
Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
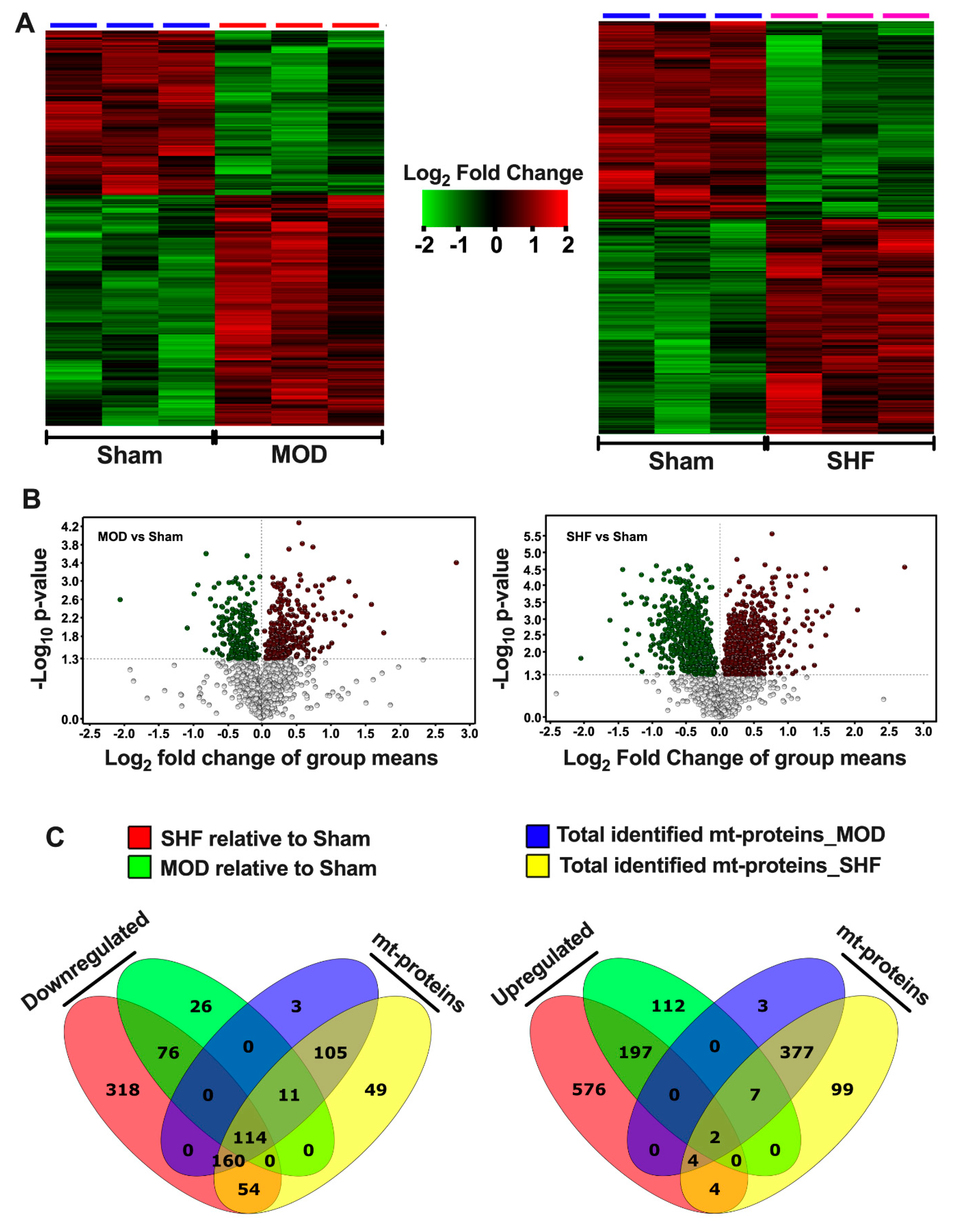
