
Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression




Abstract
:1. Introduction
2. Cancer Progression Is Driven by MMPs in the TME
2.1. Epithelial-to-Mesenchymal Transition Depends on the Activity of MMPs
2.2. The ECM-Degrading Activity of MMPs Is Involved in All Steps of the Metastasis Cascade
2.3. ECM Remodeling by MMPs Is Important for Tumor Angiogenesis
3. Molecular Biology of MMPs
3.1. MMPs Show Many Structural and Functional Similarities and Yet Great Diversity
3.2. MMPs Have Diverse Molecular Functions
3.3. Matrix Metalloproteinases Are Tightly Regulated
3.4. MMP-14 Has a Central Role among the MMPs
4. Cellular Adhesome Structures in the TME
4.1. Using Varied Adhesome Structures, Cells Can Interact Differently with the ECM
4.2. Focal Complexes Are Formed as the First Adhesive Matrix Contacts
4.3. Focal Adhesions and Fibrillary Adhesions Allow Force Exertion
4.4. Podosomes Coordinate Cell Adhesion with Focal ECM Degradation
4.5. Invadosomes Are Both Adhesive and Proteolytic Structures
4.6. A Sealing Zone Surrounds the Resorption Lacuna of Osteoclasts
4.7. Invading Cancer Cells Can Cleave the ECM at Belt-Like Compressions That Impede Cell Migration
5. MMPs and TME: More Than a Hit-and-Run Relation
5.1. MMPs Play an Essential Role in the Remodeling of Tumor Stroma ECM
5.2. MMPs Generate Bioactive Matrikines during Degradation and Remodeling of the ECM
6. Translational Perspectives: MMPs and Invadopodia Are Worthwhile Targets for Inhibiting Cancer Progression
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL2 | Abelson murine leukemia viral oncogene homolog (Arg, Abl-related gene) |
| ADAM | a disintegrin and metalloproteinase |
| ADAMTS | a disintegrin and metalloproteinase with thrombospondin motifs |
| ADGRB1 | adhesion G protein-coupled receptor B1 |
| Arp2/3 | actin related protein 2/3 (complex) |
| AP-2µ2 | adaptor protein-2 subunit µ2 |
| Ask1 | apoptosis signal-regulating kinase 1 |
| BAI1 | brain-specific angiogenesis inhibitor 1 |
| BLACAT1 | bladder cancer-associated transcript-1 |
| BM | basement membrane |
| CAF | cancer-associated fibroblast |
| CAP-G | capping actin protein, gelsolin-like |
| CCN | Cyr61-CTGF-NOV family of matricellular proteins |
| CDC42 | cell division control protein 42 homolog |
| CDCP1 | CUB-domain-containing protein 1 |
| Cdk5 | cyclin-dependent kinase 5 |
| CLCN7 | chloride voltage-gated channel 7 |
| CLIC4 | intracellular chloride channel 4 |
| COMP | cartilage oligomeric matrix protein |
| CTGF | connective tissue growth factor, CCN2 |
| CTGF-L | connective tissue growth factor ligand, CCN5 |
| Cyr61 | Cysteine-rich angiogenic inducer 61, CCN1 |
| DDR1 | discoidin domain receptor 1 |
| DKK-3 | Dickkopf-related protein-3 |
| DLL1 | Delta-like 1 |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF(R) | epidermal growth factor (receptor) |
| EGR-1 | early growth response protein-1 |
| EMT | epithelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| ER-β | estrogen receptor β |
| ESCRT | endosomal sorting complex |
| EVL | Ena/vasodilator-stimulated phosphoprotein (VASP)-like |
| FAK | focal adhesion kinase |
| FYCO1 | FYVE and coiled-coil domain-containing protein 1 |
| FIH-1 | factor inhibiting HIF-1 |
| GRASP55 | Golgi reassembly stacking protein 55 |
| HA | hyaluronic acid |
| HB-EGF | heparin-binding epidermal growth factor |
| HIC-5 | hydrogen peroxide-inducible clone 5 protein (TGFB1I1) |
| HIF | hypoxia induced factor |
| ICAM-1 | intercellular adhesion molecule-1 |
| IκBα | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IL | interleukin |
| ILK | integrin-linked kinase |
| ILP | invadopodia-like protrusion |
| IQGAP1 | Ras GTPase-activating-like protein |
| KIF | kinesin superfamily protein |
| LAMTOR1 | late endosomal/lysosomal adaptor, MAPK, and mTOR activator 1 |
| LOX(L) | lysyl oxidase (-like) |
| mDia2 | Diaphanous-related formin protein 2 |
| MAPK | mitogen-activated protein kinase |
| Mena/VASP | protein-enabled homolog/vasodilator-stimulated phosphoprotein |
| Mi-2/NuRD | nucleosome remodeling deacetylase |
| Mint3 | Munc18-1-interacting protein 3 |
| MMP | matrix-metalloproteinase |
| mTOR | mechanistic target of rapamycin |
| NC1 | non-collagenous domain-1 |
| NCAM | neural cell adhesion molecule |
| NCK1 | non-catalytic region of tyrosine kinase adaptor protein 1 |
| NET | neutrophil extracellular trap |
| NHE1 | Na+/H+ exchanger 1 |
| NHERF1 | Na+/H+ exchanger 1 regulating factor 1 |
| NOV | nephroblastoma overexpressed, CCN3 |
| NRTK | non-receptor tyrosine kinase |
| N-WASp | neural Wiskott–Aldrich syndrome protein |
| PAK | p21-activated kinase |
| PARK7 | Parkinsonism-associated deglycase |
| PCSK6 | proprotein convertase subtilisin/kexin type 6 |
| PARP | poly (ADP-ribose) polymerase |
| PDZ | post synaptic density protein, Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein domain |
| PI3K | phosphatidylinositol 3-kinase |
| PI(3,4)P2 | phosphatidylinositol-3,4-bisphosphate |
| PKC | protein kinase C |
| PTK2B | protein tyrosine kinase-2β, Pyk2 |
| PX | phox homology |
| RAB7 | Ras-related protein 7 |
| Ras | rat sarcoma virus |
| RECK | reversion-inducing cysteine-rich protein with Kazal motifs |
| RhoA | Ras homolog family member A |
| SDF | stromal cell-derived factor |
| SERPINE2 | serine proteinase inhibitor, clade E, member 2 |
| Src | sarcoma proto-oncogene tyrosine-protein kinase |
| SNAI1 | snail family transcriptional repressor-1 |
| SPARC | secreted protein acidic and rich in cysteine, osteonectin |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TGF-β | transforming growth factor β |
| TGFB1I1 | transforming growth factor β-1-induced transcript 1 protein (HIC-5) |
| TIMP | tissue inhibitor of metalloproteinase |
| TKS4/5 | tyrosine kinase substrate (scaffold) protein with four/five SH3 domains |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor-α |
| UBTD1 | ubiquitin domain-containing protein 1 |
| uPA(-R) | urokinase plasminogen activator (surface receptor) |
| VASP | vasodilator-stimulated phosphoprotein |
| WASp | Wiskott–Aldrich syndrome protein |
| WAVE | WASp and verprolin homolog |
| Wnt | Wingless-related integration site |
| YAP | Yes-associated protein 1 |
| ZEB | zinc finger E-box binding homeobox |
References
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. The aging of the 2000 and 2011 Hallmarks of Cancer reviews: A critique. J. Bio Sci. 2013, 38, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amith, S.R.; Fong, S.; Baksh, S.; Fliegel, L. Na (+)/H (+) exchange in the tumour microenvironment: Does NHE1 drive breast cancer carcinogenesis? Int. J. Dev. Biol. 2015, 59, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Kryza, T.; Lyons, N.J.; He, Y.; Hooper, J.D. The CDCP1 Signaling Hub: A Target for Cancer Detection and Therapeutic Intervention. Cancer Res. 2021, 81, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Panda, C.K.; Nandi, S.; Mukhopadhyay, A. An insight into metastasis: Random or evolving paradigms? Pathol. Res. Pract. 2018, 214, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Niland, S.; Eble, J.A. Hold on or Cut? Integrin-and MMP-Mediated Cell-Matrix Interactions in the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 22, 238. [Google Scholar] [CrossRef] [PubMed]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rosse, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Ohtsuka, T.; Okada, Y.; Kanai, Y. Stromal metalloproteinases: Crucial contributors to the tumor microenvironment. Pathol. Int. 2021, 71, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Garcia-Hernandez, A.A.; Ramos, C. Matrix Metalloproteinases’ Role in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 97–131. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Will, H.; Atkinson, S.J.; Butler, G.S.; Smith, B.; Murphy, G. The soluble catalytic domain of membrane type 1 matrix metalloproteinase cleaves the propeptide of progelatinase A and initiates autoproteolytic activation. Regulation by TIMP-2 and TIMP-3. J. Biol. Chem. 1996, 271, 17119–17123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamahana, H.; Terashima, M.; Takatsuka, R.; Asada, C.; Suzuki, T.; Uto, Y.; Takino, T. TGF-β1 facilitates MT1-MMP-mediated proMMP-9 activation and invasion in oral squamous cell carcinoma cells. Bio Chem. Biophys. Rep. 2021, 27, 101072. [Google Scholar] [CrossRef] [PubMed]
- Knäuper, V.; Will, H.; López-Otin, C.; Smith, B.; Atkinson, S.J.; Stanton, H.; Hembry, R.M.; Murphy, G. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate active enzyme. J. Biol. Chem. 1996, 271, 17124–17131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabova, L.; Chrysovergis, K.; Yamada, S.S.; Holmbeck, K. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene 2008, 27, 3274–32781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, E.K.; Courtneidge, S.A. Invadosomes are coming: New insights into function and disease relevance. FEBS J. 2018, 285, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9373. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francou, A.; Anderson, K.V. The Epithelial-to-Mesenchymal Transition (EMT) in Development and Cancer. Annu Rev. Cancer Biol. 2020, 4, 197–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Thiery, J.P.; Huang, R.Y. Epithelial-to-mesenchymal transition: Lessons from development, insights into cancer and the potential of EMT-subtype based therapeutic intervention. Phys. Biol. 2019, 16, 041004. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell. 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. EMT: An Update. Methods Mol. Biol. 2021, 2179, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Albig, A.R.; Regner, M.; Schiemann, B.J.; Schiemann, W.P. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 2008, 29, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zheng, J.; Yu, J.; Wu, Y.; Guo, J.; Xu, Z.; Sun, X. Knockdown of MMP-1 inhibits the progression of colorectal cancer by suppressing the PI3K/Akt/c-myc signaling pathway and EMT. Oncol. Rep. 2020, 43, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Qi, Y.; Zhao, L.; Chen, D.; Zhou, Y.; Zhou, H.; Lv, Y.; Zhang, L.; Jin, S.; Li, S.; et al. Matrix metalloproteinase-14 induces epithelial-to-mesenchymal transition in synovial sarcoma. Hum. Pathol. 2018, 80, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Molina, J.; Gramolelli, S.; Liao, Z.; Carlson, J.W.; Ojala, P.M.; Lehti, K. MMP14 in Sarcoma: A Regulator of Tumor Microenvironment Communication in Connective Tissues. Cells 2019, 8, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illman, S.A.; Lohi, J.; Keski-Oja, J. Epilysin (MMP-28)-structure, expression and potential functions. Exp. Dermatol. 2008, 17, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Lin, Z.; Jiang, J.; Lu, S.; Chen, M.; Que, H.; He, X.; Que, G.; Mao, J.; Xiao, J.; et al. MMP14 regulates cell migration and invasion through epithelial-mesenchymal transition in nasopharyngeal carcinoma. Am. J. Transl. Res. 2015, 7, 950–958. [Google Scholar] [PubMed]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garmon, T.; Wittling, M.; Nie, S. MMP14 Regulates Cranial Neural Crest Epithelial-to-Mesenchymal Transition and Migration. Dev. Dyn. 2018, 247, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Pekkonen, P.; Laurinavicius, S.; Sugiyama, N.; Henderson, S.; Gunther, T.; Rantanen, V.; Kaivanto, E.; Aavikko, M.; Sarek, G.; et al. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe 2011, 10, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozanov, D.V.; Deryugina, E.I.; Monosov, E.Z.; Marchenko, N.D.; Strongin, A.Y. Aberrant, persistent inclusion into lipid rafts limits the tumorigenic function of membrane type-1 matrix metalloproteinase in malignant cells. Exp. Cell Res. 2004, 293, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44–46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Taniwaki, K.; Fukamachi, H.; Komori, K.; Ohtake, Y.; Nonaka, T.; Sakamoto, T.; Shiomi, T.; Okada, Y.; Itoh, T.; Itohara, S.; et al. Stroma-derived matrix metalloproteinase (MMP)-2 promotes membrane type 1-MMP-dependent tumor growth in mice. Cancer Res. 2007, 67, 4311–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillari, G. The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process. Int. J. Mol. Sci. 2020, 21, 4526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Miyake, M.; Lawton, A.; Goodison, S.; Rosser, C.J. Matrix metalloproteinase-10 promotes tumor progression through regulation of angiogenic and apoptotic pathways in cervical tumors. BMC Cancer 2014, 14, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepeda, M.A.; Pelling, J.J.; Evered, C.L.; Williams, K.C.; Freedman, Z.; Stan, I.; Willson, J.A.; Leong, H.S.; Damjanovski, S. Less is more: Low expression of MT1-MMP is optimal to promote migration and tumourigenesis of breast cancer cells. Mol. Cancer 2016, 15, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaco, A.; Rezaei, M.; Niland, S.; Eble, J.A. Collateral Damage Intended-Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wen, G.; Zhang, L.; Lin, L.; Moore, A.; Wu, S.; Ye, S.; Xiao, Q. An important role of matrix metalloproteinase-8 in angiogenesis in vitro and in vivo. Cardiovasc. Res. 2013, 99, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Yana, I.; Sagara, H.; Takaki, S.; Takatsu, K.; Nakamura, K.; Nakao, K.; Katsuki, M.; Taniguchi, S.; Aoki, T.; Sato, H.; et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J. Cell Sci. 2007, 120 Pt 9, 1607–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Jin, G.; Cao, R.; Zhang, S.; Cao, Y.; Zhou, Z. MT1-MMP sheds LYVE-1 on lymphatic endothelial cells and suppresses VEGF-C production to inhibit lymphangiogenesis. Nat. Commun. 