring Pocket Size According to the Lock-and-Key Principle. Biophys. J. 2020, 119, 1513–1524. [Google Scholar] [CrossRef]
- Scott, A.D.; Phillips, C.; Alex, A.; Flocco, M.; Bent, A.; Randall, A.; O’Brien, R.; Damian, L.; Jones, L.H. Thermodynamic Optimisation in Drug Discovery: A Case Study using Carbonic Anhydrase Inhibitors. ChemMedChem 2009, 4, 1985–1989. [Google Scholar] [CrossRef]
- Čapkauskaitė, E.; Zubrienė, A.; Baranauskienė, L.; Tamulaitienė, G.; Manakova, E.; Kairys, V.; Gražulis, S.; Tumkevičius, S.; Matulis, D. Design of [(2-pyrimidinylthio)acetyl]benzenesulfonamides as inhibitors of human carbonic anhydrases. Eur. J. Med. Chem. 2012, 51, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Čapkauskaitė, E.; Zubrienė, A.; Smirnov, A.; Torresan, J.; Kišonaitė, M.; Kazokaitė, J.; Gylytė, J.; Michailovienė, V.; Jogaitė, V.; Manakova, E.; et al. Benzenesulfonamides with pyrimidine moiety as inhibitors of human carbonic anhydrases I, II, VI, VII, XII, and XIII. Bioorg. Med. Chem. 2013, 21, 6937–6947. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, C.; Mboge, M.; Frost, S.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.Z.; Aggarwal, M.; Kovalevsky, A.Y.; Silverman, D.N.; McKenna, R. Neutron Diffraction of Acetazolamide-Bound Human Carbonic Anhydrase II Reveals Atomic Details of Drug Binding. J. Am. Chem. Soc. 2012, 134, 14726–14729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubrienė, A.; Matulis, D. Observed Versus Intrinsic Thermodynamics of Inhibitor Binding to Carbonic Anhydrases. In Carbonic Anhydrase as Drug Target: Thermodynamics and Structure of Inhibitor Binding; Matulis, D., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 107–123. [Google Scholar]
- Dudutienė, V.; Matulienė, J.; Smirnov, A.; Timm, D.D.; Zubrienė, A.; Baranauskienė, L.; Morkūnaite, V.; Smirnovienė, J.; Michailovienė, V.; Juozapaitienė, V.; et al. Discovery and characterization of novel selective inhibitors of carbonic anhydrase IX. J. Med. Chem. 2014, 57, 9435–9446. [Google Scholar] [CrossRef] [PubMed]
- Genis, C.; Sippel, K.H.; Case, N.; Wengang Cao Avvaru, B.S.; Tartaglia, L.J.; Lakshmanan Govindasamy Tu, C.; Agbandje-McKenna, M.; Silverman, D.N.; Rosser, C.J.; McKenna, R. Design of a carbonic anhydrase IX active-site mimic to screen inhibitors for possible anticancer properties. Biochemistry 2009, 48, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Pinard, M.A.; Boone, C.D.; Rife, B.D.; Supuran, C.T.; McKenna, R. Structural study of interaction between brinzolamide and dorzolamide inhibition of human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 7210–7215. [Google Scholar] [CrossRef]
- Čapkauskaitė, E.; Linkuvienė, V.; Smirnov, A.; Milinavičiūtė, G.; Timm, D.D.; Kasiliauskaitė, A.; Manakova, E.; Gražulis, S.; Matulis, D. Combinatorial Design of Isoform-Selective N-Alkylated Benzimidazole-Based Inhibitors of Carbonic Anhydrases. ChemistrySelect 2017, 2, 5360–5371. [Google Scholar] [CrossRef]