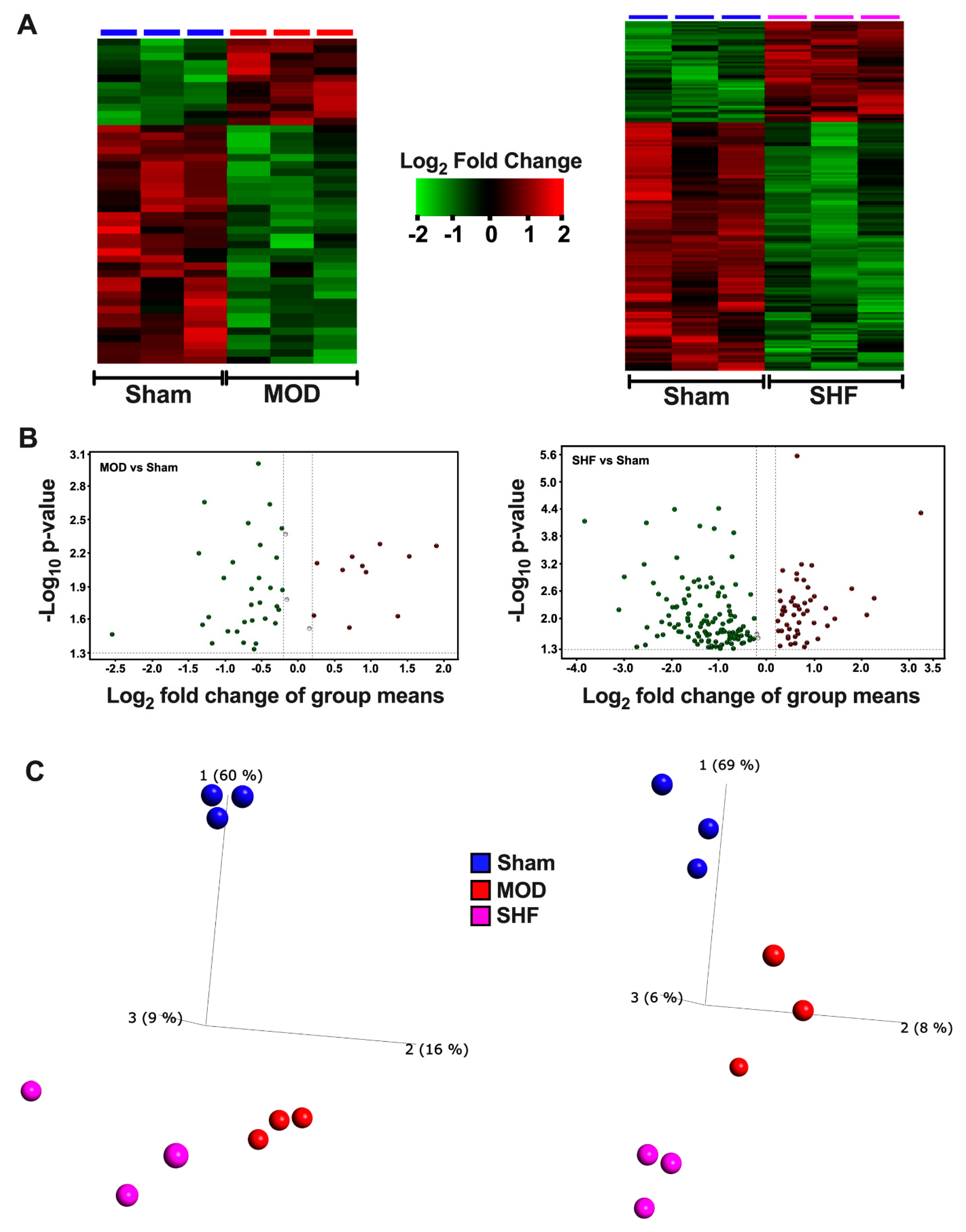
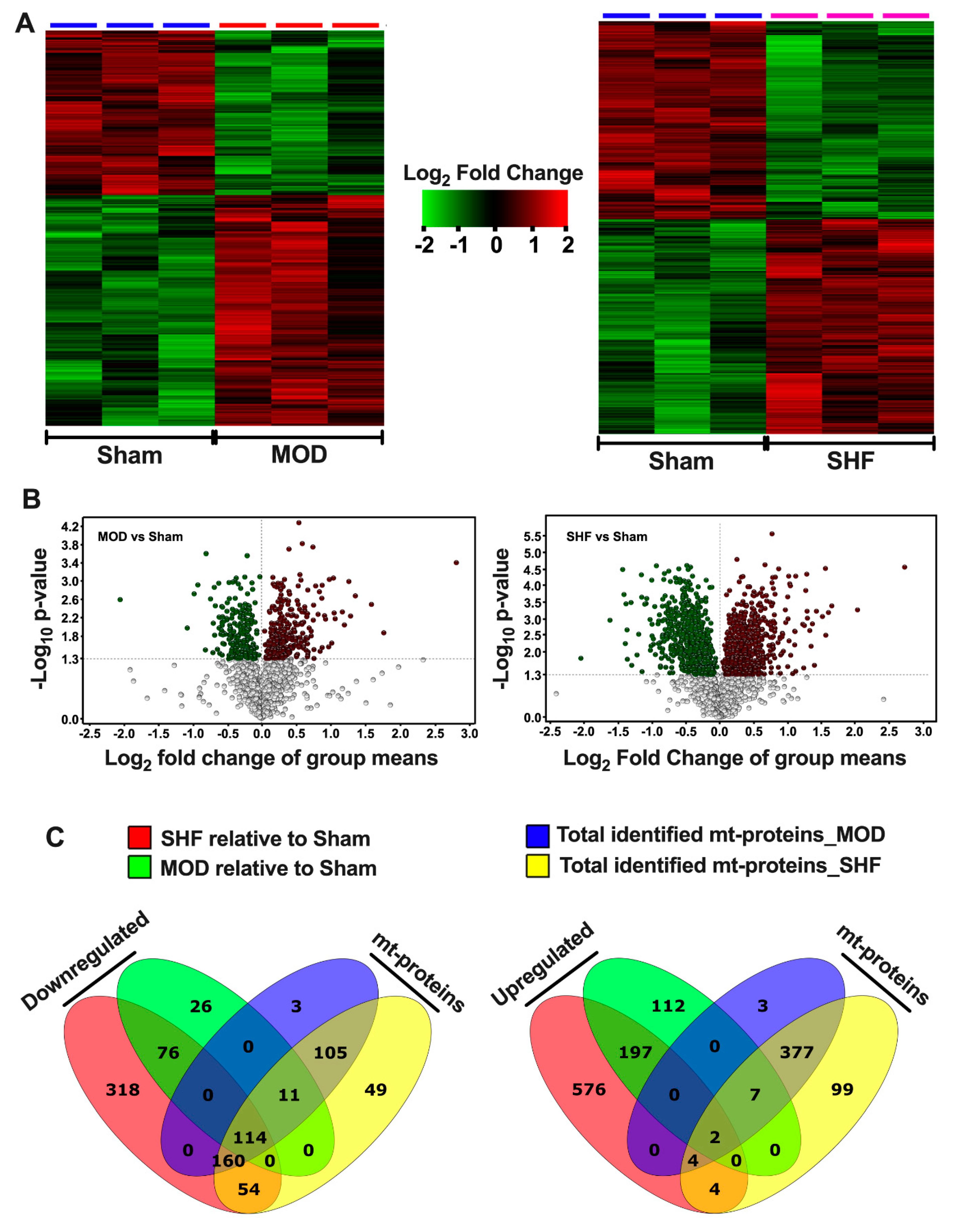
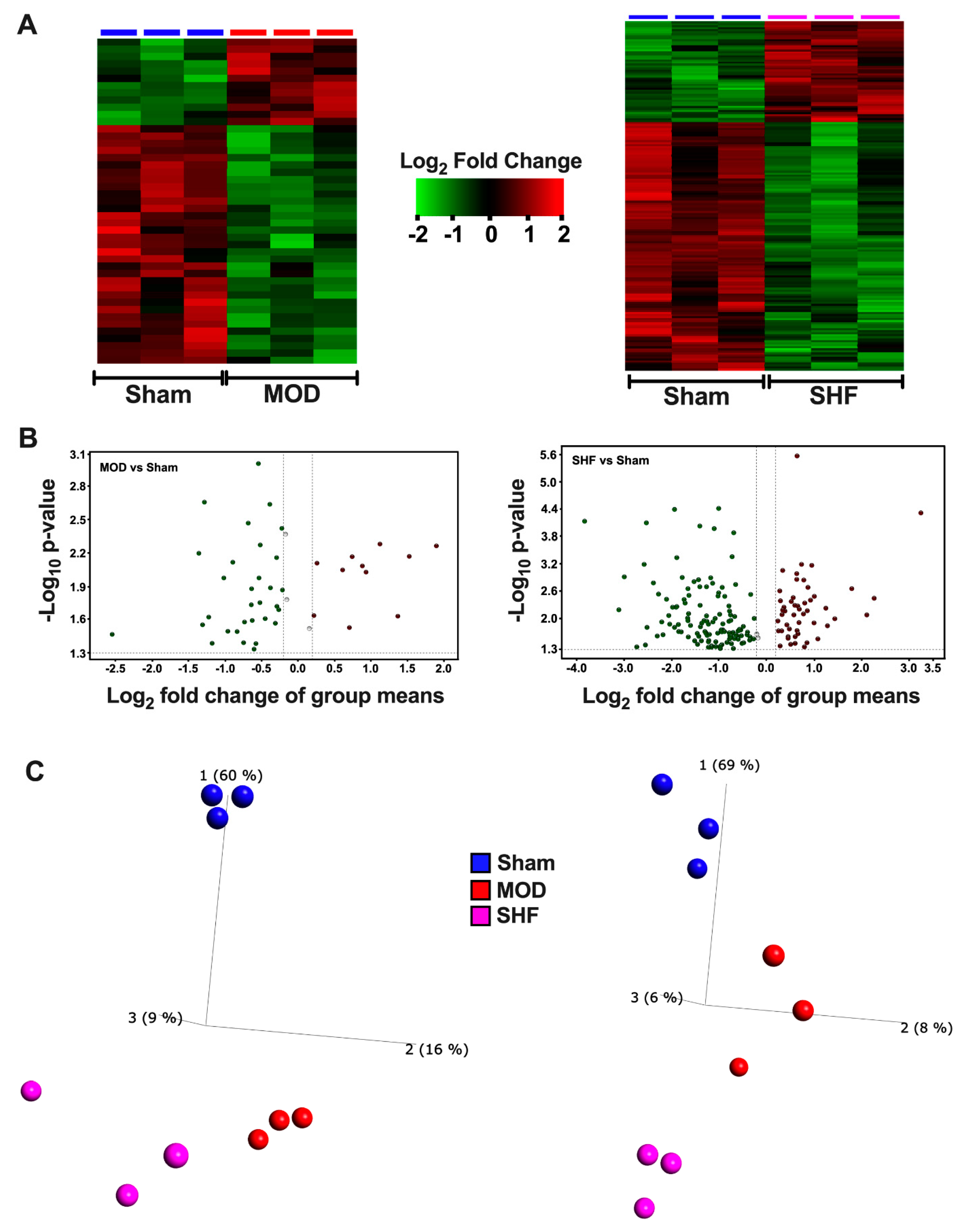