2016, 7, 10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Robey, P.G.; Poole, A.R.; Pidoux, I.; et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.Y.; Chang, J.H.; Azar, D.T. MMP14-Containing Exosomes Cleave VEGFR1 and Promote VEGFA-Induced Migration and Proliferation of Vascular Endothelial Cells. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Eisenach, P.A.; Roghi, C.; Fogarasi, M.; Murphy, G.; English, W.R. MT1-MMP regulates VEGF-A expression through a complex with VEGFR-2 and Src. J. Cell Sci. 2010, 123 Pt 23, 4182–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, J.R.; Holmbeck, K.; Bugge, T.H.; Gutkind, J.S. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J. Biol. Chem. 2007, 282, 6899–6905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.M.; Zhang, X.H.; Ye, L.Q.; Zhang, K.; Yang, N.N.; Geng, S.; Chen, J.; Zhao, S.X.; Yang, K.L.; Fan, F.F. Insufficient CD100 shedding contributes to suppression of CD8(+) T-cell activity in non-small cell lung cancer. Immunology 2020, 160, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Seeger, W.; Pullamsetti, S.S. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur.. Respir. J. 2012, 40, 766–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C.; et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.K.; Li, Q.; Parks, W.C. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am. J. Pathol. 2003, 162, 1831–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Park, J.H.; Cheon, D.H.; Kim, T.; Park, Y.E.; Oh, E.S.; Lee, J.E.; Lee, S.T. Processing of syndecan-2 by matrix metalloproteinase-14 and effect of its cleavage on VEGF-induced tube formation of HUVECs. Bio Chem. J. 2017, 474, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Holopainen, J.M.; Moilanen, J.A.; Sorsa, T.; Kivelä-Rajamäki, M.; Tervahartiala, T.; Vesaluoma, M.H.; Tervo, T.M. Activation of matrix metalloproteinase-8 by membrane type 1-MMP and their expression in human tears after photorefractive keratectomy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2550–2556. [Google Scholar] [CrossRef] [PubMed]
- Miekus, N.; Luise, C.; Sippl, W.; Baczek, T.; Schmelzer, C.E.H.; Heinz, A. MMP-14 degrades tropoelastin and elastin. Biochimie 2019, 165, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Asp. Med. 2008, 29, 290–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, B.C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse matrix metalloproteinase families. Hum. Genomics 2010, 4, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J. Cell Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.J.; Du, Y.; Zhao, X.; Ma, L.Y.; Cao, G.W. Inflammation-related factors predicting prognosis of gastric cancer. World J. Gastroenterol. 2014, 20, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qu, L.; Zhao, C.; Shou, C. Extracellular gamma-synuclein promotes tumor cell motility by activating β1 integrin-focal adhesion kinase signaling pathway and increasing matrix metalloproteinase-24,-2 protein secretion. J. Exp. Clin. Cancer Res. 2018, 37, 117. [Google Scholar] [CrossRef] [PubMed]
- Gutschalk, C.M.; Yanamandra, A.K.; Linde, N.; Meides, A.; Depner, S.; Mueller, M.M. GM-CSF enhances tumor invasion by elevated MMP-2, -9, and -26 expression. Cancer Med. 2013, 2, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl Acad Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Kinoshita, T.; Takino, T.; Nakayama, K.; Seiki, M. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996, 393, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.; Nagase, H.; Pei, D. Activation of membrane-type matrix metalloproteinase 3 zymogen by the proprotein convertase furin in the trans-Golgi network. Cancer Res. 2002, 62, 675–681. [Google Scholar] [PubMed]
- Yana, I.; Weiss, S.J. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol. Biol. Cell 2000, 11, 2387–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, D.E.; Lopez De Cicco, R.; Cenna, J.; Litwin, S.; Cukierman, E.; Klein-Szanto, A.J. PACE4 expression in mouse basal keratinocytes results in basement membrane disruption and acceleration of tumor progression. Cancer Res. 2005, 65, 7310–7319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25): Chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkowski, K.; Bielecka, E.; Thøgersen, I.B.; Bocheńska, O.; Płaza, K.; Kalińska, M.; Sąsiadek, L.; Magoch, M.; Pęcak, A.; Wiśniewska, M.; et al. Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)-A CleavEx Based Analysis. Int. J. Mol. Sci. 2020, 21, 4383. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, E.; Imai, K.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997, 272, 2446–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Morodomi, T.; Enghild, J.J.; Suzuki, K.; Yasui, A.; Nakanishi, I.; Salvesen, G.; Nagase, H. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur. J. Bio Chem. 1990, 194, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Will, H.; Atkinson, S.J.; Murphy, G. Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. Eur. J. Bio Chem. 1997, 244, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Takino, T.; Sato, H.; Shinagawa, A.; Seiki, M. Identification of the second membrane-type matrix metalloproteinase (MT-MMP-2) gene from a human placenta cDNA library. MT-MMPs form a unique membrane-type subclass in the MMP family. J. Biol. Chem. 1995, 270, 23013–23020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, D. Identification and characterization of the fifth membrane-type matrix metalloproteinase MT5-MMP. J. Biol. Chem. 1999, 274, 8925–8932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano, E.; Pendás, A.M.; Freije, J.P.; Nakano, A.; Knäuper, V.; Murphy, G.; López-Otin, C. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase a overexpressed in brain tumors. Cancer Res. 1999, 59, 2570–2576. [Google Scholar] [PubMed]
- Morrison, C.J.; Overall, C.M. TIMP independence of matrix metalloproteinase (MMP)-2 activation by membrane type 2 (MT2)-MMP is determined by contributions of both the MT2-MMP catalytic and hemopexin C domains. J. Biol. Chem. 2006, 281, 26528–26539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Nakamura, H.; Ohuchi, E.; Fujii, Y.; Murakami, Y.; Sato, H.; Seiki, M.; Okada, Y. Characterization of a truncated recombinant form of human membrane type 3 matrix metalloproteinase. Eur. J. Bio Chem. 1999, 262, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Allen, E.; Apel, I.J.; Gyetko, M.R.; Weiss, S.J. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell 1998, 95, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotary, K.B.; Yana, I.; Sabeh, F.; Li, X.Y.; Holmbeck, K.; Birkedal-Hansen, H.; Allen, E.D.; Hiraoka, N.; Weiss, S.J. Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and-independent processes. J. Exp. Med. 2002, 195, 295–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, E.; Yana, I.; Fujita, C.; Irifune, A.; Takeda, M.; Madachi, A.; Mori, S.; Hamada, Y.; Kawaguchi, N.; Matsuura, N. The role of MT2-MMP in cancer progression. Bio Chem. Biophys.. Res. Commun. 2010, 393, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Valdés-Sánchez, T.; Llano, E.; Menéndez, L.; Baamonde, A.; Denlinger, B.L.; Belmonte, C.; Juárez, L.; Lastra, A.; García-Suárez, O.; et al. Metalloproteinase MT5-MMP is an essential modulator of neuro-immune interactions in thermal pain stimulation. Proc. Natl. Acad. Sci. USA 2009, 106, 16451–16456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauwe, B.; Opdenakker, G. Intracellular substrate cleavage: A novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit. Rev. Bio. Chem. Mol. Biol. 2010, 45, 351–423. [Google Scholar] [CrossRef] [PubMed]
- Shofuda, T.; Shofuda, K.; Ferri, N.; Kenagy, R.D.; Raines, E.W.; Clowes, A.W. Cleavage of focal adhesion kinase in vascular smooth muscle cells overexpressing membrane-type matrix metalloproteinases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, T.; Seiki, M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 2010, 285, 29951–29964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo, P.; Guadamillas, M.C.; Hernández-Riquer, M.V.; Pollán, A.; Grande-García, A.; Bartolomé, R.A.; Vasanji, A.; Ambrogio, C.; Chiarle, R.; Teixidó, J.; et al. MT1-MMP is required for myeloid cell fusion via regulation of Rac1 signaling. Dev. Cell 2010, 18, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingras, D.; Michaud, M.; Di Tomasso, G.; Béliveau, E.; Nyalendo, C.; Béliveau, R. Sphingosine-1-phosphate induces the association of membrane-type 1 matrix metalloproteinase with p130Cas in endothelial cells. FEBS Lett. 2008, 582, 399–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, D.; Tomari, T.; Nagano, M.; Koshikawa, N.; Seiki, M. A novel protein associated with membrane-type 1 matrix metalloproteinase binds p27(kip1) and regulates RhoA activation, actin remodeling, and matrigel invasion. J. Biol. Chem. 2009, 284, 27315–27326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Wang, C.Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44–46, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, M.P.; Brinckerhoff, C.E. Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: Can MMPs be good for you? J. Cell Physiol. 2007, 213, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Jana, S.; Choudhary, P.; Swarnakar, S. Triumph and tumult of matrix metalloproteinases and their crosstalk with eicosanoids in cancer. Cancer Metastasis Rev. 2018, 37, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol. Ther. 2012, 20, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.L.; Pillai, S.; Pernazza, D.; Sebti, S.M.; Lawrence, N.J.; Chellappan, S.P. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb-Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012, 72, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kuscu, C.; Banach, A.; Zhang, Q.; Pulkoski-Gross, A.; Kim, D.; Liu, J.; Roth, E.; Li, E.; Shroyer, K.R.; et al. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer Res. 2015, 75, 2674–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Wang, L.; Lv, W.; Lv, Z. Effects of miR-7 on Hcy-induced rat cerebral arterial vascular smooth muscle cell proliferation, migration and inflammatory factor expression by targeting MMP-14 to regulate TLR4/NF-κB signaling pathway. Cell Mol. Biol. 2020, 66, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Feng, X.D.; Zhu, W.Q.; Bao, Y.N. LncRNA BLACAT1 regulates the viability, migration and invasion of oral squamous cell carcinoma cells by targeting miR-142-5p. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10313–10323. [Google Scholar] [CrossRef] [PubMed]
- He, J.H.; Han, Z.P.; Luo, J.G.; Jiang, J.W.; Zhou, J.B.; Chen, W.M.; Lv, Y.B.; He, M.L.; Zheng, L.; Li, Y.G.; et al. Hsa_Circ_0007843 Acts as a mIR-518c-5p Sponge to Regulate the Migration and Invasion of Colon Cancer SW480 Cells. Front. Genet. 2020, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Djuric, T.; Zivkovic, M. Overview of MMP Biology and Gene Associations in Human Diseases. In The Role of Matrix Metalloproteinase in Human Body Pathologies; Travascio, F., Ed.; IntechOpen: London, UK, 2017; pp. 3–33. [Google Scholar]