- Smirnov, A.; Zubrienė, A.; Manakova, E.; Gražulis, S.; Matulis, D. Crystal structure correlations with the intrinsic thermodynamics of human carbonic anhydrase inhibitor binding. PeerJ 2018, 6, e4412. [Google Scholar] [CrossRef] [Green Version]
- Matulis, D. Thermodynamics of the hydrophobic effect. III. Condensation and aggregation of alkanes, alcohols, and alkylamines. Biophys. Chem. 2001, 93, 67–82. [Google Scholar] [CrossRef]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef] [Green Version]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Sūdžius, J.; Baranauskienė, L.; Golovenko, D.; Matulienė, J.; Michailovienė, V.; Torresan, J.; Jachno, J.; Sukackaitė, R.; Manakova, E.; Gražulis, S.; et al. 4-[N-(Substituted 4-pyrimidinyl)amino]benzenesulfonamides as inhibitors of carbonic anhydrase isozymes I, II, VII, and XIII. Bioorg. Med. Chem. 2010, 18, 7413–7421. [Google Scholar] [CrossRef]
- Jogaitė, V.; Zubrienė, A.; Michailovienė, V.; Gylytė, J.; Morkūnaitė, V.; Matulis, D. Characterization of human carbonic anhydrase XII stability and inhibitor binding. Bioorg. Med. Chem. 2013, 21, 1431–1436. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Kazlauskas, E.; Petrauskas, V.; Paketurytė, V.; Matulis, D. Standard operating procedure for fluorescent thermal shift assay (FTSA) for determination of protein-ligand binding and protein stability. Eur. Biophys. J. 2021, 50, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Matulis, D.; Baumann, C.G.; Bloomfield, V.A.; Lovrien, R.E. 1-Anilino-8-naphthalene sulfonate as a protein conformational tightening agent. Biopolymers 1999, 49, 451–458. [Google Scholar] [CrossRef]
- Baranauskienė, L.; Hilvo, M.; Matulienė, J.; Golovenko, D.; Manakova, E.; Dudutienė, V.; Michailovienė, V.; Torresan, J.; Jachno, J.; Parkkila, S.; et al. Inhibition and binding studies of carbonic anhydrase isozymes I, II and IX with benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides. J. Enzyme Inhib. Med. Chem. 2010, 25, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Falconer, R.J. Applications of isothermal titration calorimetry—The research and technical developments from 2011 to 2015: Review of Isothermal Titration Calorimetry from 2011 to 2015. J. Mol. Recognit. 2016, 29, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Callies, O.; Daranas, A.H. Application of isothermal titration calorimetry as a tool to study natural product interactions. Nat. Prod. Rep. 2016, 33, 881–904. [Google Scholar] [CrossRef]
- Linkuvienė, V.; Zubrienė, A.; Manakova, E.; Petrauskas, V.; Baranauskienė, L.; Zakšauskas, A.; Smirnov, A.; Gražulis, S.; Ladbury, J.E.; Matulis, D. Thermodynamic, kinetic, and structural parameterization of human carbonic anhydrase interactions toward enhanced inhibitor design. Q. Rev. Biophys. 2018, 51, 1–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, P.W.; Mecinović, J.; Moustakas, D.T.; Thomas, S.W.; Harder, M.; Mack, E.T.; Lockett, M.R.; Héroux, A.; Sherman, W.; Whitesides, G.M. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc. Natl. Acad. Sci. USA 2011, 108, 17889–17894. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assta quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Bruker. SAINT (Version 8.18c). In Bruker Advanced X-ray Solutions; Bruker AXS: Madison, WI, USA, 2012. [Google Scholar]