2.1. Visualization of the MOD and SHF Proteomic Datasets
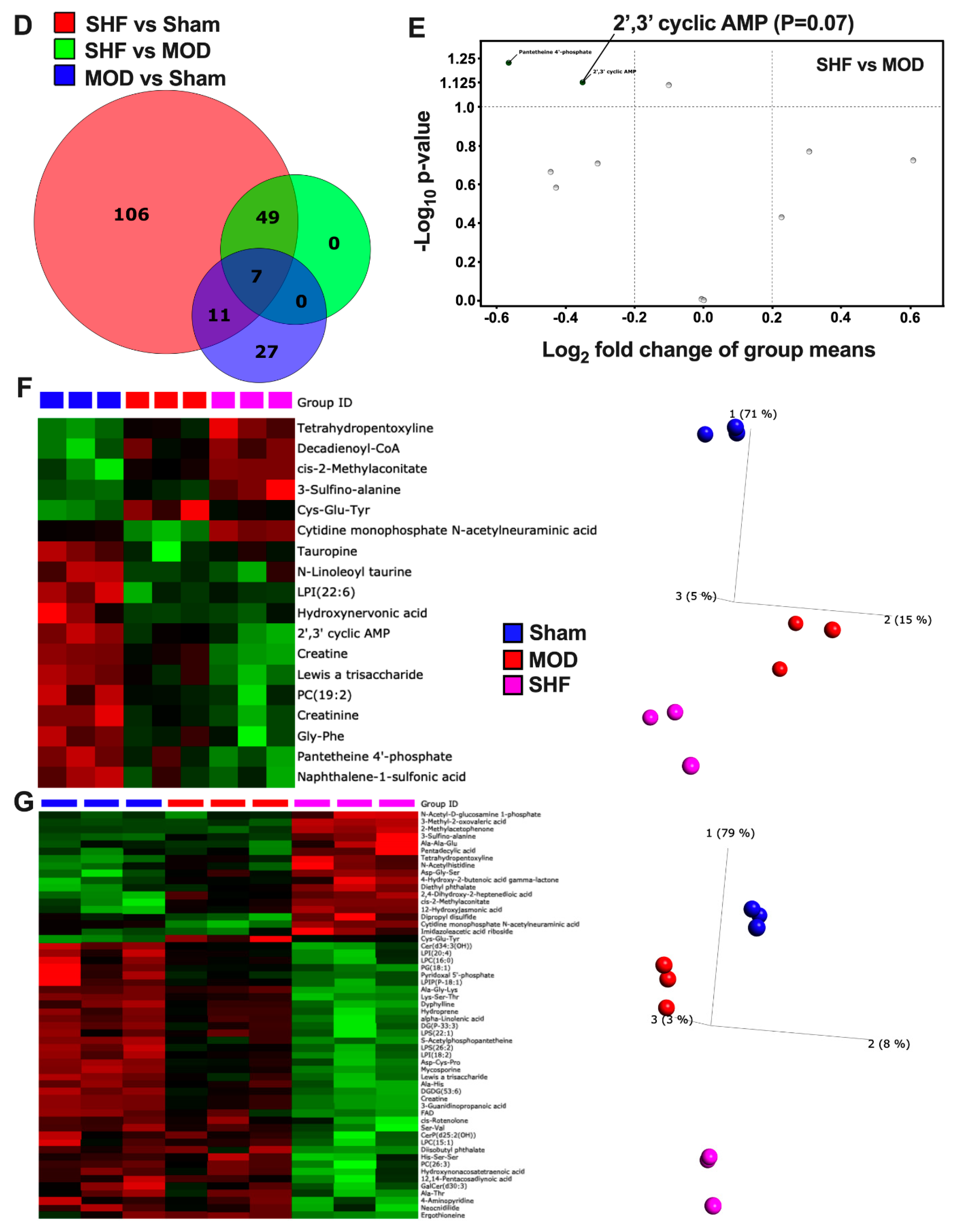