- Buache, E.; Thai, R.; Wendling, C.; Alpy, F.; Page, A.; Chenard, M.P.; Dive, V.; Ruff, M.; Dejaegere, A.; Tomasetto, C.; et al. Functional relationship between matrix metalloproteinase-11 and matrix metalloproteinase-14. Cancer Med. 2014, 3, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.; Hernandez-Barrantes, S.; Osenkowski, P.; Bernardo, M.M.; Gervasi, D.C.; Shimura, Y.; Meroueh, O.; Kotra, L.P.; Gálvez, B.G.; Arroyo, A.G.; et al. Complex pattern of membrane type 1 matrix metalloproteinase shedding. Regulation by autocatalytic cells surface inactivation of active enzyme. J. Biol. Chem. 2002, 277, 26340–26350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Strongin, A.Y.; Collier, I.; Bannikov, G.; Marmer, B.L.; Grant, G.A.; Goldberg, G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995, 270, 5331–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, Y.; Itoh, Y.; Nagase, H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J. Biol. Chem. 1995, 270, 18506–18511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Seiki, M. MT1-MMP: A potent modifier of pericellular microenvironment. J. Cell Physiol. 2006, 206, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.; Drews, M.; Conner, C.; Foda, H.D.; DeClerck, Y.A.; Langley, K.E.; Bahou, W.F.; Docherty, A.J.; Cao, J. Tissue inhibitor of metalloproteinase-2 (TIMP-2) binds to the catalytic domain of the cell surface receptor, membrane type 1-matrix metalloproteinase 1 (MT1-MMP). J. Biol. Chem. 1998, 273, 1216–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ito, N.; Nagase, H.; Seiki, M. The second dimer interface of MT1-MMP, the transmembrane domain, is essential for ProMMP-2 activation on the cell surface. J. Biol. Chem. 2008, 283, 13053–13062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knäuper, V.; Bailey, L.; Worley, J.R.; Soloway, P.; Patterson, M.L.; Murphy, G. Cellular activation of proMMP-13 by MT1-MMP depends on the C-terminal domain of MMP-13. FEBS Lett. 2002, 532, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, V.; Itoh, Y. MT1-MMP-dependent cell migration: Proteolytic and non-proteolytic mechanisms. Bio. Chem. Soc. Trans. 2019, 47, 811–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernov, A.V.; Sounni, N.E.; Remacle, A.G.; Strongin, A.Y. Epigenetic control of the invasion-promoting MT1-MMP/MMP-2/TIMP-2 axis in cancer cells. J. Biol. Chem. 2009, 284, 12727–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; He, Z.; Wang, C.; Zhou, Y.; Li, F.; Pu, W.; Zhang, X.; Feng, X.; Zhang, M.; Yecheng, X.; et al. Epigenetic silencing of ZNF132 mediated by methylation-sensitive Sp1 binding promotes cancer progression in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Lohi, J.; Lehti, K.; Valtanen, H.; Parks, W.C.; Keski-Oja, J. Structural analysis and promoter characterization of the human membrane-type matrix metalloproteinase-1 (MT1-MMP) gene. Gene 2000, 242, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Li, X.Y.; Hu, Y.; Saunders, T.L.; Virtanen, I.; Garcia de Herreros, A.; Becker, K.F.; Ingvarsen, S.; Engelholm, L.H.; Bommer, G.T.; et al. Mesenchymal cells reactivate Snail1 expression to drive three-dimensional invasion programs. J. Cell Biol. 2009, 184, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, H.J.; Okada, A.; Kim, K.W.; Sato, H.; Seiki, M. Identification of cis-acting promoter elements that support expression of membrane-type 1 matrix metalloproteinase (MT1-MMP) in v-src transformed Madin-Darby canine kidney cells. Clin. Exp. Metastasis 2000, 18, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Gramolelli, S.; Cheng, J.; Martinez-Corral, I.; Vähä-Koskela, M.; Elbasani, E.; Kaivanto, E.; Rantanen, V.; Tuohinto, K.; Hautaniemi, S.; Bower, M.; et al. PROX1 is a transcriptional regulator of MMP14. Sci. Rep. 2018, 8, 9531. [Google Scholar] [CrossRef] [PubMed]
- Lohi, J.; Lehti, K.; Westermarck, J.; Kähäri, V.M.; Keski-Oja, J. Regulation of membrane-type matrix metalloproteinase-1 expression by growth factors and phorbol 12-myristate 13-acetate. Eur. J. Bio Chem. 1996, 239, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Elsir, T.; Smits, A.; Lindström, M.S.; Nistér, M. Transcription factor PROX1: Its role in development and cancer. Cancer Metastasis Rev. 2012, 31, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrirukwanit, K.; Pavasant, P.; Blick, T.; Lafleur, M.A.; Thompson, E.W. High threshold of β1 integrin inhibition required to block collagen I-induced membrane type-1 matrix metalloproteinase (MT1-MMP) activation of matrix metalloproteinase 2 (MMP-2). Cancer Cell Int. 2014, 14, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigrino, P.; Drescher, C.; Mauch, C. Collagen-induced proMMP-2 activation by MT1-MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur. J. Cell Biol. 2001, 80, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Nakamura, T.; Suzuki, Y.; Imizu, T.; Matsumoto, K. 3-D collagen-dependent cell surface expression of MT1-MMP and MMP-2 activation regardless of integrin β1 function and matrix stiffness. Bio. Chem. Biophys. Res. Commun. 2011, 412, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, T.L.; Stitelman, D.; Davis, S.J.; Apte, S.S.; Madri, J.A. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J. Biol. Chem. 1999, 274, 22679–22685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Roghi, C.; Jones, L.; Gratian, M.; English, W.R.; Murphy, G. Golgi reassembly stacking protein 55 interacts with membrane-type (MT) 1-matrix metalloprotease (MMP) and furin and plays a role in the activation of the MT1-MMP zymogen. FEBS J. 2010, 277, 3158–3175. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Wu, K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012, 19, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Bio Chem. Mol. Biol. 2012, 3, 165–178. [Google Scholar] [PubMed]
- Kuscu, C.; Evensen, N.; Cao, J. MMP14 (matrix metallopeptidase 14 (membrane-inserted)). Atlas Genet. Cytogenet. Oncol. Haematol. 2011. [Google Scholar] [CrossRef]
- Yan, S.F.; Lu, J.; Zou, Y.S.; Soh-Won, J.; Cohen, D.M.; Buttrick, P.M.; Cooper, D.R.; Steinberg, S.F.; Mackman, N.; Pinsky, D.J.; et al. Hypoxia-associated induction of early growth response-1 gene expression. J. Biol. Chem. 1999, 274, 15030–15040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, A.; Kitajima, Y.; Ide, T.; Ohtaka, K.; Nagasawa, H.; Uto, Y.; Hori, H.; Miyazaki, K. Hypoxia accelerates cancer invasion of hepatoma cells by upregulating MMP expression in an HIF-1alpha-independent manner. Int. J. Oncol. 2006, 29, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C.E. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene 2005, 24, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Nájar, U.M.; Neurath, K.M.; Vumbaca, F.; Claffey, K.P. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene 2006, 25, 2379–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, T.; Seiki, M. Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: Beyond a proteinase. Cancer Sci. 2017, 108, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhou, Y.; Wang, L.; Zhang, J.; Wu, H.; Xiong, J.; Zhang, J.; Tian, Y.; Wang, C.; Wu, H. Transcriptional upregulation of MT2-MMP in response to hypoxia is promoted by HIF-1α in cancer cells. Mol. Carcinog. 2011, 50, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, E.; Castagnino, A.; Ferrari, R.; Monteiro, P.; Agüera-González, S.; Paul-Gilloteaux, P.; Domingues, M.J.; Maiuri, P.; Raab, M.; Shanahan, C.M.; et al. LINC complex-Lis1 interplay controls MT1-MMP matrix digest-on-demand response for confined tumor cell migration. Nat. Commun. 2018, 9, 2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Han, M.; Chen, W.; He, Y.; Huang, B.; Zhao, P.; Huang, Q.; Gao, L.; Qu, X.; Li, X. KIF1B promotes glioma migration and invasion via cell surface localization of MT1-MMP. Oncol. Rep. 2016, 35, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Marrero-Diaz, R.; Megías, D.; Genís, L.; García-Grande, A.; García, M.A.; Arroyo, A.G.; Montoya, M.C. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 2007, 26, 1499–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122 Pt 17, 3015–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Lo, A.T.; Inman, J.L.; Alcaraz, J.; Ghajar, C.M.; Mott, J.D.; Nelson, C.M.; Chen, C.S.; Zhang, H.; Bascom, J.L.; et al. Transmembrane/cytoplasmic, rather than catalytic, domains of Mmp14 signal to MAPK activation and mammary branching morphogenesis via binding to integrin β1. Development 2013, 140, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, N.M.; Wenzel, E.M.; Wang, L.; Antoine, S.; Chavrier, P.; Stenmark, H.; Raiborg, C. Protrudin-mediated ER-endosome contact sites promote MT1-MMP exocytosis and cell invasion. J. Cell Biol. 2020, 219, e202003063. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.S.; Otsu, W.; Li, Y.; Wang, H.C.; Chen, S.; Tsang, S.H.; Chuang, J.Z.; Sung, C.H. CLIC4 regulates late endosomal trafficking and matrix degradation activity of MMP14 at focal adhesions in RPE cells. Sci. Rep. 2019, 9, 12247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grafinger, O.R.; Gorshtein, G.; Stirling, T.; Brasher, M.I.; Coppolino, M.G. beta1 integrin-mediated signaling regulates MT1-MMP phosphorylation to promote tumor cell invasion. J. Cell Sci. 2020, 133, jcs239152. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Wu, Y.I.; Overall, C.M.; Stack, M.S. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J. Biol. Chem. 2001, 276, 24833–24842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.A.; Wolters, B.; Ito, N.; Woskowicz, A.M.; Kaneko, K.; Shitomi, Y.; Seiki, M.; Itoh, Y. Basal localization of MT1-MMP is essential for epithelial cell morphogenesis in 3D collagen matrix. J. Cell Sci. 2014, 127 Pt 6, 1203–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y. MT1-MMP: A key regulator of cell migration in tissue. IUBMB Life 2006, 58, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Yamada, A.; Takino, T.; Miyamori, H.; Takahashi, T.; Yamashita, J.; Sato, H. Suppression of membrane-type 1 matrix metalloproteinase (MMP)-mediated MMP-2 activation and tumor invasion by testican 3 and its splicing variant gene product, N-Tes. Cancer Res. 2001, 61, 8896–8902. [Google Scholar] [PubMed]
- Leco, K.J.; Waterhouse, P.; Sanchez, O.H.; Gowing, K.L.; Poole, A.R.; Wakeham, A.; Mak, T.W.; Khokha, R. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J. Clin. Investig. 2001, 108, 817–829. [Google Scholar] [CrossRef] [PubMed]
- English, J.L.; Kassiri, Z.; Koskivirta, I.; Atkinson, S.J.; Di Grappa, M.; Soloway, P.D.; Nagase, H.; Vuorio, E.; Murphy, G.; Khokha, R. Individual Timp deficiencies differentially impact pro-MMP-2 activation. J. Biol. Chem. 2006, 281, 10337–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrechtsen, R.; Kveiborg, M.; Stautz, D.; Vikeså, J.; Noer, J.B.; Kotzsh, A.; Nielsen, F.C.; Wewer, U.M.; Fröhlich, C. ADAM12 redistributes and activates MMP-14, resulting in gelatin degradation, reduced apoptosis and increased tumor growth. J. Cell Sci. 2013, 126 Pt 20, 4707–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ito, N.; Nagase, H.; Evans, R.D.; Bird, S.A.; Seiki, M. Cell surface collagenolysis requires homodimerization of the membrane-bound collagenase MT1-MMP. Mol. Biol. Cell 2006, 17, 5390–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahwa, S.; Stawikowski, M.J.; Fields, G.B. Monitoring and inhibiting MT1-MMP during cancer initiation and progression. Cancers 2014, 6, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Pahwa, S.; Bhowmick, M.; Amar, S.; Cao, J.; Strongin, A.Y.; Fridman, R.; Weiss, S.J.; Fields, G.B. Characterization and regulation of MT1-MMP cell surface-associated activity. Chem. Biol. Drug Des. 2019, 93, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Chia, J.; Ros, M.; Hui, K.M.; Saltel, F.; Bard, F. Organelle Specific O-Glycosylation Drives MMP14 Activation, Tumor Growth, and Metastasis. Cancer Cell 2017, 32, 639–653.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Seiki, M. MT1-MMP: An enzyme with multidimensional regulation. Trends Bio Chem. Sci. 2004, 29, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.; Murphy, G.; Roghi, C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J. Cell Sci. 2003, 116 Pt 19, 3905–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uekita, T.; Itoh, Y.; Yana, I.; Ohno, H.; Seiki, M. Cytoplasmic tail-dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity. J. Cell Biol. 2001, 155, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; Uekita, T.; Couchman, J.R.; Nagase, H.; Seiki, M.; Itoh, Y. Palmitoylation at Cys574 is essential for MT1-MMP to promote cell migration. FASEB J. 2005, 19, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- Kruglikov, I.L.; Joffin, N.; Scherer, P.E. The MMP14-caveolin axis and its potential relevance for lipoedema. Nat. Rev. Endocrinol. 2020, 16, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Planchon, D.; Rios Morris, E.; Genest, M.; Comunale, F.; Vacher, S.; Bièche, I.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Linder, S.; et al. MT1-MMP targeting to endolysosomes is mediated by upregulation of flotillins. J. Cell Sci. 2018, 131, jcs218925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrecque, L.; Nyalendo, C.; Langlois, S.; Durocher, Y.; Roghi, C.; Murphy, G.; Gingras, D.; Béliveau, R. Src-mediated tyrosine phosphorylation of caveolin-1 induces its association with membrane type 1 matrix metalloproteinase. J. Biol. Chem. 2004, 279, 52132–52140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassarre, T.; Watt, K.; Truesdell, P.; Meens, J.; Schneider, M.M.; Sengupta, S.K.; Craig, A.W. Endophilin A2 Promotes TNBC Cell Invasion and Tumor Metastasis. Mol. Cancer Res. 2015, 13, 1044–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ma, D.; Keski-Oja, J.; Pei, D. Co-recycling of MT1-MMP and MT3-MMP through the trans-Golgi network. Identification of DKV582 as a recycling signal. J. Biol. Chem. 2004, 279, 9331–9336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Azzouzi, K.; Wiesner, C.; Linder, S. Metalloproteinase MT1-MMP islets act as memory devices for podosome reemergence. J. Cell Biol. 2016, 213, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehti, K.; Lohi, J.; Valtanen, H.; Keski-Oja, J. Proteolytic processing of membrane-type-1 matrix metalloproteinase is associated with gelatinase A activation at the cell surface. Bio Chem. J. 1998, 334 Pt 2, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, H.; Gavrilovic, J.; Atkinson, S.J.; d’Ortho, M.P.; Yamada, K.M.; Zardi, L.; Murphy, G. The activation of ProMMP-2 (gelatinase A) by HT1080 fibrosarcoma cells is promoted by culture on a fibronectin substrate and is concomitant with an increase in processing of MT1-MMP (MMP-14) to a 45 kDa form. J. Cell Sci. 1998, 111 Pt 18, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Toth, M.; Fridman, R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP). J. Cell Physiol. 