- Bruker. SADABS (Version 2.11). In Bruker Advanced X-ray Solutions; Bruker AXS: Madison, WI, USA, 2012. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of ıt Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Compound | Thiol | Conversion (%) | 2-Substituted (%) | 4-Substituted (%) | 2,4-Disubstituted (%) |
|---|---|---|---|---|---|
| 1 |  | 30.07 28.77 | 18.70 17.38 | 81.30 82.62 | - |
| 2 | 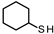 | 15.71 13.92 | 9.09 14.11 | 90.91 85.89 | - |
| 1 | 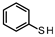 | 89.47 96.67 | 58.82 | 35.29 | 5.88 11.22 |
| 88.78 * | |||||
| 2 | 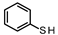 | 95.07 96.92 | 51.81 | 40.42 | 7.77 14.45 |
| 85.55 * | |||||
| 1 | 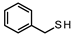 | 72.73 71.55 | 23.53 22.91 | 73.53 72.79 | 2.94 4.30 |
| 1 | 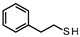 | 72.15 73.60 | 12.28 12.05 | 87.72 87.95 | - |
| 1 | 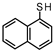 | 90.97 95.25 | 76.34 78.84 | 19.08 15.72 | 4.58 5.44 |
| 1 | 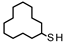 | 13.70 18.19 | - | 100.00 100.00 | - |
| Compound | Chemical Structure | ortho Substituent | para Substituent | pKa | Kd (nM) | CAI | CAII | CAIII | CAIV | CAVA | CAVB | CAVI | CAVII | C A IX | CAXII | CAXIII | CAXIV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Lab. Name) | |||||||||||||||||
| 1 | 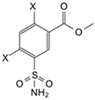 | Cl | Cl | 8.9 | Observed | 14,000 | 71 | 50,000 | 180 | 6700 | 310 | 170 | 67 | 59 | 560 | 83 | 72 |
| (EA2-1) | Intrinsic | 160 | 0.39 | 150 | 0.65 | 55 | 1.9 | 0.19 | 0.32 | 0.21 | 2.7 | 0.94 | 0.35 | ||||
| 2 | Br | Br | 8.8 | Observed | 20,000 | 91 | ≥2 × 105 | 140 | 3700 | 33 | 290 | 56 | 120 | 330 | 63 | 40 | |
| (LJ14-4) | Intrinsic | 290 | 0.63 | ≥750 | 0.63 | 38 | 0.26 | 0.41 | 0.34 | 0.52 | 2 | 0.89 | 0.24 | ||||
| 3b | cyclohexyl | Cl | 9.6 | Observed | 17,000 | 250 | 40,000 | 400 | 100,000 | 11,000 | 330 | 150 | 0.33 | 33 | 50 | 13 | |
| (EA2-3) | Intrinsic | 39 | 0.28 | 24 | 0.29 | 170 | 14 | 0.080 | 0.15 | 0.00020 | 0.030 | 0.11 | 0.010 | ||||
| 3c |  | benzyl | Cl | 9.5 | Observed | 6900 | 110 | 80,000 | 120 | ≥2 × 105 | 5000 | 360 | 150 | 2.9 | 22 | 38 | 40 |
| (EA2-4) | Intrinsic | 20 | 0.16 | 61 | 0.11 | ≥420 | 7.9 | 0.10 | 0.19 | 0.0030 | 0.030 | 0.11 | 0.050 | ||||
| 3d | 2-phenylethyl | Cl | 9.6 | Observed | 13,000 | 170 | 15,000 | 430 | 110,000 | 10,000 | 630 | 100 | 13 | 53 | 38 | 43 | |
| (EA2-5) | Intrinsic | 30 | 0.18 | 9.3 | 0.30 | 190 | 13 | 0.14 | 0.10 | 0.0090 | 0.050 | 0.090 | 0.040 | ||||
| 3e | cyclododecyl | Cl | Observed | 12,000 | 400 | ≥2 × 105 | 200 | ≥2 × 105 | 67,000 | 2000 | 3300 | 0.44 | 41 | 27 | 37 | ||