2.2. Visualization of the Untargeted Metabolomics Dataset in Sham, MOD and SHF Groups
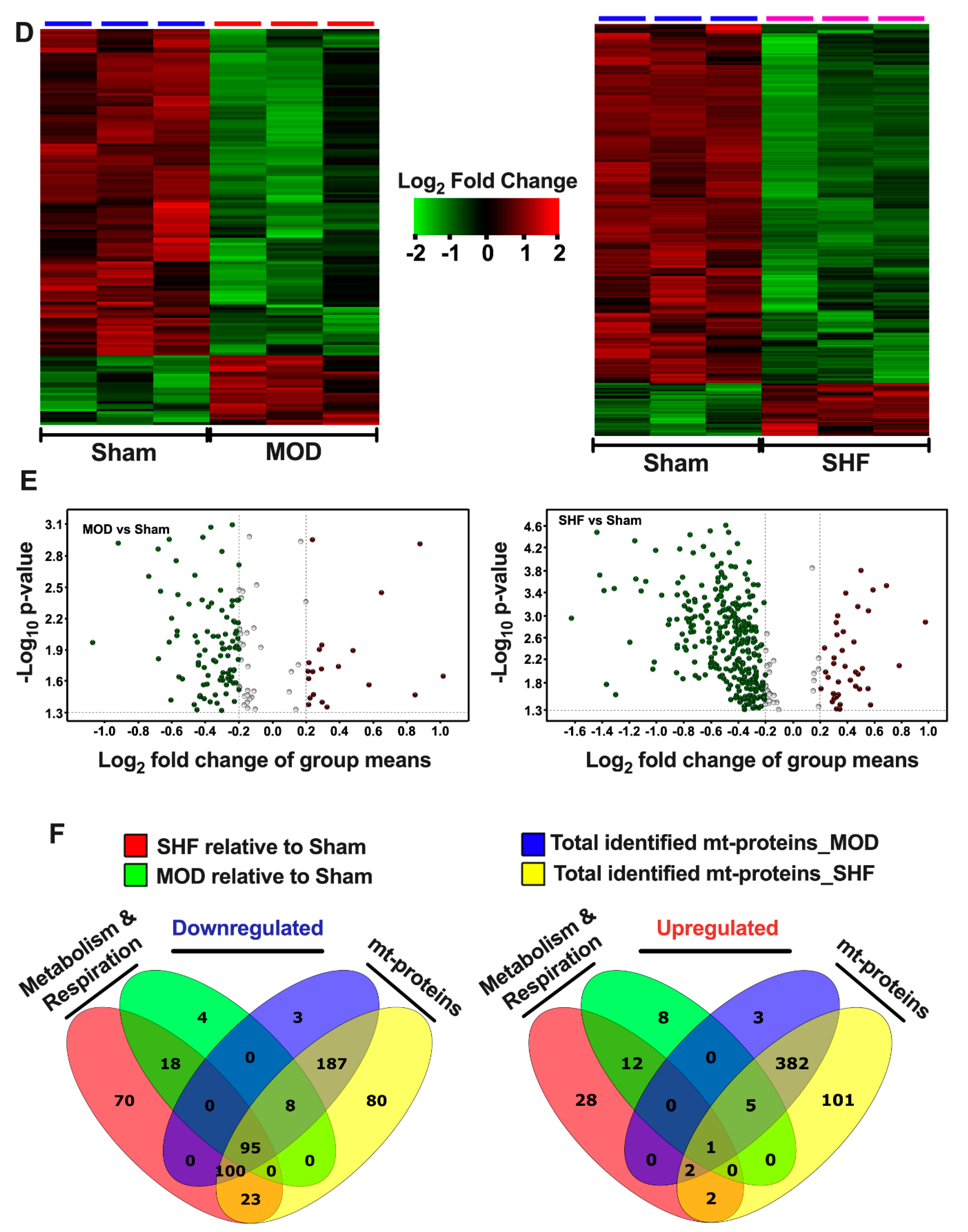