2004, 200, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Tobar, N.; Avalos, M.C.; Méndez, N.; Smith, P.C.; Bernabeu, C.; Quintanilla, M.; Martínez, J. Soluble MMP-14 produced by bone marrow-derived stromal cells sheds epithelial endoglin modulating the migratory properties of human breast cancer cells. Carcinogenesis 2014, 35, 1770–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudanski, P.; Swiatecka, J.; Kozlowski, L.; Lesniewska, M.; Wojtukiewicz, M.; Wolczynski, S. Increased serum level of membrane type 1-matrix metalloproteinase (MT1-MMP/MMP-14) in patients with breast cancer. Folia Histo Chem. Cyto Biol. 2010, 48, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotary, K.; Allen, E.; Punturieri, A.; Yana, I.; Weiss, S.J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. Cell Biol. 2000, 149, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Apte, S.S.; Soininen, R.; Cao, R.; Baaklini, G.Y.; Rauser, R.W.; Wang, J.; Cao, Y.; Tryggvason, K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA 2000, 97, 4052–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Fernández, A.; Soria-Valles, C.; Osorio, F.G.; Gutiérrez-Abril, J.; Garabaya, C.; Aguirre, A.; Fueyo, A.; Fernández-García, M.S.; Puente, X.S.; López-Otín, C. Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 2015, 34, 1875–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapinska, A.M.; Fields, G.B. The Expanding Role of MT1-MMP in Cancer Progression. Pharmaceuticals 2019, 12, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaisse, J.M.; Foged, N.T. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatti, O.; Vehviläinen, P.; Lehti, K.; Keski-Oja, J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell Res. 2008, 314, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- McQuibban, G.A.; Butler, G.S.; Gong, J.H.; Bendall, L.; Power, C.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001, 276, 43503–43508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbolina, M.V.; Stack, M.S. Membrane type 1-matrix metalloproteinase: Substrate diversity in pericellular proteolysis. Semin. Cell Dev. Biol. 2008, 19, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotary, K.; Li, X.Y.; Allen, E.; Stevens, S.L.; Weiss, S.J. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 2006, 20, 2673–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K. Tube travel: The role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008, 68, 7247–7249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Marcink, T.C.; Simoncic, J.A.; An, B.; Knapinska, A.M.; Fulcher, Y.G.; Akkaladevi, N.; Fields, G.B.; Van Doren, S.R. MT1-MMP Binds Membranes by Opposite Tips of Its beta Propeller to Position It for Pericellular Proteolysis. Structure 2019, 27, 281–292.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Okamoto, I.; Araki, N.; Kawano, Y.; Nakao, M.; Fujiyama, S.; Tomita, K.; Mimori, T.; Saya, H. Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell-cell contacts. Oncogene 1999, 18, 7080–7090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhusen, U.; Weiske, J.; Badock, V.; Tauber, R.; Bommert, K.; Huber, O. Cleavage and shedding of E-cadherin after induction of apoptosis. J. Biol. Chem. 2001, 276, 4972–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, Y.; Uekita, T.; Ito, Y.; Seiki, M.; Yamaguchi, H.; Sakai, R. CDCP1 regulates the function of MT1-MMP and invadopodia-mediated invasion of cancer cells. Mol. Cancer Res. 2013, 11, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.M.; Rajasekaran, A.K. Dishonorable discharge: The oncogenic roles of cleaved E-cadherin fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Mizushima, H.; Minegishi, T.; Iwamoto, R.; Mekada, E.; Seiki, M. Membrane type 1-matrix metalloproteinase cleaves off the NH2-terminal portion of heparin-binding epidermal growth factor and converts it into a heparin-independent growth factor. Cancer Res. 2010, 70, 6093–6103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, K. Ectodomain shedding of HB-EGF: A potential target for cancer therapy. J. Bio Chem. 2012, 151, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.M.; Wong, H.L.; Jin, G.; Liu, B.; Cao, R.; Cao, Y.; Lehti, K.; Tryggvason, K.; Zhou, Z. MT1-MMP inactivates ADAM9 to regulate FGFR2 signaling and calvarial osteogenesis. Dev. Cell 2012, 22, 1176–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, O.; Murakami, D.; Hartmann, D.; De Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deryugina, E.I.; Ratnikov, B.I.; Postnova, T.I.; Rozanov, D.V.; Strongin, A.Y. Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J. Biol. Chem. 2002, 277, 9749–9756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubkov, V.S.; Chekanov, A.V.; Cieplak, P.; Aleshin, A.E.; Chernov, A.V.; Zhu, W.; Radichev, I.A.; Zhang, D.; Dong, P.D.; Strongin, A.Y. The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: Implications in cancer and embryogenesis. J. Biol. Chem. 2010, 285, 35740–35749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubkov, V.S.; Boyd, S.; Savinov, A.Y.; Chekanov, A.V.; Osterman, A.L.; Remacle, A.; Rozanov, D.V.; Doxsey, S.J.; Strongin, A.Y. Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J. Biol. Chem. 2005, 280, 25079–25086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wali, N.; Hosokawa, K.; Malik, S.; Saito, H.; Miyaguchi, K.; Imajoh-Ohmi, S.; Miki, Y.; Nakanishi, A. Centrosomal BRCA2 is a target protein of membrane type-1 matrix metalloproteinase (MT1-MMP). Bio. Chem. Biophys. Res. Commun. 2014, 443, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Bhat, R.; Bruni-Cardoso, A.; Chen, E.I.; Jorgens, D.M.; Coutinho, K.; Louie, K.; Bowen, B.B.; Inman, J.L.; Tecca, V.; et al. New insight into the role of MMP14 in metabolic balance. Peer J. 2016, 4, e2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, B.H.; Kubo, T.; Healey, J.H.; Yang, R.; Nathan, S.S.; Kolb, E.A.; Mazza, B.; Meyers, P.A.; Gorlick, R. Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the Wnt-beta-catenin pathway. Cancer Res. 2004, 64, 2734–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeb-Parsy, K.; Veerakumarasivam, A.; Wallard, M.J.; Thorne, N.; Kawano, Y.; Murphy, G.; Neal, D.E.; Mills, I.G.; Kelly, J.D. MT1-MMP regulates urothelial cell invasion via transcriptional regulation of Dickkopf-3. Br. J. Cancer 2008, 99, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Hirota, R.; Xiong, W.; Baxter, B.T.; Kunkel, S.L.; Maillard, I.; Chen, X.W.; Sabeh, F.; Liu, R.; Li, X.Y.; Weiss, S.J. MT1-MMP regulates the PI3Kδ·Mi-2/NuRD-dependent control of macrophage immune function. Genes Dev. 2012, 26, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunen, S.P.; Tatti-Bugaeva, O.; Lehti, K. Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt A, 1974–1988. [Google Scholar] [CrossRef] [PubMed]
- Ota, I.; Li, X.Y.; Hu, Y.; Weiss, S.J. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc. Natl. Acad. Sci. USA 2009, 106, 20318–20323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolopoulou, P.A.; Koufaki, M.A.; Kostourou, V. The Adhesome Network: Key Components Shaping the Tumour Stroma. Cancers 2021, 13, 525. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Branch, K.M.; Hoshino, D.; Weaver, A.M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 2012, 1, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, R.; Pariente, A.; Perez-Sala, A.; Larrayoz, I.M. Integrins: Moonlighting Proteins in Invadosome Formation. Cancers (Basel) 2019, 11, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, Y.G.; Manavathi, B. Focal adhesion dynamics in cellular function and disease. Cell Signal. 2021, 85, 110046. [Google Scholar] [CrossRef] [PubMed]
- Revach, O.Y.; Grosheva, I.; Geiger, B. Biomechanical regulation of focal adhesion and invadopodia formation. J. Cell Sci. 2020, 133, jcs244848. [Google Scholar] [CrossRef] [PubMed]
- Barber-Perez, N.; Georgiadou, M.; Guzman, C.; Isomursu, A.; Hamidi, H.; Ivaska, J. Mechano-responsiveness of fibrillar adhesions on stiffness-gradient gels. J. Cell Sci. 2020, 133, jcs242909. [Google Scholar] [CrossRef] [PubMed]
- Linder, S. Invadosomes at a glance. J. Cell Sci. 2009, 122 Pt 17, 3009–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, S.; Cervero, P. The podosome cap: Past, present, perspective. Eur. J. Cell Biol. 2020, 99, 151087. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Spuul, P.; Genot, E. Podosomes in endothelial cell-microenvironment interactions. Curr. Opin. Hematol. 2020, 27, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Martinelli, R. T Lymphocyte-Endothelial Interactions: Emerging Understanding of Trafficking and Antigen-Specific Immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci. 2018, 19, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Invadopodia: Clearing the way for cancer cell invasion. Ann. Transl. Med. 2020, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Sage, P.T.; Sciuto, T.E.; de la Fuente, M.A.; Geha, R.S.; Ochs, H.D.; Dvorak, H.F.; Dvorak, A.M.; Springer, T.A. Transcellular diapedesis is initiated by invasive podosomes. Immunity 2007, 26, 784–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemesh, M.; Addadi, L.; Geiger, B. Surface microtopography modulates sealing zone development in osteoclasts cultured on bone. J. R. Soc. Interface 2017, 14, 20160958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesbitt, S.A.; Horton, M.A. Trafficking of matrix collagens through bone-resorbing osteoclasts. Science 1997, 276, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Salo, J.; Lehenkari, P.; Mulari, M.; Metsikko, K.; Vaananen, H.K. Removal of osteoclast bone resorption products by transcytosis. Science 1997, 276, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Chabadel, A.; Bañon-Rodríguez, I.; Cluet, D.; Rudkin, B.B.; Wehrle-Haller, B.; Genot, E.; Jurdic, P.; Anton, I.M.; Saltel, F. CD44 and beta3 integrin organize two functionally distinct actin-based domains in osteoclasts. Mol. Biol. Cell 2007, 18, 4899–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carman, C.V. Mechanisms for transcellular diapedesis: Probing and pathfinding by ‘invadosome-like protrusions. J. Cell Sci. 2009, 122 Pt 17, 3025–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, I.; Duong, L.T.; Rodan, S.B.; Rodan, G.A. Involvement of alpha(v)beta3 integrins in osteoclast function. J. Bone Miner. Metab. 2007, 25, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bati, G.; Okvur, D.P. Invadopodia: Proteolytic feet of cancer cells. Turk. J. Biol. 2014, 38, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Koçer, G.; Jonkheijm, P. About Chemical Strategies to Fabricate Cell-Instructive Biointerfaces with Static and Dynamic Complexity. Adv. Healthc. Mater. 2018, 7, e1701192. [Google Scholar] [CrossRef] [PubMed]
- Buccione, R.; Orth, J.D.; McNiven, M.A. Foot and mouth: Podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 2004, 5, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Aepfelbacher, M. Podosomes: Adhesion hot-spots of invasive cells. Trends Cell Biol. 2003, 13, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Wiesner, C.; Himmel, M. Degrading devices: Invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 2011, 27, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legerstee, K.; Geverts, B.; Slotman, J.A.; Houtsmuller, A.B. Dynamics and distribution of paxillin, vinculin, zyxin and VASP depend on focal adhesion location and orientation. Sci. Rep. 2019, 9, 10460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adh. Migr. 2014, 8, 191–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geblinger, D.; Addadi, L.; Geiger, B. Nano-topography sensing by osteoclasts. J. Cell Sci. 2010, 123 Pt 9, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Wirtz, D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013, 27, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Veillat, V.; Spuul, P.; Daubon, T.; Egana, I.; Kramer, I.; Genot, E. Podosomes: Multipurpose organelles? Int. J. Bio Chem. Cell Biol. 2015, 65, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Schachtner, H.; Calaminus, S.D.; Thomas, S.G.; Machesky, L.M. Podosomes in adhesion, migration, mechanosensing and matrix remodeling. Cytoskeleton 2013, 70, 572–589. [Google Scholar] [CrossRef] [PubMed]