| (EA2-12) | Intrinsic | 27 | 0.44 | ≥120 | 0.14 | ≥330 | 84 | 0.46 | 3.2 | 0.00030 | 0.040 | 0.060 | 0.040 | ||||
| 4b | cyclohexyl | Br | 9.6 * | Observed | 6700 | 200 | 48,000 | 330 | ≥2 × 105 | 3300 | 560 | 220 | 0.12 | 16 | 33 | 20 | |
| (LJ15-12) | Intrinsic | 15 | 0.22 | 29 | 0.24 | ≥330 | 4.2 | 0.13 | 0.22 | 0.000080 | 0.020 | 0.080 | 0.020 | ||||
| 5a | 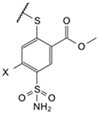 | Cl | phenyl | 9.4 * LS | Observed | 3300 | 36 | 17,000 | 14 | ≥2 × 105 | 250 | 200 | 50 | 2.5 | 5.7 | 20 | 19 |
| (EA2-2) | Intrinsic | 12 | 0.060 | 16 | 0.020 | ≥530 | 0.50 | 0.070 | 0.080 | 0.0030 | 0.0090 | 0.070 | 0.030 | ||||
| 5b | Cl | cyclohexyl | 9.4 | Observed | 330 | 20 | ≥2 × 105 | 67 | 4000 | 5.7 | 210 | 21 | 7.5 | 13 | 10 | 5.1 | |
| (EA1-3N) | Intrinsic | 1.2 | 0.040 | ≥190 | 0.080 | 11 | 0.010 | 0.070 | 0.030 | 0.0080 | 0.020 | 0.040 | 0.0080 | ||||
| 5c | Cl | benzyl | 9.4 * LS | Observed | 2000 | 33 | ≥2 × 105 | 120 | 1800 | 470 | 290 | 130 | 14 | 400 | 100 | 47 | |
| (EA1-4N) | Intrinsic | 7.3 | 0.060 | ≥190 | 0.13 | 4.8 | 0.92 | 0.10 | 0.19 | 0.020 | 0.61 | 0.36 | 0.070 | ||||
| 5d | Cl | 2-phenylethyl | 9.3 | Observed | 3400 | 27 | 89,000 | 1000 | 3800 | 4.4 | 850 | 24 | 12 | 180 | 7.4 | 17 | |
| (EA1-5N) | Intrinsic | 16 | 0.060 | 110 | 1.5 | 13 | 0.010 | 0.38 | 0.050 | 0.020 | 0.34 | 0.030 | 0.030 | ||||
| 5f | Cl | 1-naphthyl | 9.4 * LS | Observed | 27,000 | 110 | ≥2 × 105 | 66,000 | 96,000 | 510 | 14,000 | 1100 | 22 | 66 | 57 | 63,000 | |
| (EA2-NF) | Intrinsic | 98 | 0.20 | ≥190 | 75 | 250 | 1.0 | 5.0 | 1.7 | 0.020 | 0.10 | 0.21 | 97 | ||||
| 6a | Br | phenyl | 9.4 * LS | Observed | 2500 | 80 | ≥2 × 105 | 40 | 86,000 | 31 | 420 | 210 | 9.8 | 20 | 16 | 13 | |
| (LJ15-11) | Intrinsic | 9.2 | 0.14 | ≥190 | 0.050 | 230 | 0.060 | 0.15 | 0.32 | 0.010 | 0.030 | 0.060 | 0.020 | ||||
| 7b | 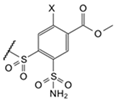 | cyclohexyl | Cl | 9.8 | Observed | 100,000 | 50,000 | 100,000 | 41,000 | 100,000 | 100,000 | 190,000 | ≥2 × 105 | 9200 | 67,000 | 69,000 | 69,000 |
| (EA2-3o) | Intrinsic | 150 | 35 | 38 | 19 | 110 | 79 | 28 | ≥120 | 4.2 | 41 | 99 | 42 | ||||
| 7c | benzyl | Cl | 9.8* LS | Observed | ≥2 × 105 | 190,000 | ≥2 × 105 | 5900 | ≥2 × 105 | ≥2 × 105 | 130,000 | ≥2 × 105 | 790 | 6700 | 140,000 | 6700 | |
| (EA2-4o) | Intrinsic | ≥290 | 130 | ≥76 | 2.7 | ≥210 | ≥160 | 19 | ≥120 | 0.36 | 4.1 | 200 | 4.1 | ||||
| 7d | 2-phenylethyl | Cl | 9.8 * LS | Observed | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | 130,000 | ≥2 × 105 | 6700 | 34,000 | 22,000 | ≥2 × 105 | |
| (EA2-5o) | Intrinsic | ≥300 | ≥140 | ≥76 | ≥90 | ≥210 | ≥160 | 19 | ≥120 | 3.0 | 21 | 32 | ≥120 | ||||
| 8b | cyclohexyl | Br | 9.9 | Observed | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | ≥2 × 105 | 6700 | ≥2 × 105 | 71,000 | ≥2 × 105 | |
| (LJ15-12o) | Intrinsic | ≥230 | ≥110 | ≥60 | ≥16 | ≥170 | ≥130 | ≥23 | ≥97 | 2.4 | ≥97 | 82 | ≥97 | ||||
| 9a | 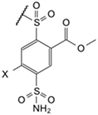 | Cl | phenyl | 8.2 | Observed | 140 | 0.91 | 3300 | 25 | 770 | 13 | 56 | 0.45 | 3.3 | 22 | 5.6 | 0.83 |