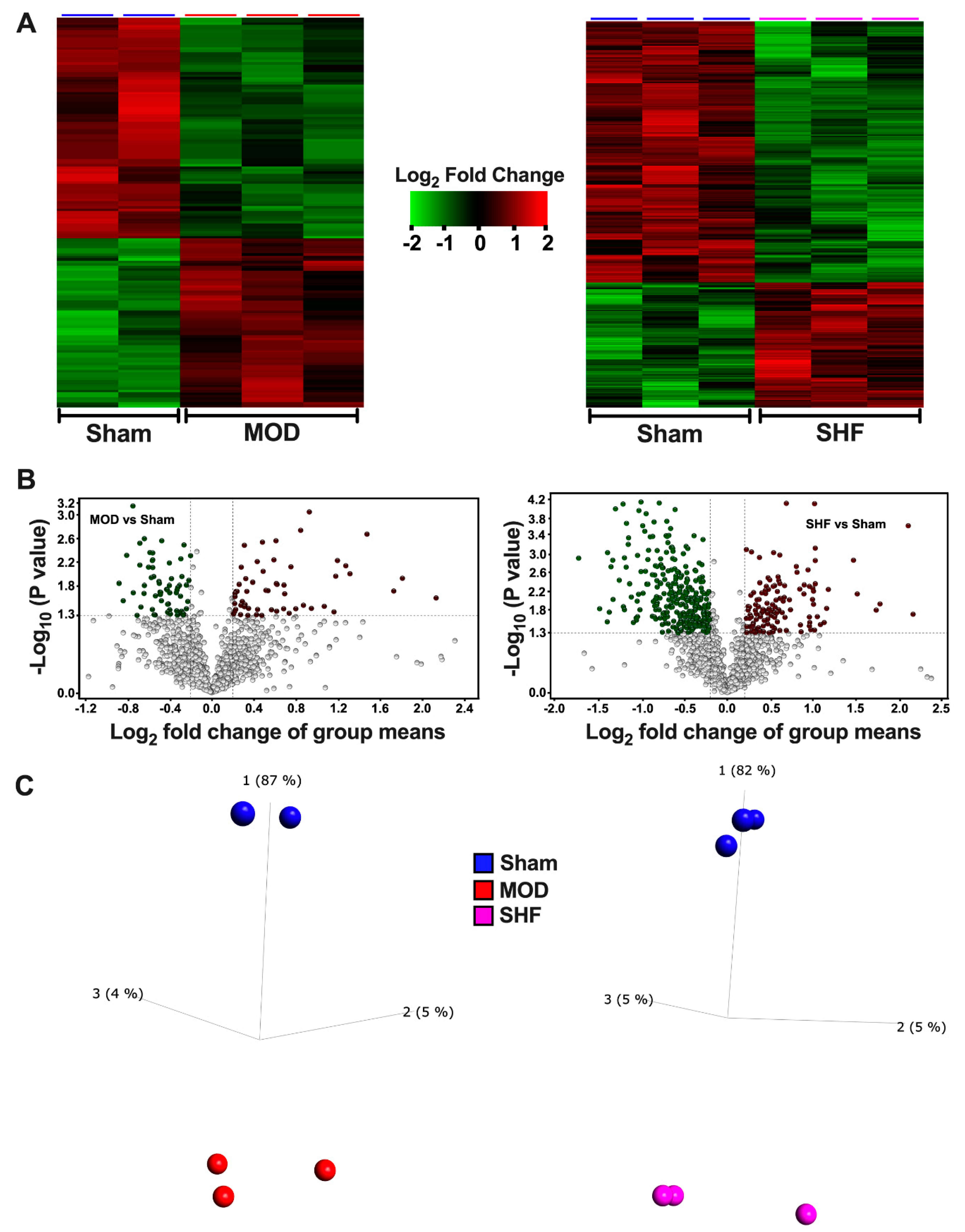
2.3. Visualization of the MOD and SHF p-Proteomic Datasets