- Artym, V.V.; Matsumoto, K.; Mueller, S.C.; Yamada, K.M. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur. J. Cell Biol. 2011, 90, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamir, E.; Katz, B.Z.; Aota, S.; Yamada, K.M.; Geiger, B.; Kam, Z. Molecular diversity of cell-matrix adhesions. J. Cell Sci. 1999, 112 Pt 11, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Saltel, F.; Destaing, O.; Bard, F.; Eichert, D.; Jurdic, P. Apatite-mediated actin dynamics in resorbing osteoclasts. Mol. Biol. Cell 2004, 15, 5231–5241. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Delon, I.; Brown, N.H. Integrins and the actin cytoskeleton. Curr. Opin. Cell Biol. 2007, 19, 43–50. [Google Scholar] [CrossRef]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix and the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef]
- Paolillo, M.; Galiazzo, M.C.; Daga, A.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int. J. Oncol. 2018, 53, 2683–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Cai, J.; Zuo, Z.; Li, J. Collagen facilitates the colorectal cancer stemness and metastasis through an integrin/PI3K/AKT/Snail signaling pathway. Biomed. Pharmacother. 2019, 114, 108708. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Jiang, S.; Wang, R.; Zhang, Y.; Dong, J.; Li, Y. Mechanistic Insight Into the Roles of Integrins in Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 693484. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Parsons, M. New perspectives on integrin-dependent adhesions. Curr. Opin. Cell Biol. 2020, 63, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Soe, Z.Y.; Park, E.J.; Shimaoka, M. Integrin Regulation in Immunological and Cancerous Cells and Exosomes. Int. J. Mol. Sci. 2021, 22, 2193. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Manso, A.M.; Ross, R.S. Talin and Kindlin as Integrin-Activating Proteins: Focus on the Heart. Pediatr. Cardiol. 2019, 40, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Costell, M.; Fassler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Carman, C.V.; Kim, M.; Shimaoka, M.; Springer, T.A.; Luo, B.H. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood 2007, 110, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarzija, I.; Dekanic, A.; Humphries, J.D.; Paradzik, M.; Stojanovic, N.; Humphries, M.J.; Ambriovic-Ristov, A. Integrin Crosstalk Contributes to the Complexity of Signalling and Unpredictable Cancer Cell Fates. Cancers 2020, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122 Pt 2, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.A.; Hemler, M.E. Interaction of the integrin beta1 cytoplasmic domain with ICAP-1 protein. J. Biol. Chem. 1999, 274, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidel-Bar, R.; Geiger, B. The switchable integrin adhesome. J. Cell Sci. 2010, 123 Pt 9, 1385–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Fremillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C. Focal adhesion: A focal point in current cell biology and molecular medicine. Cell Adh. Migr. 2007, 1, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.S.; Su, Y.A.; Chuang, M.C.; Liu, Y.W. Probing invadosomes: Technologies for the analysis of invadosomes. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, K.M.; Adebowale, K.; Chang, J.; Lee, J.Y.; Nam, S.; Desai, R.; Rossen, N.S.; Rafat, M.; West, R.B.; Hodgson, L.; et al. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nat. Commun. 2018, 9, 4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Small, J.V.; Stradal, T.; Vignal, E.; Rottner, K. The lamellipodium: Where motility begins. Trends Cell Biol. 2002, 12, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Palmisano, R.; Anilkumar, N.; Nagase, H.; Miyawaki, A.; Seiki, M. Dimerization of MT1-MMP during cellular invasion detected by fluorescence resonance energy transfer. Bio Chem. J. 2011, 440, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; del Carmen Ovejero, M.; Hou, P.; Heegaard, A.M.; Kumegawa, M.; Foged, N.T.; Delaissé, J.M. Identification of the membrane-type matrix metalloproteinase MT1-MMP in osteoclasts. J. Cell Sci. 1997, 110 Pt 5, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Takamura, A.; Ito, N.; Maru, Y.; Sato, H.; Suenaga, N.; Aoki, T.; Seiki, M. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2001, 20, 4782–4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Tomari, T.; Koshikawa, N.; Kajita, M.; Itoh, Y.; Sato, H.; Tojo, H.; Yana, I.; Seiki, M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002, 21, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, N.; Mori, H.; Itoh, Y.; Seiki, M. CD44 binding through the hemopexin-like domain is critical for its shedding by membrane-type 1 matrix metalloproteinase. Oncogene 2005, 24, 859–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell, P.T.; Zech, T. Actin-Based Cell Protrusion in a 3D Matrix. Trends Cell Biol. 2018, 28, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Friedl, P. Mapping proteolytic cancer cell-extracellular matrix interfaces. Clin. Exp. Metastasis 2009, 26, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takino, T.; Saeki, H.; Miyamori, H.; Kudo, T.; Sato, H. Inhibition of Membrane-Type 1 Matrix Metalloproteinase at Cell-Matrix Adhesions. Cancer Res. 2007, 67, 11621–11629. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvez, B.G.; Matias-Roman, S.; Yanez-Mo, M.; Sanchez-Madrid, F.; Arroyo, A.G. ECM regulates MT1-MMP localization with beta1 or alphavbeta3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J. Cell Biol. 2002, 159, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Puleo, J.I.; Parker, S.S.; Roman, M.R.; Watson, A.W.; Eliato, K.R.; Peng, L.; Saboda, K.; Roe, D.J.; Ros, R.; Gertler, F.B.; et al. Mechanosensing during directed cell migration requires dynamic actin polymerization at focal adhesions. J. Cell Biol. 2019, 218, 4215–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Ota, I.; Yana, I.; Sabeh, F.; Weiss, S.J. Molecular dissection of the structural machinery underlying the tissue-invasive activity of membrane type-1 matrix metalloproteinase. Mol. Biol. Cell 2008, 19, 3221–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotary, K.B.; Allen, E.D.; Brooks, P.C.; Datta, N.S.; Long, M.W.; Weiss, S.J. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 2003, 114, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woskowicz, A.M.; Weaver, S.A.; Shitomi, Y.; Ito, N.; Itoh, Y. MT-LOOP-dependent localization of membrane type I matrix metalloproteinase (MT1-MMP) to the cell adhesion complexes promotes cancer cell invasion. J. Biol. Chem. 2013, 288, 35126–35137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K. Proteolytic interstitial cell migration: A five-step process. Cancer Metastasis Rev. 2009, 28, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seano, G.; Primo, L. Podosomes and invadopodia: Tools to breach vascular basement membrane. Cell Cycle 2015, 14, 1370–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; Jones, G.E.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2011, 9, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.P.; Eddy, R.; Entenberg, D.; Kai, M.; Gertler, F.B.; Condeelis, J. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr. Biol. 2013, 23, 2079–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.M. Invadopodia. Curr. Biol. 2008, 18, R362–R364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimona, M.; Buccione, R.; Courtneidge, S.A.; Linder, S. Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 2008, 20, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, P.P.; Freeman, S.A.; Fairn, G.; Grinstein, S. Dynamic Podosome-Like Structures in Nascent Phagosomes Are Coordinated by Phosphoinositides. Dev. Cell 2019, 50, 397–410.e3. [Google Scholar] [CrossRef] [PubMed]
- Pacini, S.; Fazzi, R.; Montali, M.; Carnicelli, V.; Lazzarini, E.; Petrini, M. Specific integrin expression is associated with podosome-like structures on mesodermal progenitor cells. Stem Cells Dev. 2013, 22, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Chen, Y.; Guo, Y.; Bock, C.E.; Hagan, J.P.; Kim, D.H.; Xu, Z. Podosome formation impairs endothelial barrier function by sequestering zonula occludens proteins. J. Cell Physiol. 2020, 235, 4655–4666. [Google Scholar] [CrossRef] [PubMed]
- Chuang, M.C.; Lin, S.S.; Ohniwa, R.L.; Lee, G.H.; Su, Y.A.; Chang, Y.C.; Tang, M.J.; Liu, Y.W. Tks5 and Dynamin-2 enhance actin bundle rigidity in invadosomes to promote myoblast fusion. J. Cell Biol. 2019, 218, 1670–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, J.J.; Balabiyev, A.; Heddleston, J.M.; Podolnikova, N.P.; Baluch, D.P.; Chew, T.L.; Ugarova, T.P. An actin-based protrusion originating from a podosome-enriched region initiates macrophage fusion. Mol. Biol. Cell 2019, 30, 2254–2267. [Google Scholar] [CrossRef] [PubMed]
- Calle, Y.; Chou, H.C.; Thrasher, A.J.; Jones, G.E. Wiskott-Aldrich syndrome protein and the cytoskeletal dynamics of dendritic cells. J. Pathol. 2004, 204, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Panzer, L.; Trübe, L.; Klose, M.; Joosten, B.; Slotman, J.; Cambi, A.; Linder, S. The formins FHOD1 and INF2 regulate inter-and intra-structural contractility of podosomes. J. Cell Sci. 2016, 129, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foxall, E.; Staszowska, A.; Hirvonen, L.M.; Georgouli, M.; Ciccioli, M.; Rimmer, A.; Williams, L.; Calle, Y.; Sanz-Moreno, V.; Cox, S.; et al. PAK4 Kinase Activity Plays a Crucial Role in the Podosome Ring of Myeloid Cells. Cell Rep. 2019, 29, 3385–3393.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akisaka, T.; Yoshida, H.; Suzuki, R.; Takama, K. Adhesion structures and their cytoskeleton-membrane interactions at podosomes of osteoclasts in culture. Cell Tissue Res. 2008, 331, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Van den Dries, K.; Nahidiazar, L.; Slotman, J.A.; Meddens, M.B.M.; Pandzic, E.; Joosten, B.; Ansems, M.; Schouwstra, J.; Meijer, A.; Steen, R.; et al. Modular actin nano-architecture enables podosome protrusion and mechanosensing. Nat. Commun. 2019, 10, 5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Dries, K.; Linder, S.; Maridonneau-Parini, I.; Poincloux, R. Probing the mechanical landscape—New insights into podosome architecture and mechanics. J. Cell Sci. 2019, 132, jcs236828. [Google Scholar] [CrossRef] [PubMed]
- Linder, S. The matrix corroded: Podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Cervero, P.; Wiesner, C.; Bouissou, A.; Poincloux, R.; Linder, S. Lymphocyte-specific protein 1 regulates mechanosensory oscillation of podosomes and actin isoform-based actomyosin symmetry breaking. Nat. Commun. 2018, 9, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labernadie, A.; Thibault, C.; Vieu, C.; Maridonneau-Parini, I.; Charriere, G.M. Dynamics of podosome stiffness revealed by atomic force microscopy. Proc. Natl. Acad. Sci. USA 2010, 107, 21016–21021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, O.; Tracqui, P.; Stephanou, A.; Usson, Y.; Clement-Lacroix, J.; Planus, E. Spatiotemporal dynamics of actin-rich adhesion microdomains: Influence of substrate flexibility. J. Cell Sci. 2006, 119 Pt 9, 1914–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouissou, A.; Proag, A.; Bourg, N.; Pingris, K.; Cabriel, C.; Balor, S.; Mangeat, T.; Thibault, C.; Vieu, C.; Dupuis, G.; et al. Podosome Force Generation Machinery: A Local Balance between Protrusion at the Core and Traction at the Ring. ACS Nano 2017, 11, 4028–4040. [Google Scholar] [CrossRef] [PubMed]
- Glazier, R.; Brockman, J.M.; Bartle, E.; Mattheyses, A.L.; Destaing, O.; Salaita, K. DNA mechanotechnology reveals that integrin receptors apply pN forces in podosomes on fluid substrates. Nat. Commun. 2019, 10, 4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Rowe, R.G.; Botvinick, E.L.; Kurup, A.; Putnam, A.J.; Seiki, M.; Weaver, V.M.; Keller, E.T.; Goldstein, S.; Dai, J.; et al. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell 2013, 25, 402–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrino, S.; Roustan, F.R.; Kaminski, L.; Bertero, T.; Pisano, S.; Ambrosetti, D.; Dufies, M.; Uhler, J.P.; Lemichez, E.; Mettouchi, A.; et al. UBTD1 is a mechano-regulator controlling cancer aggressiveness. EMBO Rep. 2019, 20, e46570. [Google Scholar] [CrossRef] [PubMed]
- Meddens, M.B.; Pandzic, E.; Slotman, J.A.; Guillet, D.; Joosten, B.; Mennens, S.; Paardekooper, L.M.; Houtsmuller, A.B.; van den Dries, K.; Wiseman, P.W.; et al. Actomyosin-dependent dynamic spatial patterns of cytoskeletal components drive mesoscale podosome organization. Nat. Commun. 2016, 7, 13127. [Google Scholar] [CrossRef] [PubMed]