| (EA2-2o) | Intrinsic | 7.9 | 0.020 | 48 | 0.42 | 30 | 0.40 | 0.30 | 0.010 | 0.060 | 0.51 | 0.30 | 0.020 | ||||
| 9b | Cl | cyclohexyl | 8.2 | Observed | 67 | 0.67 | 2800 | 200 | 500 | 0.75 | 42 | 0.18 | 1 | 14 | 2.7 | 0.64 | |
| (EA1-3No) | Intrinsic | 3.7 | 0.020 | 40 | 3.4 | 20 | 0.020 | 0.22 | 0.0040 | 0.020 | 0.33 | 0.15 | 0.010 | ||||
| 9c | Cl | benzyl | 8.2 | Observed | 560 | 5 | 450 | 100 | 830 | 1.1 | 14 | 2.5 | 2.5 | 20 | 5 | 2.6 | |
| (EA1-4No) | Intrinsic | 31 | 0.13 | 6.5 | 1.7 | 33 | 0.030 | 0.080 | 0.060 | 0.040 | 0.46 | 0.27 | 0.060 | ||||
| 9d | Cl | 2-phenylethyl | 8.4 | Observed | 330 | 1.7 | 920 | 140 | 200 | 1.1 | 26 | 0.85 | 2.5 | 48 | 3 | 1.3 | |
| (EA1-5No) | Intrinsic | 12 | 0.030 | 8.5 | 1.6 | 5.1 | 0.020 | 0.090 | 0.010 | 0.030 | 0.72 | 0.10 | 0.020 | ||||
| 9f | Cl | 1-naphthyl | 8.4 | Observed | 140 | 1.3 | 14,000 | 50 | 1400 | 3.4 | 100 | 0.67 | 10 | 50 | 1.3 | 4 | |
| (EA2-Nfo) | Intrinsic | 5.1 | 0.020 | 130 | 0.55 | 36 | 0.060 | 0.35 | 0.010 | 0.11 | 0.74 | 0.040 | 0.060 | ||||
| 10a | Br | phenyl | 8.4 | Observed | 330 | 2.3 | 4800 | 370 | 1600 | 5.3 | 110 | 0.67 | 3.3 | 16 | 4.3 | 2 | |
| (LJ15-11o) | Intrinsic | 12 | 0.040 | 44 | 4.0 | 41 | 0.10 | 0.37 | 0.010 | 0.040 | 0.24 | 0.15 | 0.030 | ||||
| Acetazolamide | 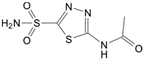 | 7.0 | Observed | 2400 | 46 | 40,000 | 87 | 840 | 140 | 220 | 13 | 21 | 130 | 120 | 63 | ||
| Intrinsic | 1100 | 9.8 | 4600 | 12 | 270 | 34 | 9.8 | 2.4 | 2.9 | 24 | 53 | 12 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakšauskas, A.; Čapkauskaitė, E.; Paketurytė-Latvė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Stančaitis, L.; Mickevičiūtė, A.; Jachno, J.; et al. Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX. Int. J. Mol. Sci. 2022, 23, 130. https://doi.org/10.3390/ijms23010130
Zakšauskas A, Čapkauskaitė E, Paketurytė-Latvė V, Smirnov A, Leitans J, Kazaks A, Dvinskis E, Stančaitis L, Mickevičiūtė A, Jachno J, et al. Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX. International Journal of Molecular Sciences. 2022; 23(1):130. https://doi.org/10.3390/ijms23010130
Chicago/Turabian StyleZakšauskas, Audrius, Edita Čapkauskaitė, Vaida Paketurytė-Latvė, Alexey Smirnov, Janis Leitans, Andris Kazaks, Elviss Dvinskis, Laimonas Stančaitis, Aurelija Mickevičiūtė, Jelena Jachno, and et al. 2022. "Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX" International Journal of Molecular Sciences 23, no. 1: 130. https://doi.org/10.3390/ijms23010130
APA StyleZakšauskas, A., Čapkauskaitė, E., Paketurytė-Latvė, V., Smirnov, A., Leitans, J., Kazaks, A., Dvinskis, E., Stančaitis, L., Mickevičiūtė, A., Jachno, J., Jezepčikas, L., Linkuvienė, V., Sakalauskas, A., Manakova, E., Gražulis, S., Matulienė, J., Tars, K., & Matulis, D. (2022). Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX. International Journal of Molecular Sciences, 23(1), 130. https://doi.org/10.3390/ijms23010130