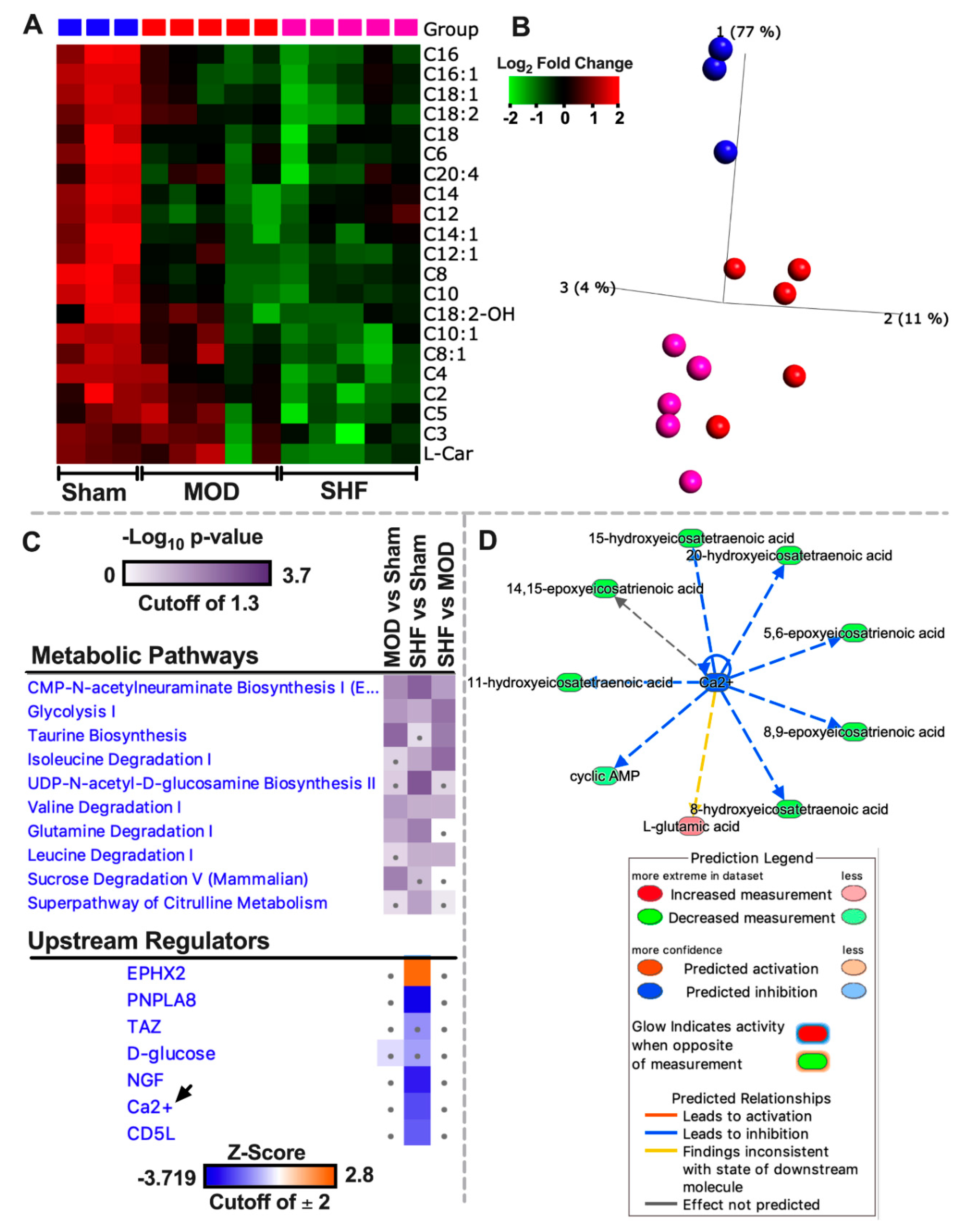
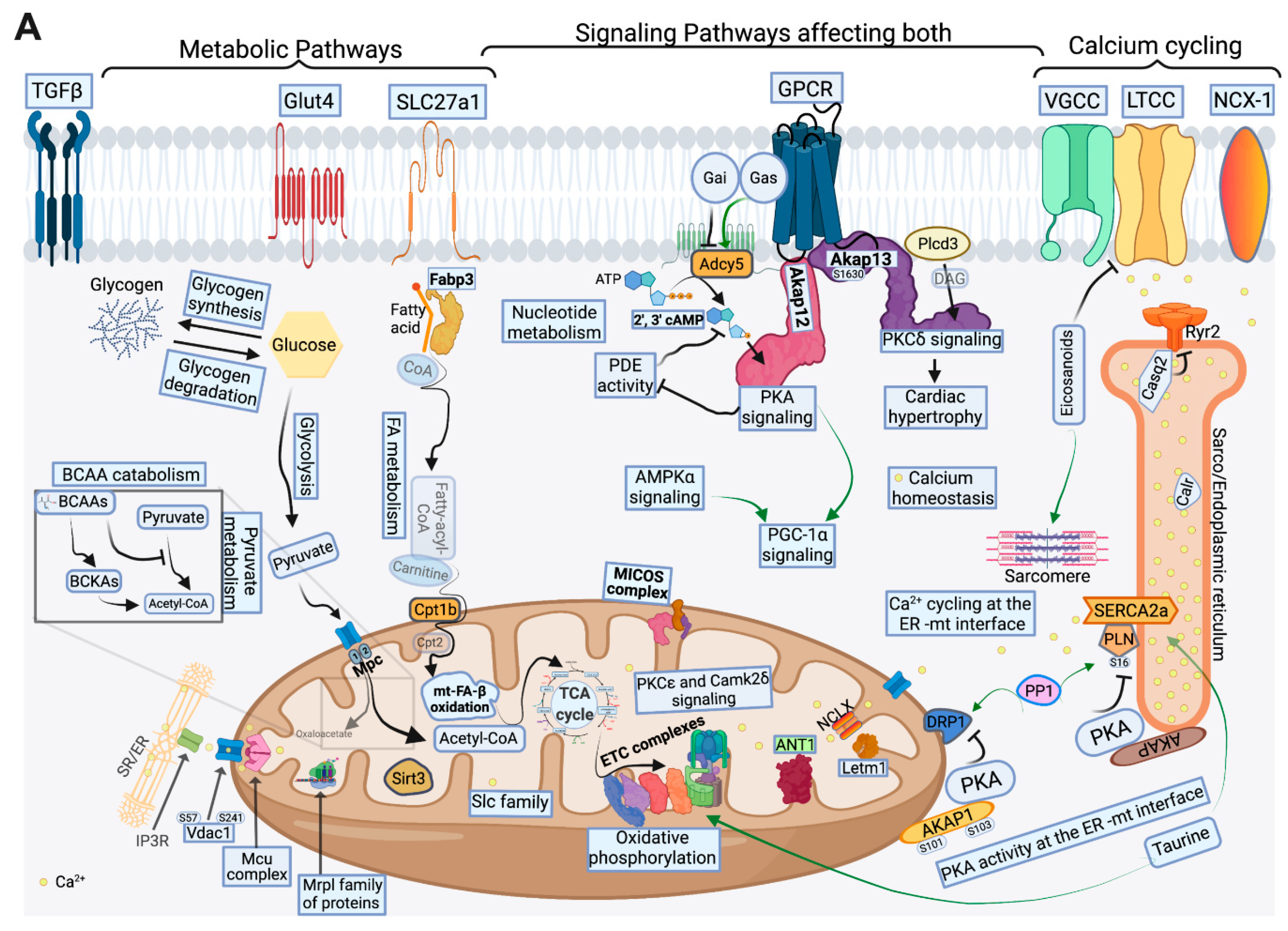
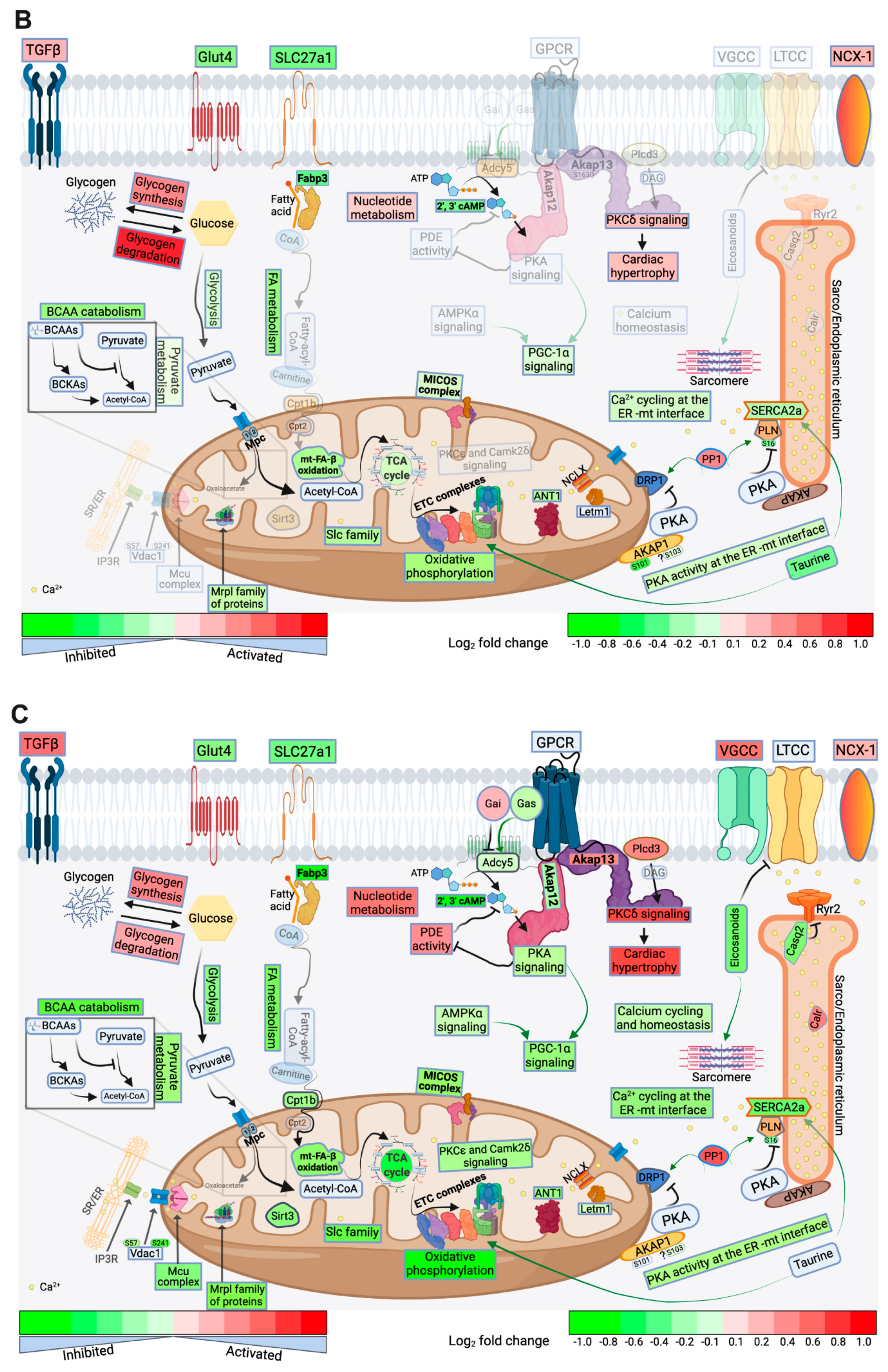