- Revach, O.Y.; Weiner, A.; Rechav, K.; Sabanay, I.; Livne, A.; Geiger, B. Mechanical interplay between invadopodia and the nucleus in cultured cancer cells. Sci. Rep. 2015, 5, 9466. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Chavrier, P. Tissue remodeling by invadosomes. FAC Rev. 2021, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Yamaguchi, H.; Mader, C.C.; Bravo-Cordero, J.J.; Arias, M.; Chen, X.; Desmarais, V.; van Rheenen, J.; Koleske, A.J.; Condeelis, J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 2009, 186, 571–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulvady, A.C.; Forsythe, I.J.; Turner, C.E. Hic-5 regulates Src-induced invadopodia rosette formation and organization. Mol. Biol. Cell 2019, 30, 1298–1313. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, P.; Rossé, C.; Castro-Castro, A.; Irondelle, M.; Lagoutte, E.; Paul-Gilloteaux, P.; Desnos, C.; Formstecher, E.; Darchen, F.; Perrais, D.; et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J. Cell Biol. 2013, 203, 1063–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesin, V.; Castro-Castro, A.; Lodillinsky, C.; Castagnino, A.; Cyrta, J.; Bonsang-Kitzis, H.; Fuhrmann, L.; Irondelle, M.; Infante, E.; Montagnac, G.; et al. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J. Cell Biol. 2015, 211, 339–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagnino, A.; Castro-Castro, A.; Irondelle, M.; Guichard, A.; Lodillinsky, C.; Fuhrmann, L.; Vacher, S.; Agüera-González, S.; Zagryazhskaya-Masson, A.; Romao, M.; et al. Coronin 1C promotes triple-negative breast cancer invasiveness through regulation of MT1-MMP traffic and invadopodia function. Oncogene 2018, 37, 6425–6441. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Parveen, S.; Shah, L.V.; Mukherjee, M.; Kalaidzidis, Y.; Kozielski, A.J.; Rosato, R.; Chang, J.C.; Datta, S. SNX27-retromer assembly recycles MT1-MMP to invadopodia and promotes breast cancer metastasis. J. Cell Biol. 2020, 219, e201812098. [Google Scholar] [CrossRef] [PubMed]
- Beaty, B.T.; Sharma, V.P.; Bravo-Cordero, J.J.; Simpson, M.A.; Eddy, R.J.; Koleske, A.J.; Condeelis, J. β1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol. Biol. Cell 2013, 24, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H.; Mueller, S.C.; Nomizu, M.; Yamada, Y.; Yeh, Y.; Chen, W.T. Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J. Biol. Chem. 1998, 273, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.C.; Coppolino, M.G. SNARE-dependent interaction of Src, EGFR and β1 integrin regulates invadopodia formation and tumor cell invasion. J. Cell Sci. 2014, 127 Pt 8, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalski, M.; Yi, Q.; Kean, M.J.; Myers, D.W.; Williams, K.C.; Burtnik, A.; Coppolino, M.G. Lamellipodium extension and membrane ruffling require different SNARE-mediated trafficking pathways. BMC Cell Biol. 2010, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalski, M.; Sharma, N.; Williams, K.; Kruspe, A.; Coppolino, M.G. SNARE-mediated membrane traffic is required for focal adhesion kinase signaling and Src-regulated focal adhesion turnover. Biochim. Biophys. Acta 2011, 1813, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kean, M.J.; Williams, K.C.; Skalski, M.; Myers, D.; Burtnik, A.; Foster, D.; Coppolino, M.G. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J. Cell Sci. 2009, 122 Pt 22, 4089–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.C.; McNeilly, R.E.; Coppolino, M.G. SNAP23, Syntaxin4, and vesicle-associated membrane protein 7 (VAMP7) mediate trafficking of membrane type 1-matrix metalloproteinase (MT1-MMP) during invadopodium formation and tumor cell invasion. Mol. Biol. Cell 2014, 25, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Gorshtein, G.; Grafinger, O.; Coppolino, M.G. Targeting SNARE-Mediated Vesicle Transport to Block Invadopodium-Based Cancer Cell Invasion. Front. Oncol. 2021, 11, 679955. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013, 5, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell Bio. Chem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Gil-Henn, H. Targeting invadopodia for blocking breast cancer metastasis. Drug Resist. Updat 2018, 39, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.L.; Learman, B.S.; Boucherle, A.K.; Sirintrapun, S.J.; Isom, S.; Diaz, B.; Courtneidge, S.A.; Seals, D.F. Src-dependent Tks5 phosphorylation regulates invadopodia-associated invasion in prostate cancer cells. Prostate 2014, 74, 134–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylli, S.S.; Stacey, T.T.; Kaye, A.H.; Lock, P. Prognostic significance of Tks5 expression in gliomas. J. Clin. Neuro Sci. 2012, 19, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Chen, G.; Dayton, T.L.; Kim-Kiselak, C.; Hoersch, S.; Whittaker, C.A.; Bronson, R.T.; Beer, D.G.; Winslow, M.M.; Jacks, T. Differential Tks5 isoform expression contributes to metastatic invasion of lung adenocarcinoma. Genes Dev. 2013, 27, 1557–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, E.L.; Matrisian, L.M. Matrix metalloproteases in head and neck cancer. Head Neck 2006, 28, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, E.T.; Barth, M.; Thomas, D.; Glazer, R.I.; Mueller, S.C. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 1999, 18, 4440–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tondravi, M.; Liu, J.; Smith, E.; Haudenschild, C.C.; Kaczmarek, M.; Zhan, X. Cortactin potentiates bone metastasis of breast cancer cells. Cancer Res. 2001, 61, 6906–6911. [Google Scholar] [PubMed]
- Chen, W.T.; Wang, J.Y. Specialized surface protrusions of invasive cells, invadopodia and lamellipodia, have differential MT1-MMP, MMP-2, and TIMP-2 localization. Ann. N. Y. Acad. Sci. 1999, 878, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Baldassarre, M.; Beznoussenko, G.; Giacchetti, G.; Cao, J.; Zucker, S.; Luini, A.; Buccione, R. Intracellular processing and activation of membrane type 1 matrix metalloprotease depends on its partitioning into lipid domains. J. Cell Sci. 2004, 117 Pt 26, 6275–6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, H.; Howard, L.; Thompson, E.W.; Sato, H.; Seiki, M.; Yeh, Y.; Chen, W.T. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc. Natl. Acad. Sci. USA 1997, 94, 7959–7964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revach, O.Y.; Geiger, B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adh. Migr. 2014, 8, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.M.; Hedman, A.C.; Sacks, D.B. IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 2015, 25, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Dawson, J.C.; Forero-Vargas, M.; Spence, H.J.; Yu, X.; Konig, I.; Anderson, K.; Machesky, L.M. The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr. Biol. 2010, 20, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Destaing, O.; Saltel, F.; Geminard, J.C.; Jurdic, P.; Bard, F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol. Biol. Cell 2003, 14, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Das, A.; Barai, A.; Sen, S. MMP Secretion Rate and Inter-invadopodia Spacing Collectively Govern Cancer Invasiveness. Biophys. J. 2018, 114, 650–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuwania, R.; Cornfine, S.; Fang, Z.Y.; Kruger, M.; Luna, E.J.; Linder, S. Supervillin couples myosin-dependent contractility to podosomes and enables their turnover. J. Cell Sci. 2012, 125, 2300–2314. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Dash, P.R. Formation of atypical podosomes in extravillous trophoblasts regulates extracellular matrix degradation. Eur. J. Cell Biol. 2012, 91, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsky, W.L.; Lin, C.Y.; Aoyama, A.; Kelly, T.; Akiyama, S.K.; Mueller, S.C.; Chen, W.T. A potential marker protease of invasiveness, seprase, is localized on invadopodia of human malignant melanoma cells. Cancer Res. 1994, 54, 5702–5710. [Google Scholar] [PubMed]
- Yu, X.; Zech, T.; McDonald, L.; Gonzalez, E.G.; Li, A.; Macpherson, I.; Schwarz, J.P.; Spence, H.; Futó, K.; Timpson, P.; et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J. Cell Biol. 2012, 199, 527–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmer, L.L.; Kelley, L.C.; Hagedorn, E.J.; Sherwood, D.R. Invadopodia and basement membrane invasion in vivo. Cell Adh. Migr. 2014, 8, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Eschenbruch, J.; Dreissen, G.; Springer, R.; Konrad, J.; Merkel, R.; Hoffmann, B.; Noetzel, E. From Microspikes to Stress Fibers: Actin Remodeling in Breast Acini Drives Myosin II-Mediated Basement Membrane Invasion. Cells 2021, 10, 1979. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.; Weaver, A.M. Regulation of invadopodia by mechanical signaling. Exp. Cell Res. 2016, 343, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, J.; Henriet, E.; Ezzoukhry, Z.; Goetz, J.G.; Moreau, V.; Saltel, F. The microenvironment controls invadosome plasticity. J. Cell Sci. 2016, 129, 1759–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaty, B.T.; Condeelis, J. Digging a little deeper: The stages of invadopodium formation and maturation. Eur. J. Cell Biol. 2014, 93, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siqueira, A.S.; Pinto, M.P.; Cruz, M.C.; Smuczek, B.; Cruz, K.S.; Barbuto, J.A.; Hoshino, D.; Weaver, A.M.; Freitas, V.M.; Jaeger, R.G. Laminin-111 peptide C16 regulates invadopodia activity of malignant cells through β1 integrin, Src and ERK 1/2. Oncotarget 2016, 7, 47904–47917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seals, D.F.; Azucena, E.F., Jr.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naegeli, K.M.; Hastie, E.; Garde, A.; Wang, Z.; Keeley, D.P.; Gordon, K.L.; Pani, A.M.; Kelley, L.C.; Morrissey, M.A.; Chi, Q.; et al. Cell Invasion In Vivo via Rapid Exocytosis of a Transient Lysosome-Derived Membrane Domain. Dev. Cell 2017, 43, 403–417.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedziora, K.M.; Isogai, T.; Jalink, K.; Innocenti, M. Invadosomes—Shaping actin networks to follow mechanical cues. Front. BioSci. 2016, 21, 1092–1117. [Google Scholar] [CrossRef] [PubMed]
- Pourfarhangi, K.E.; Bergman, A.; Gligorijevic, B. ECM Cross-Linking Regulates Invadopodia Dynamics. Biophys. J. 2018, 114, 1455–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Gil-Henn, H. Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases. Cells 2021, 10, 2037. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Cortesio, C.L.; Huttenlocher, A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 2009, 185, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genna, A.; Lapetina, S.; Lukic, N.; Twafra, S.; Meirson, T.; Sharma, V.P.; Condeelis, J.S.; Gil-Henn, H. Pyk2 and FAK differentially regulate invadopodia formation and function in breast cancer cells. J. Cell Biol. 2018, 217, 375–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, T.; Oyama, M.; Kozuka-Hata, H.; Uehara, S.; Udagawa, N.; Saya, H.; Matsuo, K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J. Cell Biol. 2012, 197, 553–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylli, S.S.; Stacey, T.T.; Verhagen, A.M.; Xu, S.S.; Pass, I.; Courtneidge, S.A.; Lock, P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J. Cell Sci. 2009, 122 Pt 15, 2727–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Henn, H.; Patsialou, A.; Wang, Y.; Warren, M.S.; Condeelis, J.S.; Koleske, A.J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparski, A.N.; Wilson, J.T.; Banerjee, A.; Beningo, K.A. The Role of PAK1 in the Maturation of Invadopodia During Transient Mechanical Stimulation. Front. Cell Dev. Biol. 2019, 7, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Hong, Z.; Zhong, M.; Klomp, J.; Bayless, K.J.; Mehta, D.; Karginov, A.V.; Hu, G.; Malik, A.B. Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am. J. Physiol. Cell Physiol. 2019, 316, C92–C103. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Rezania, S.; Kammerer, S.; Sokolowski, A.; Devaney, T.; Gorischek, A.; Jahn, S.; Hackl, H.; Groschner, K.; Windpassinger, C.; et al. Piezo1 forms mechanosensitive ion channels in the human MCF-7 breast cancer cell line. Sci. Rep. 2015, 5, 8364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.N.; Lu, Y.P.; Liu, J.J.; Huang, J.K.; Liu, Y.P.; Xiao, C.X.; Jazag, A.; Ren, J.L.; Guleng, B. Piezo1 is as a novel trefoil factor family 1 binding protein that promotes gastric cancer cell mobility in vitro. Dig. Dis. Sci. 2014, 59, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Pineau, J.; Saez, P.J.; Maurin, M.; Lankar, D.; San Roman, M.; Hennig, K.; Boura, V.F.; Voituriez, R.; Karlsson, M.C.I.; et al. Actomyosin-driven force patterning controls endocytosis at the immune synapse. Nat. Commun. 2019, 10, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettmann, J.; Santos, A.M.; Dushek, O.; Davis, S.J. Membrane Ultrastructure and T Cell Activation. Front. Immunol. 2018, 9, 2152. [Google Scholar] [CrossRef] [PubMed]
- Juin, A.; Billottet, C.; Moreau, V.; Destaing, O.; Albiges-Rizo, C.; Rosenbaum, J.; Génot, E.; Saltel, F. Physiological type I collagen organization induces the formation of a novel class of linear invadosomes. Mol. Biol. Cell 2012, 23, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Juin, A.; Di Martino, J.; Leitinger, B.; Henriet, E.; Gary, A.S.; Paysan, L.; Bomo, J.; Baffet, G.; Gauthier-Rouvière, C.; Rosenbaum, J.; et al. Discoidin domain receptor 1 controls linear invadosome formation via a Cdc42-Tuba pathway. J. Cell Biol. 2014, 207, 517–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxenburg, C.; Geblinger, D.; Klein, E.; Anderson, K.; Hanein, D.; Geiger, B.; Addadi, L. The architecture of the adhesive apparatus of cultured osteoclasts: From podosome formation to sealing zone assembly. PLoS ONE 2007, 2, e179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxenburg, C.; Addadi, L.; Geiger, B. The molecular dynamics of osteoclast adhesions. Eur. J. Cell Biol. 2006, 85, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Luxenburg, C.; Parsons, J.T.; Addadi, L.; Geiger, B. Involvement of the Src-cortactin pathway in podosome formation and turnover during polarization of cultured osteoclasts. J. Cell Sci. 2006, 119 Pt 23, 4878–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.P.H.; Xu, J.; Xue, M.; Jackson, C. Matrix metalloproteinases in bone development and pathology: Current knowledge and potential clinical utility. Met. Med. 2016, 3, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Tang, Y.; Li, X.Y.; Keller, E.T.; Yang, J.; Cho, J.S.; Feinberg, T.Y.; Weiss, S.J. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lou, J.; Jin, Z.; Yang, X.; Shan, W.; Du, Q.; Liao, Q.; Xu, J.; Xie, R. Advances in research on the regulatory mechanism of NHE1 in tumors. Oncol. Lett. 2021, 21, 273. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, T.; Addison, W.N.; Kokabu, S.; Neff, L.; Horne, W.; Gori, F.; Baron, R. Characterization of unique functionalities in c-Src domains required for osteoclast podosome belt formation. J. Biol. Chem. 2021, 296, 100790. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, E.; Weigert, A. Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues. Int. J. Mol. Sci. 2021, 22, 11636. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Obermann, W.M.J.; Brockhaus, K.; Eble, J.A. Platelets, Constant and Cooperative Companions of Sessile and Disseminating Tumor Cells, Crucially Contribute to the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 674553. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, M.M.; Fusenig, N.E. Friends or foes-bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Modulation of MMP-2 and MMP-9 secretion by cytokines, inducers and inhibitors in human glioblastoma T-98G cells. Oncol. Rep. 2017, 37, 1907–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Lan, F.; Wang, X.; Yu, Y.; Ouyang, X.; Zheng, F.; Han, J.; Lin, Y.; Xie, Y.; Xie, F.; et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 2014, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Park, S.S.; Kim, W.J.; Moon, S.K. IL-5-induced migration via ERK1/2-mediated MMP-9 expression by inducing activation of NF-κB in HT1376 cells. Oncol. Rep. 2012, 28, 1084–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Lee, S.J.; Kim, S.; Cho, S.C.; Choi, Y.H.; Kim, W.J.; Moon, S.K. Interleukin-5 enhances the migration and invasion of bladder cancer cells via ERK1/2-mediated MMP-9/NF-kappaB/AP-1 pathway: Involvement of the p21WAF1 expression. Cell Signal. 2013, 25, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Li, J.; Lu, Z.; Li, X.; Yu, J.; Li, N. IL-22 promotes the migration and invasion of gastric cancer cells via IL-22R1/AKT/MMP-9 signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 3694–3703. [Google Scholar] [PubMed]
- Fukui, H.; Zhang, X.; Sun, C.; Hara, K.; Kikuchi, S.; Yamasaki, T.; Kondo, T.; Tomita, T.; Oshima, T.; Watari, J.; et al. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br. J. Cancer 2014, 111, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019, 39, BSR20182361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 2016, 7, 62425–62438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 11 Pt A, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Blázquez-Prieto, J.; Amado-Rodriguez, L.; López-Alonso, I.; Batalla-Solís, E.; González-López, A.; Sánchez-Pérez, M.; Mayoral-Garcia, C.; Gutiérrez-Fernández, A.; Albaiceta, G.M. Matrix metalloproteinase-14 triggers an anti-inflammatory proteolytic cascade in endotoxemia. J. Mol. Med. 2017, 95, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, K.J.; Corry, D.B.; Engler, D.A.; Matsunami, R.K.; Tessier, P.; Cook, R.G.; Werb, Z.; Kheradmand, F. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J. Immunol. 2006, 177, 7312–7321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sithu, S.D.; English, W.R.; Olson, P.; Krubasik, D.; Baker, A.H.; Murphy, G.; D’Souza, S.E. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J. Biol. Chem. 2007, 282, 25010–25019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salnikov, A.V.; Iversen, V.V.; Koisti, M.; Sundberg, C.; Johansson, L.; Stuhr, L.B.; Sjoquist, M.; Ahlstrom, H.; Reed, R.K.; Rubin, K. Lowering of tumor interstitial fluid pressure specifically augments efficacy of chemotherapy. FASEB J. 2003, 17, 1756–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purkayastha, P.; Jaiswal, M.K.; Lele, T.P. Molecular cancer cell responses to solid compressive stress and interstitial fluid pressure. Cytoskeleton 2021, 78, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Boddupalli, A.; Koelbl, J.; Nam, D.H.; Ge, X.; Bratlie, K.M.; Schneider, I.C. Degradation and Remodeling of Epitaxially Grown Collagen Fibrils. Cell Mol. Bioeng. 2019, 12, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, S.R. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015, 44–46, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Biophysical studies of matrix metalloproteinase/triple-helix complexes. Adv. Protein Chem. Struct. Biol. 2014, 97, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Oncology 2020, 10, 641. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Mishra, D.P. Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteoglycan neofunctions: Regulation of inflammation and autophagy in cancer biology. FEBS J. 2017, 284, 10–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofeu Feugaing, D.D.; Gotte, M.; Viola, M. More than matrix: The multifaceted role of decorin in cancer. Eur. J. Cell Biol. 2013, 92, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-C and integrins in cancer. Cell Adh. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brellier, F.; Martina, E.; Degen, M.; Heuzé-Vourc’h, N.; Petit, A.; Kryza, T.; Courty, Y.; Terracciano, L.; Ruiz, C.; Chiquet-Ehrismann, R. Tenascin-W is a better cancer biomarker than tenascin-C for most human solid tumors. BMC Clin. Pathol. 2012, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiquet-Ehrismann, R.; Hagios, C.; Matsumoto, K. The tenascin gene family. Perspect. Dev. NeuroBiol. 1994, 2, 3–7. [Google Scholar] [PubMed]
- Bellahcène, A.; Castronovo, V.; Ogbureke, K.U.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 2008, 8, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamili, N.A.; Arthur, C.M.; Gerner-Smidt, C.; Tafesse, E.; Blenda, A.; Dias-Baruffi, M.; Stowell, S.R. Key regulators of galectin-glycan interactions. Proteomics 2016, 16, 3111–3125. [Google Scholar] [CrossRef] [PubMed]
- Naschberger, E.; Liebl, A.; Schellerer, V.S.; Schütz, M.; Britzen-Laurent, N.; Kölbel, P.; Schaal, U.; Haep, L.; Regensburger, D.; Wittmann, T.; et al. Matricellular protein SPARCL1 regulates tumor microenvironment-dependent endothelial cell heterogeneity in colorectal carcinoma. J. Clin. Investig. 2016, 126, 4187–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.D.; Kaur, S.; Isenberg, J.S. Regulation of Cellular Redox Signaling by Matricellular Proteins in Vascular Biology, Immunology, and Cancer. Antioxid. Redox Signal. 2017, 27, 874–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.M.; Postovit, L.M. Matricellular proteins in cancer: A focus on secreted Frizzled-related proteins. J. Cell Commun. Signal 2018, 12, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, R.P.; Degen, M. The Expression and Possible Functions of Tenascin-W During Development and Disease. Front. Cell Dev. Biol. 2019, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.; Tham, C.L.; Harith, H.H.; Firdaus, N.; Israf, D.A. TGF-beta-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur. J. Pharmacol. 2021, 911, 174510. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Lyu, C.; Liao, H.; Du, Y. Collagen crosslinking: Effect on structure, mechanics and fibrosis progression. Biomed. Mater. 2021, 16, 062005. [Google Scholar] [CrossRef] [PubMed]
- Setargew, Y.F.I.; Wyllie, K.; Grant, R.D.; Chitty, J.L.; Cox, T.R. Targeting Lysyl Oxidase Family Meditated Matrix Cross-Linking as an Anti-Stromal Therapy in Solid Tumours. Cancers 2021, 13, 491. [Google Scholar] [CrossRef] [PubMed]
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The Biological and Biomechanical Role of Transglutaminase-2 in the Tumour Microenvironment. Cancers 2021, 13, 2788. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Hintermann, E.; Bilban, M.; Koshikawa, N.; Hojilla, C.; Khokha, R.; Quaranta, V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 2003, 161, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, C.; Polette, M.; Coraux, C.; Tournier, J.M.; Meneguzzi, G.; Munaut, C.; Volders, L.; Rousselle, P.; Birembaut, P.; Foidart, J.M. Contribution of MT1-MMP and of human laminin-5 gamma2 chain degradation to mammary epithelial cell migration. J. Cell Sci. 2001, 114 Pt 16, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Giannelli, G.; Cirulli, V.; Miyazaki, K.; Quaranta, V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol. 2000, 148, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, K.; Mayernik, L.; Moin, K.; Sloane, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.R.; Antelmi, E.; Busco, G.; Guerra, L.; Rubino, R.; Casavola, V.; Reshkin, S.J.; Cardone, R.A. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol. Rep. 2014, 31, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chang, G.; Wang, J.; Jin, W.; Wang, L.; Li, H.; Ma, L.; Li, Q.; Pang, T. NHE1 mediates MDA-MB-231 cells invasion through the regulation of MT1-MMP. Exp. Cell Res. 2011, 317, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.R.; Moro, L.; Forciniti, S.; Alfarouk, K.; Cannone, S.; Cardone, R.A.; Reshkin, S.J. Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway. Int. J. Mol. Sci. 2021, 22, 2162. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Helvert, S.; Storm, C.; Friedl, P. Mechanoreciprocity in cell migration. Nat. Cell Biol. 2018, 20, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, A.; Mak, M.; Kamm, R.D.; Moeendarbary, E. Complex mechanics of the heterogeneous extracellular matrix in cancer. Extreme Mech. Lett. 2018, 21, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Chen, C.G.; Iozzo, R.V. Endorepellin evokes an angiostatic stress signaling cascade in endothelial cells. J. Biol. Chem. 2020, 295, 6344–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, G.; Malmsten, M.; Ermilov, E. Anionic biopolyelectrolytes of the syndecan/perlecan superfamily: Physicochemical properties and medical significance. Adv. Colloid Interface Sci. 2014, 205, 275–318. [Google Scholar] [CrossRef] [PubMed]
- Cork, S.M.; Kaur, B.; Devi, N.S.; Cooper, L.; Saltz, J.H.; Sandberg, E.M.; Kaluz, S.; Van Meir, E.G. A proprotein convertase/MMP-14 proteolytic cascade releases a novel 40 kDa vasculostatin from tumor suppressor BAI1. Oncogene 2012, 31, 5144–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasco, S.; Han, J.; Gillery, P.; Bellon, G.; Maquart, F.X.; Borel, J.P.; Kefalides, N.A.; Monboisse, J.C. A specific sequence of the noncollagenous domain of the alpha3(IV) chain of type IV collagen inhibits expression and activation of matrix metalloproteinases by tumor cells. Cancer Res. 2000, 60, 467–473. [Google Scholar] [PubMed]
- Taylor, K.R.; Trowbridge, J.M.; Rudisill, J.A.; Termeer, C.C.; Simon, J.C.; Gallo, R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004, 279, 17079–17084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelcker, V.; Gebhardt, C.; Averbeck, M.; Saalbach, A.; Wolf, V.; Weih, F.; Sleeman, J.; Anderegg, U.; Simon, J. Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp. Dermatol. 2008, 17, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumour. Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Kou, L.; Tu, Y.; Zhu, L. MMP-Responsive ‘Smart’ Drug Delivery and Tumor Targeting. Trends Pharmacol. Sci. 2018, 39, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muvva, C.; Patra, S.; Venkatesan, S. MMpI: A WideRange of Available Compounds of Matrix Metalloproteinase Inhibitors. PLoS ONE 2016, 11, e0159321. [Google Scholar] [CrossRef] [PubMed]
- BT1718 in Patients with Advanced Solid Tumours. 3 April 2018 (Cited 29 November 2021), 1 p. ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US), 12 Nov 2020. Identifier: NCT03486730. Available online: https://www.clinicaltrials.gov/ct2/show/study/NCT03486730?term=MMP14&draw=2&rank=4 (accessed on 29 November 2021).
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaimal, R.; Aljumaily, R.; Tressel, S.L.; Pradhan, R.V.; Covic, L.; Kuliopulos, A.; Zarwan, C.; Kim, Y.B.; Sharifi, S.; Agarwal, A. Selective blockade of matrix metalloprotease-14 with a monoclonal antibody abrogates invasion, angiogenesis, and tumor growth in ovarian cancer. Cancer Res. 2013, 73, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletov, K.; Lewis, J.D. Invadopodia: A new therapeutic target to block cancer metastasis. Expert Rev. Anticancer Ther. 2015, 15, 733–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngan, E.; Stoletov, K.; Smith, H.W.; Common, J.; Muller, W.J.; Lewis, J.D.; Siegel, P.M. LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun. 2017, 8, 15059. [Google Scholar] [CrossRef] [PubMed]
- Sutoh Yoneyama, M.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, B.; Wyckoff, J.; Yamaguchi, H.; Wang, Y.; Roussos, E.T.; Condeelis, J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 2012, 125 Pt 3, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blouw, B.; Patel, M.; Iizuka, S.; Abdullah, C.; You, W.K.; Huang, X.; Li, J.L.; Diaz, B.; Stallcup, W.B.; Courtneidge, S.A. The invadopodia scaffold protein Tks5 is required for the growth of human breast cancer cells in vitro and in vivo. PLoS ONE 2015, 10, e0121003. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirson, T.; Genna, A.; Lukic, N.; Makhnii, T.; Alter, J.; Sharma, V.P.; Wang, Y.; Samson, A.O.; Condeelis, J.S.; Gil-Henn, H. Targeting invadopodia-mediated breast cancer metastasis by using ABL kinase inhibitors. Oncotarget 2018, 9, 22158–22183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammer, A.G.; Kelley, L.C.; Hayes, K.E.; Evans, J.V.; Lopez-Skinner, L.A.; Martin, K.H.; Frederick, B.; Rothschild, B.L.; Raben, D.; Elvin, P.; et al. Saracatinib Impairs Head and Neck Squamous Cell Carcinoma Invasion by Disrupting Invadopodia Function. J. Cancer Sci. Ther. 2009, 1, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintavalle, M.; Elia, L.; Price, J.H.; Heynen-Genel, S.; Courtneidge, S.A. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci. Signal. 2011, 4, ra49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanos, N.K.; Piperigkou, Z.; Passi, A.; Gotte, M.; Rousselle, P.; Vlodavsky, I. Extracellular matrix-based cancer targeting. Trends Mol. Med. 2021, 27, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Pancreatic cancer provides testbed for first mechanotherapeutics. Nat. Biotechnol 2019, 37, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Abdollahiyan, P.; Oroojalian, F.; Baradaran, B.; de la Guardia, M.; Mokhtarzadeh, A. Advanced mechanotherapy: Biotensegrity for governing metastatic tumor cell fate via modulating the extracellular matrix. J. Control. Release 2021, 335, 596–618. [Google Scholar] [CrossRef] [PubMed]
- Pietraszek, K.; Chatron-Colliet, A.; Brézillon, S.; Perreau, C.; Jakubiak-Augustyn, A.; Krotkiewski, H.; Maquart, F.X.; Wegrowski, Y. Lumican: A new inhibitor of matrix metalloproteinase-14 activity. FEBS Lett. 2014, 588, 4319–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appunni, S.; Rubens, M.; Ramamoorthy, V.; Anand, V.; Khandelwal, M.; Saxena, A.; McGranaghan, P.; Odia, Y.; Kotecha, R.; Sharma, A. Lumican, pro-tumorigenic or anti-tumorigenic: A conundrum. Clin. Chim Acta 2021, 514, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Kang, K.A. MMP-14 Triggered Fluorescence Contrast Agent. Adv. Exp. Med. Biol. 2016, 923, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, F.; Ma, Y.; Liu, G.; Kim, K.; Fang, X.; Lee, S.; Chen, X. In vivo optical imaging of membrane-type matrix metalloproteinase (MT-MMP) activity. Mol. Pharm. 2011, 8, 2331–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, Y.P. Fluorescent and Bioluminescent Nanoprobes for In Vitro and In Vivo Detection of Matrix Metalloproteinase Activity. BMB Rep. 2015, 48, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, E.J.; Crawford, B.D. The epitope-mediated MMP activation assay: Detection and quantification of the activation of Mmp2 in vivo in the zebrafish embryo. Histochem. Chem. Cell Biol. 2018, 149, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, S.; Ji, X.; Zhang, J.; Wang, D.; Lee, S.J.; Lee, H.; He, H.; Yang, V.C. MMP-2-responsive fluorescent nanoprobes for enhanced selectivity of tumor cell uptake and imaging. Biomater. Sci. 2018, 6, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
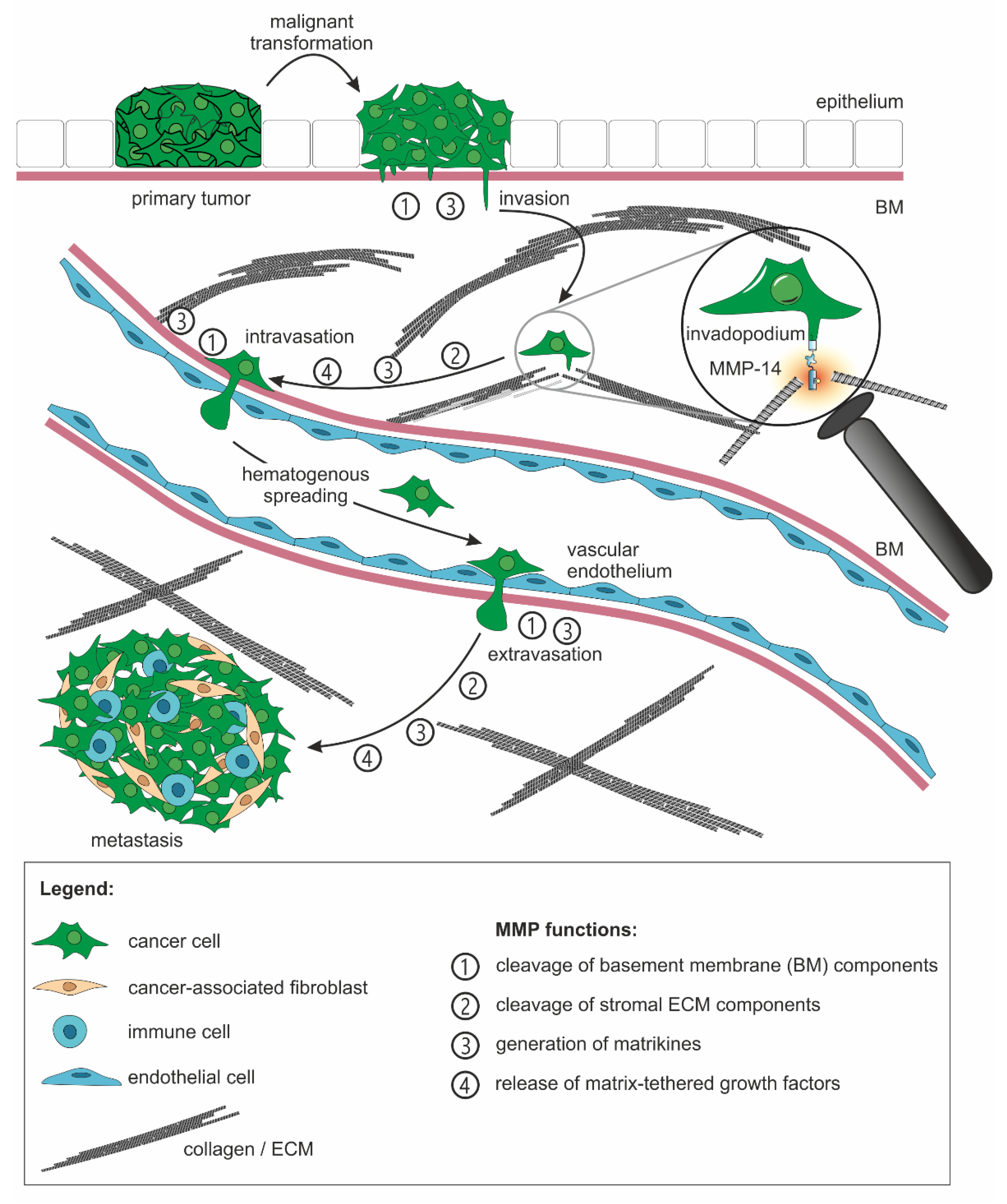
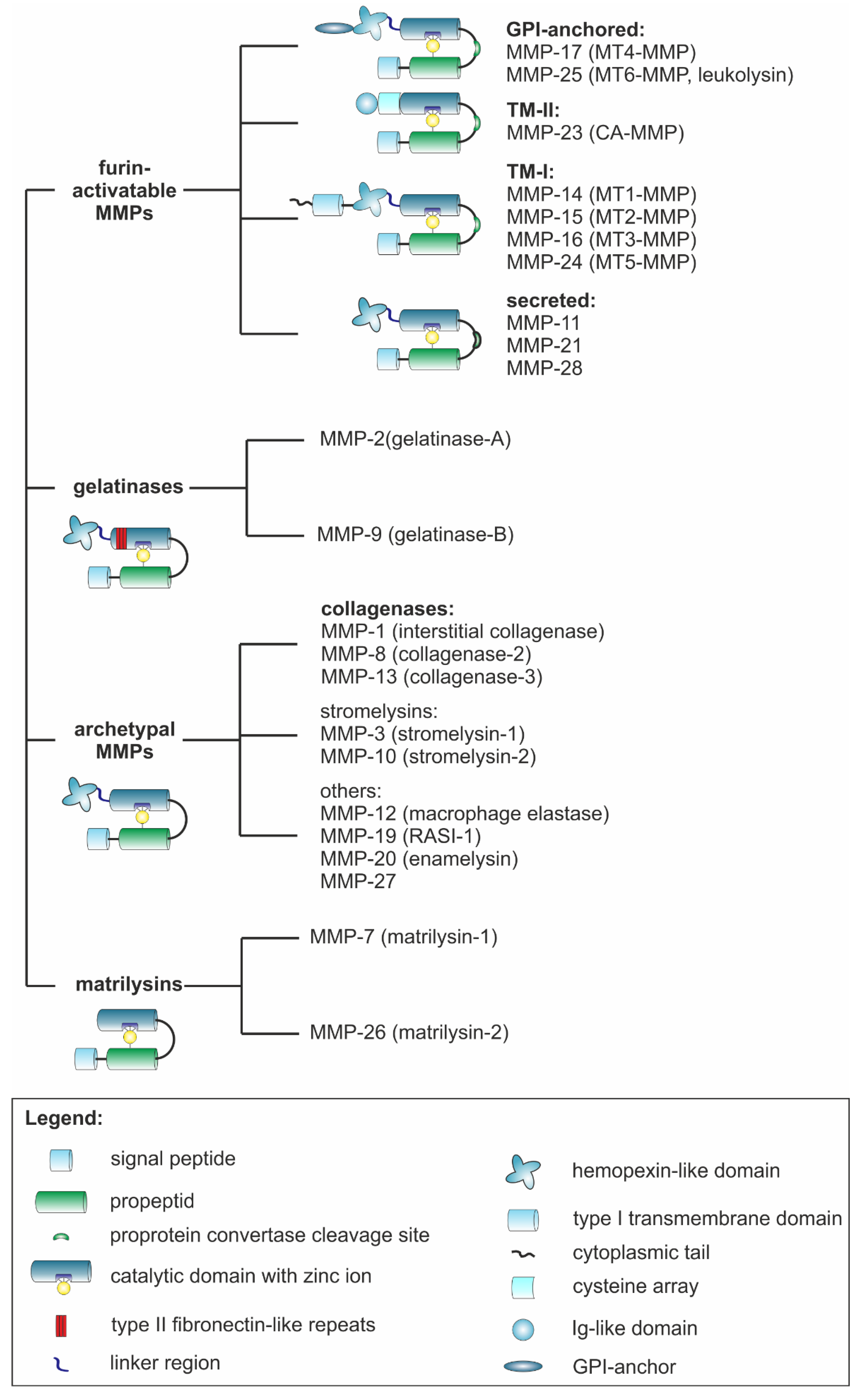


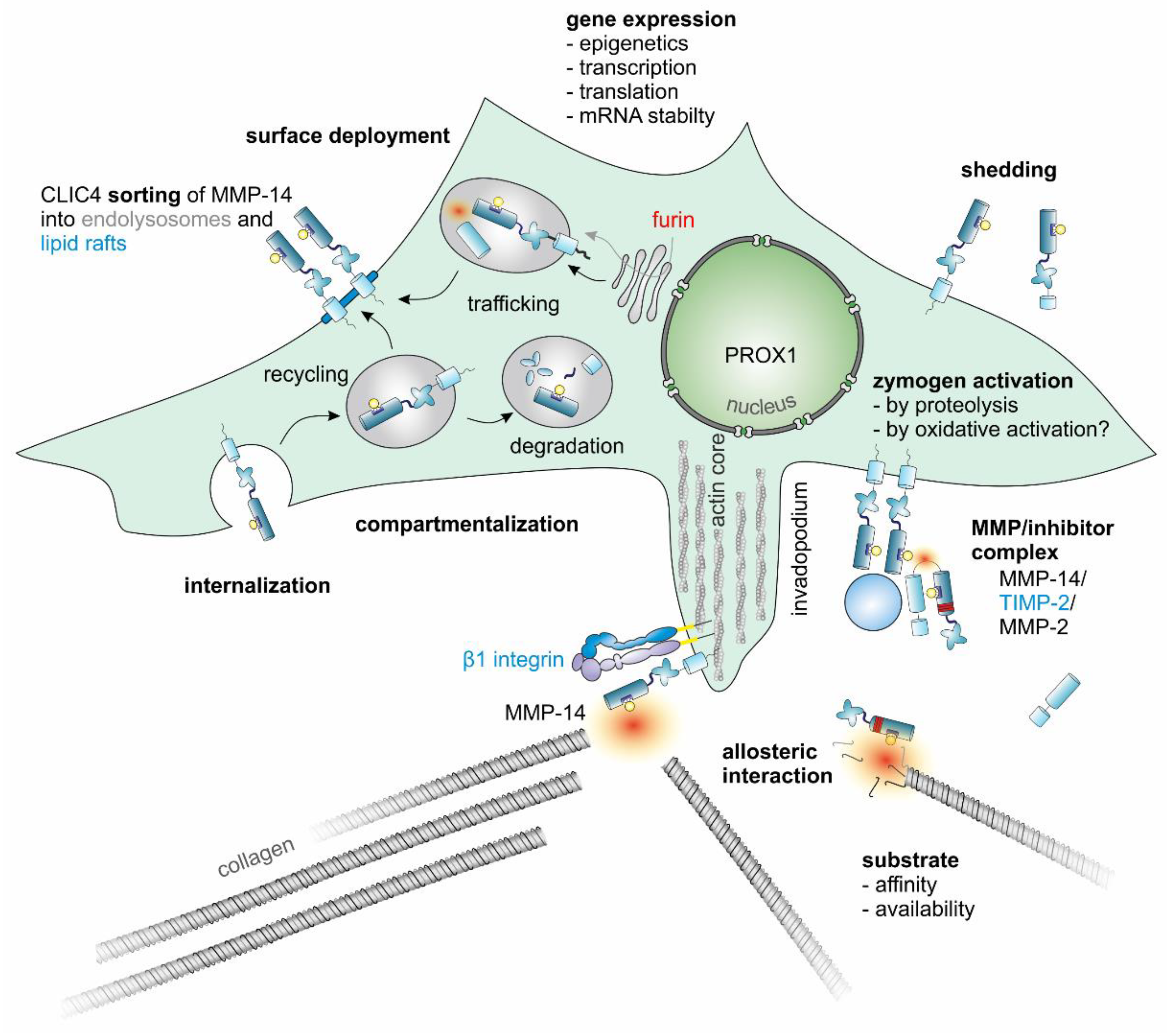
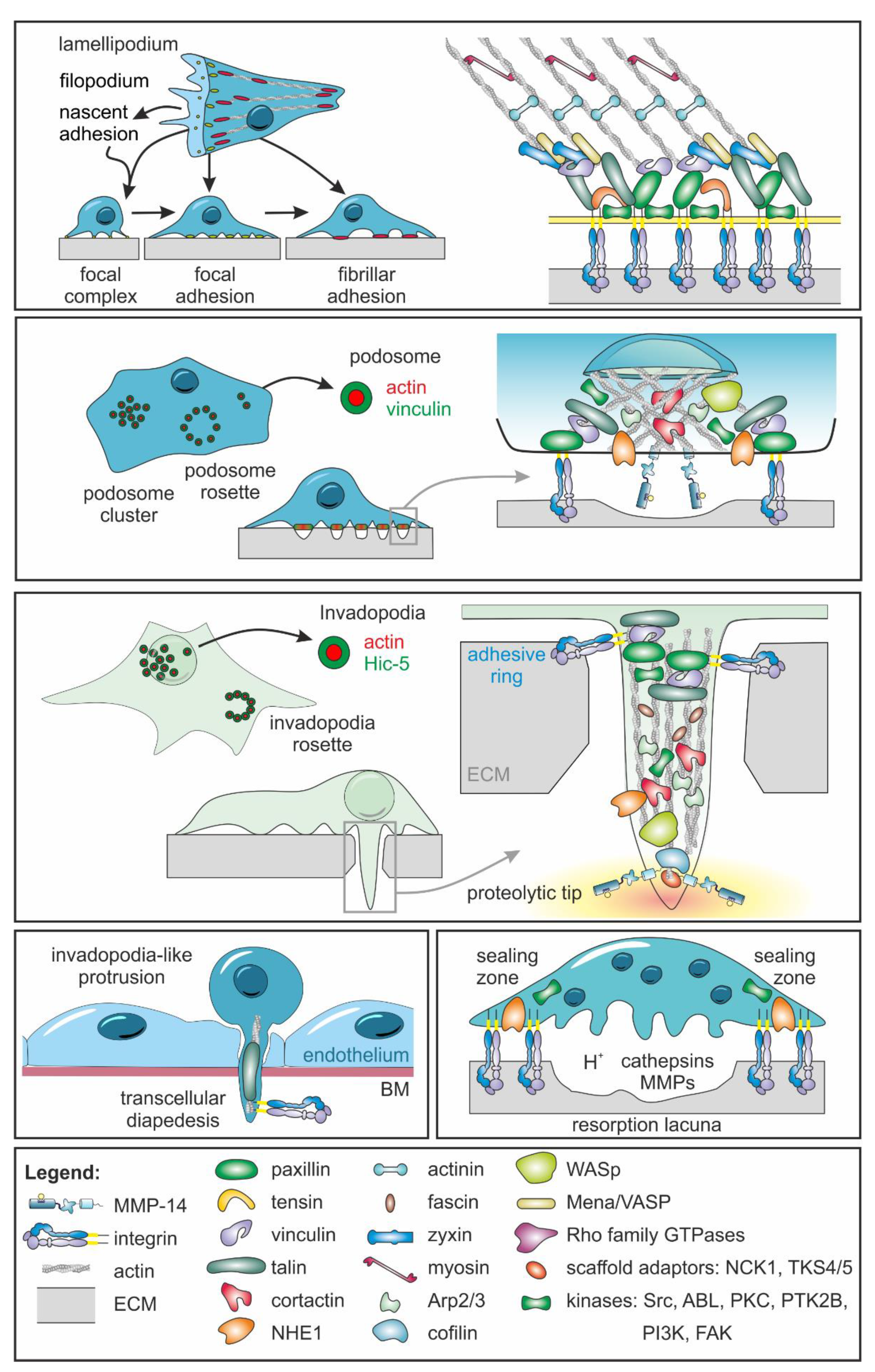
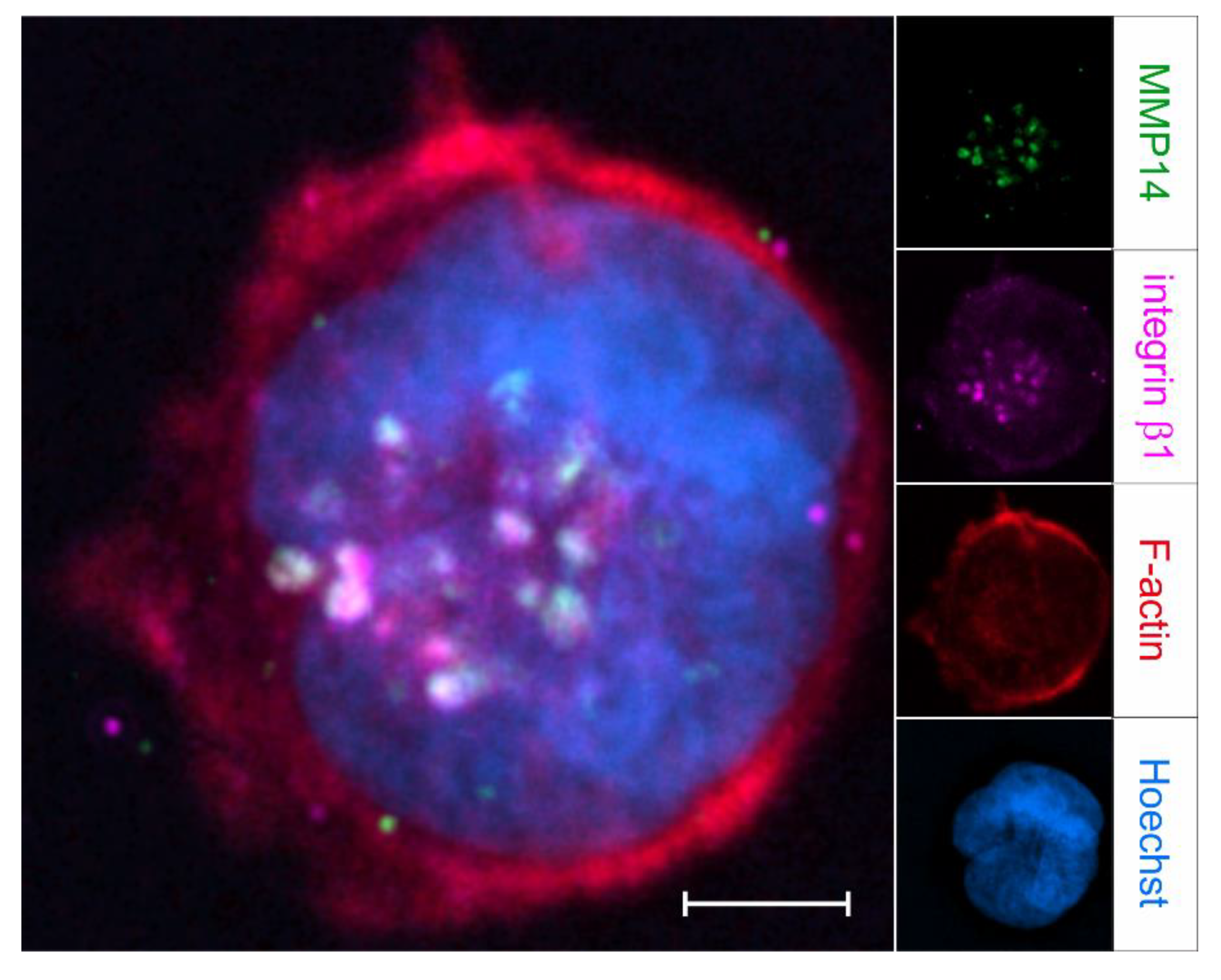
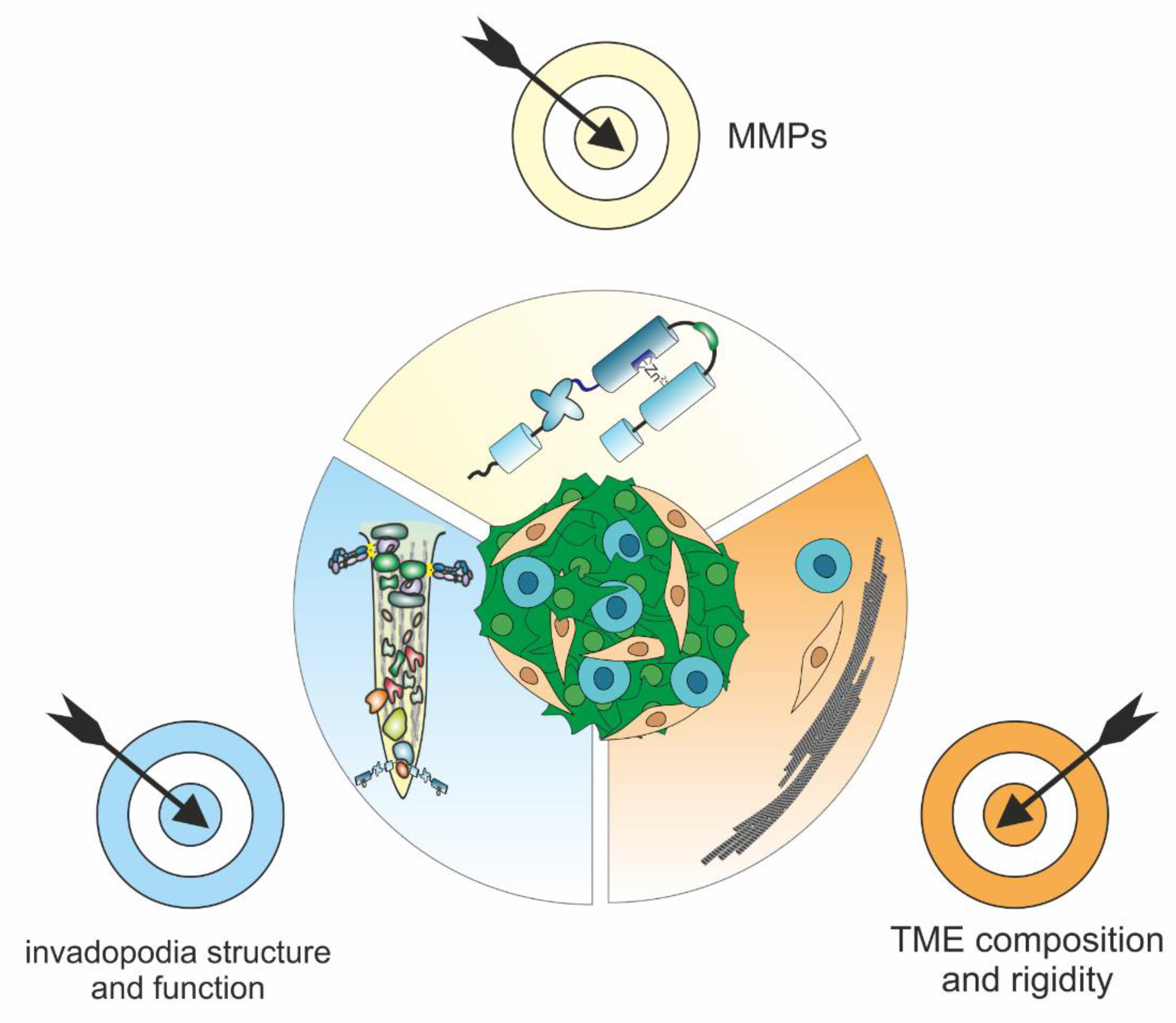

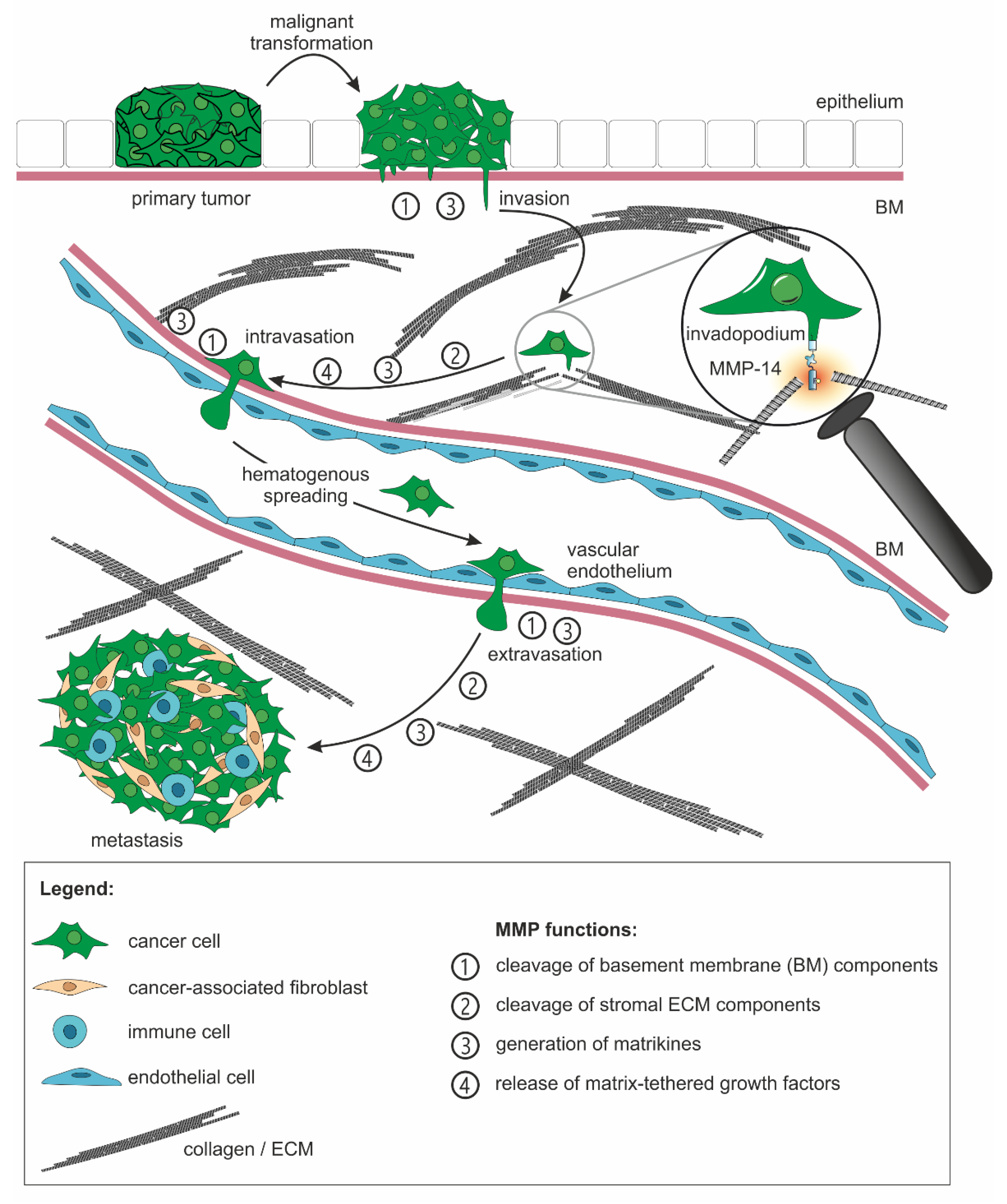{kind=link}
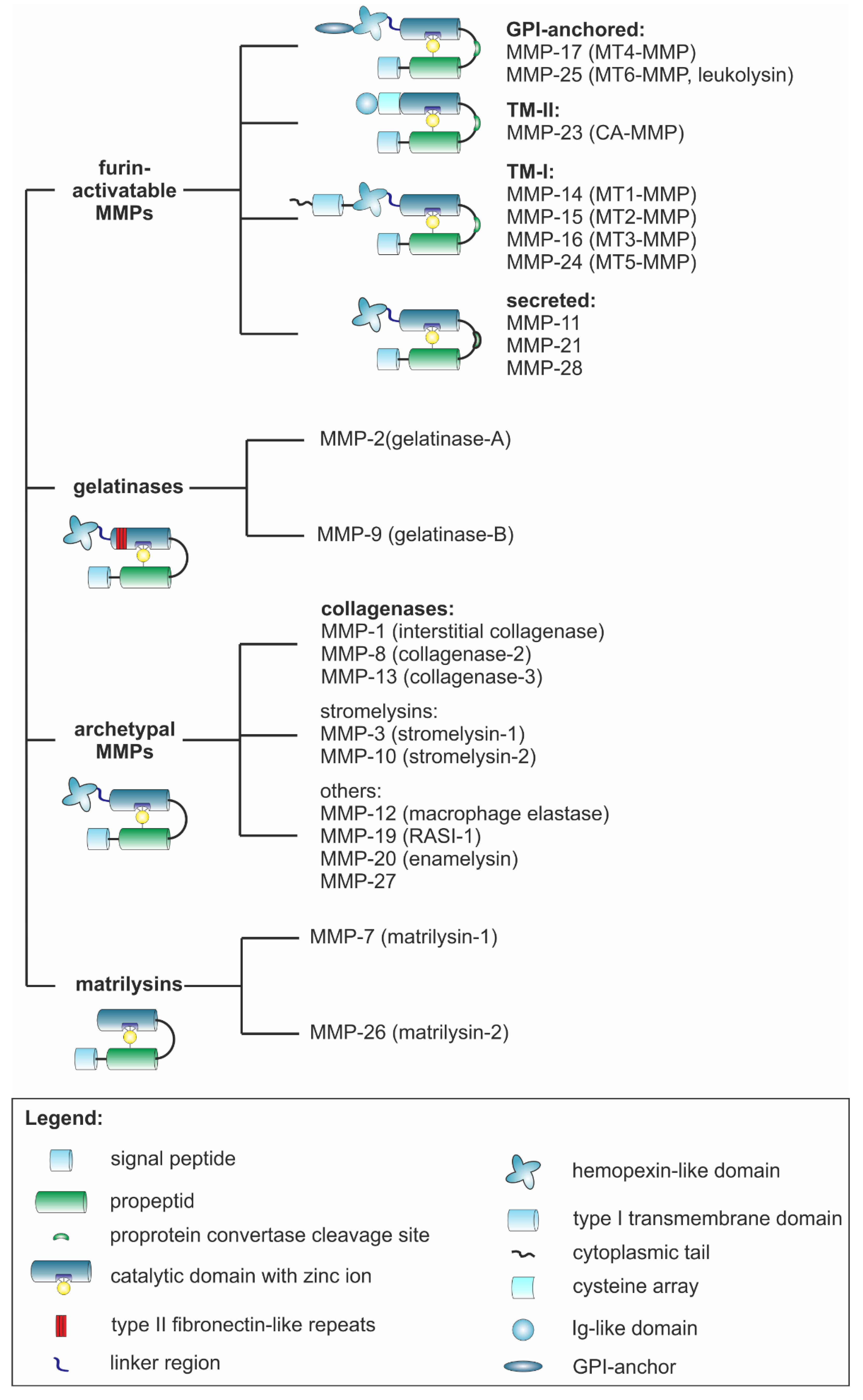{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMP | Involved In | Substrates | ||
|---|---|---|---|---|
| MMPs [47,57] | ECM [15,47,57] | Cell–Matrix and Cell–Cell Receptors [47,57] | ||
| MMP-1 | EMT [17,35], invasion and metastasis [36] | proMMPs -1, -2, -9 | collagens I, II, III, VII, VIII, X, XI, gelatin, elastin, fibronectin, vitronectin, aggrecan, neurocan, brevican, decorin, perlecan, laminin-5, nidogen, CTGF (CCN2), tenascin, SPARC, fibrinogen, fibrin, link protein | |
| MMP-2 | EMT [36] | proMMPs -1, -2, -9, -13, MMP-12 | collagens I (a), III (a), IV (a), V (a), VII (a), X (a), XI (a), gelatin, elastin, fibrillin, fibronectin, vitronectin, aggrecan, laminin, nidogen, tenascin, fibrinogen, fibrin, decorin, link protein | dystroglycan |
| MMP-3 | EMT [34,37], invasion and metastasis [36] | proMMPs -1, -2, -3, -7, -8, -9, -13 | non-triple-helical regions of collagens III, IV, V, VII, IX, X, XI, collagen telopeptides, gelatin, elastin, fibrillin, fibronectin, vitronectin, aggrecan, versican, decorin, biglycan, perlecan, laminin, nidogen, fibulin, tenascin, SPARC, osteopontin, fibrinogen, fibrin, link protein, myelin basic protein | E-cadherin [58] |
| MMP-7 | EMT [38], invasion and metastasis [36] | proMMPs -1, -2, -7, -9 | collagen IV (a), non-triple-helical regions of collagens IV, V, IX, X, XI, gelatin, elastin, fibronectin, vitronectin, aggrecan, brevican, versican, decorin, laminin, nidogen, fibulin, tenascin, SPARC, osteopontin, galectin-3, fibrinogen, fibrin, link protein, myelin basic protein | E-cadherin [59], β4 integrin, syndecans -1, -2 [60] |
| MMP-8 | tumor angiogenesis [54] | proMMP-8 | collagens I, II, III, gelatin, aggrecan, link protein | |
| MMP-9 | EMT [36], invasion and metastasis [54], tumor angiogenesis [50] | proMMPs -2, -9, -13, ADAMTS-4 (b) | non-triple-helical regions of collagens I, IV, V, XI, XIV, collagens III (a), IV (a), V (a), gelatin, elastin, fibrillin, fibronectin, vitronectin, aggrecan, versican, decorin, biglycan, laminin, nidogen, SPARC, galectins -1 and -3, fibrinogen, fibrin, link protein, myelin basic protein | E-cadherin, β2 integrin, dystroglycan |
| MMP-10 | invasion and metastasis [51], tumor angiogenesis [51] | proMMPs -1, -2, -7, -8, -9, -10, -13 | collagens I (a), III (a), IV (a), V (a), gelatin, elastin, fibronectin, aggrecan, brevican, laminin-5, link protein, fibrinogen | |
| MMP-11 | invasion and metastasis [51] | proMMPs -2, -11 | collagen IV (a), gelatin, fibronectin, aggrecan, laminin | |
| MMP-12 | collagens (a) I, IV, V, gelatin, elastin fibrillin, fibronectin, vitronectin, aggrecan, decorin, biglycan, laminin, nidogen, SPARC, fibrinogen, fibrin, myelin basic protein | |||
| MMP-13 | invasion and metastasis [36] | proMMPs -2, -9, -13 | collagens I, II, III, VI, VII, IX, X, XIV, gelatin, fibrillin, fibronectin, aggrecan, brevican core protein precursor, biglycan, perlecan, laminin-γ2, nidogen, CTGF (CCN2), tenascin, large tenascin C, SPARC, fibrinogen | |
| MMP-14 | EMT [39,40], invasion and metastasis [36], tumor angiogenesis [17,55] | proMMPs -2, -8 [61], -13, -14, MMP-14, ADAM9 (b) | collagens I, II, III, gelatin, tropoelastin [62], elastin [62], fibrillin, fibronectin, vitronectin, aggrecan, perlecan, lumican, nidogen, laminins -1, -2, -4, -5, CTGF, CTGF-L (CCN5), Cyr61 (CCN1), tenascin, galectins -1 and -3, fibrinogen, fibrin, myelin basic protein | E-cadherin, N-cadherin, ICAM-1, αV integrin, syndecan-1, syndecan-2 [60], CD44, ICAM-1, DLL1, EMMPRIN |
| MMP-15 | proMMPs -2, -13 | collagen (a) I, NC1 (collagen IV), fibronectin, aggrecan, perlecan, laminin-1, nidogen, tenascin, fibrinogen, fibrin, myelin basic protein | ||
| MMP-16 | invasion and metastasis [36] | proMMP-2 | collagen III (a), gelatin, fibronectin, vitronectin, laminin-1, fibrin, myelin basic protein | |
| MMP-17 | proMMP-2, ADAMTS4 (b) | gelatin, fibronectin, laminin-1, chondroitin sulfate proteoglycan, dermatan sulfate proteoglycan, fibrinogen, fibrin, myelin basic protein | N-cadherin | |
| MMP-19 | proMMP-19, MMP-9 | collagen IV (a), gelatin, fibronectin, aggrecan, laminin, nidogen-1, tenascin, large tenascin-C, COMP, fibrinogen, fibrin | ||
| MMP-20 | proMMP-20 (autolysis) | collagen XVIII (a), gelatin, aggrecan, laminin, COMP, amelogenin, ameloblastin | ||
| MMP-21 | gelatin, aggrecan | |||
| MMP-23 | gelatin, fibronectin | |||
| MMP-24 | proMMP-2, ADAMTS4 (b) | gelatin, fibronectin, chondroitin sulfate proteoglycan, dermatan sulfate proteoglycan, fibrinogen, fibrin | ||
| MMP-25 | proMMPs -2, -9 | collagen IV (a), gelatin, fibronectin, laminin-1, chondroitin sulfate proteoglycan, dermatan sulfate proteoglycan, SPARC, galectin-1, fibrinogen, fibrin, myelin basic protein | ||
| MMP-26 | invasion and metastasis [36] | proMMPs -9, -26 | collagen IV (a), gelatin, fibronectin, vitronectin, fibrinogen | |
| MMP-27 | proMMP-27 (autolysis) | gelatin | ||
| MMP-28 | EMT [41], invasion and metastasis [36] | NCAM | ||
| Adhesome Structure | Focal Complex [229] | Focal Adhesion [229,230] | Fibrillar Adhesion [231] | Podosome (a) [229,232,233,234] | Invadopodium (a) [229,230,232] | Invadosome-like Protrusion [235] | Sealing Zone of Resorption Lacuna [236] |
|---|---|---|---|---|---|---|---|
| Occurrence | adherent cells | adherent cells | adherent cells | rat sarcoma virus (Ras)-transformed fibroblasts, macrophages, immature dendritic cells, osteoclasts, ECs, myoblasts, neural crest cells [233] | invasive cancer cells [237] | lymphocytes [238] | osteoclasts, macrophages, dendritic cells [239] |
| Proteolytic activity | no | no | no | yes: MMP-14 [233] | yes (b) | no? (c) | yes: lysosomal enzymes [240,241] |
| Matrix receptors | β1 and β3 integrins, αV integrins | β1 and β3 integrins, αV integrins | β1 and β3 integrins, αV integrins | β1 and β2 integrins: α2β1, α3β1, α4β1, α5β1, α6β1, αVβ1, αLβ2, αMβ2, αXβ2, αDβ2, αVβ3, β4, β5 [229], CD44 [242] | β1 and β2 integrins: α2, α2β1, α3β1, α4β1, α5β1, α6β1, αVβ1,β2, αLβ2, αMβ2, αXβ2, αDβ2, αVβ3, β4, β5 [229] | integrin αLβ2 [243] | CD44, β3 integrins, αvβ3 [242,244] |
| Essential structural components | phospho-paxillin, FAK, α-actinin, talin [245] | actin, paxillin, FAK, talin, zyxin, vinculin, VASP [245] | dephospho-paxillin, FAK, talin, vinculin, VASP, α-actinin, tensin [245] | actin, vinculin, talin, Arp2/3, myosin IIa, capping protein, TKS4/5, Cdc42, Src [234] | actin, Arp2/3, cortactin, N-WASp, Nck1, cofilin, TKS5 [246] | actin, talin, vinculin [243] | actin, vinculin, talin, paxillin, zyxin, Arp2/3, N-WASp, myosin X, Arp2/3, capping protein, TKS4/5 [236] |
| Diameter [µm] | 0.5–1 [247] | 1–5 [247] | >5 [247] (d) | 0.2–2 [248,249,250] | 0.5–2 [251],8 [232] | 0.2–1 [238] | >14 [239], 95–130 [242] (e) |
| Protrusion depth [µm] | - | - | - | 0.2–0.5 [248,249,250] | >10 [243],>60 [232] | <10 [243] | - (f) |
| Lifetime [min] | 2–3 [247] | 20–90 [252] | very stable [247] | 2–12 [232,253] | >10 [243],>60 [232] | <10 [243] | 8–360 [254] |
| Number per cell | <400 [246], variable (g) [255] | 20–100 [232] | 1–10 [232] | 10–100 (h) [243] | variable (i) [255] | ||
| Function | Cell–matrix contact | cell–matrix contact | Cell–matrix contact | Cell–matrix contact, ECM degradation, sensing of substrate rigidity and topography, antigen sampling, myoblast fusion [233] | cell–matrix contact, ECM degradation, sensing of substrate rigidity and topography | biomechanical scanning, cell–cell interaction, diapedesis [235] | cell–matrix adhesion, sealing of the bone resorption lacuna [239] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2022, 23, 146. https://doi.org/10.3390/ijms23010146
Niland S, Riscanevo AX, Eble JA. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. International Journal of Molecular Sciences. 2022; 23(1):146. https://doi.org/10.3390/ijms23010146
Chicago/Turabian StyleNiland, Stephan, Andrea Ximena Riscanevo, and Johannes Andreas Eble. 2022. "Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression" International Journal of Molecular Sciences 23, no. 1: 146. https://doi.org/10.3390/ijms23010146
APA StyleNiland, S., Riscanevo, A. X., & Eble, J. A. (2022). Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. International Journal of Molecular Sciences, 23(1), 146. https://doi.org/10.3390/ijms23010146