2.4. FA Metabolism and mt FA β-Oxidation and Triacylglycerol (TAG) Metabolism
2.5. Glucose, Pyruvate Metabolism, and BCAA Catabolism
2.6. AA, Ketone Body and Nucleotide Metabolism
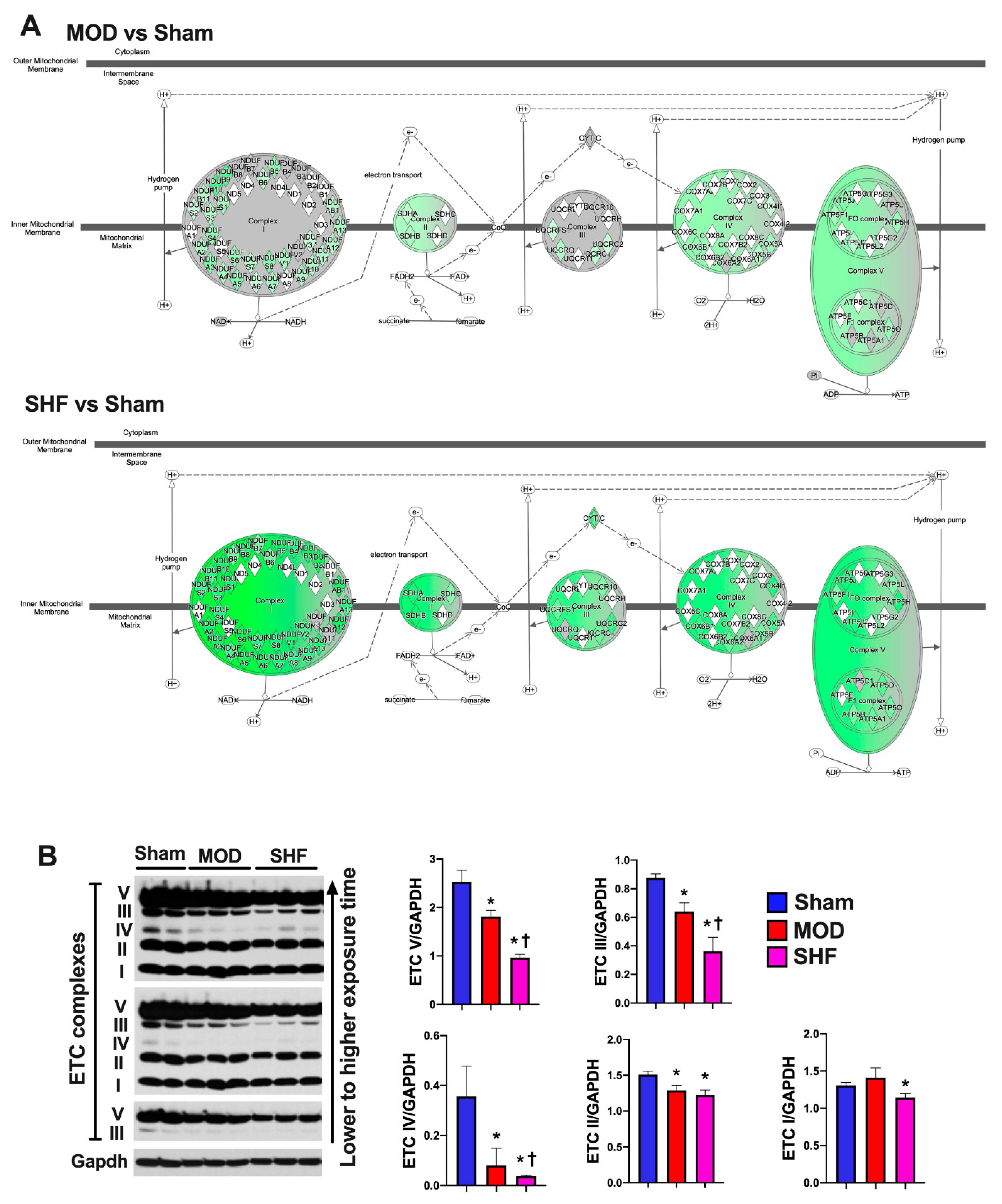
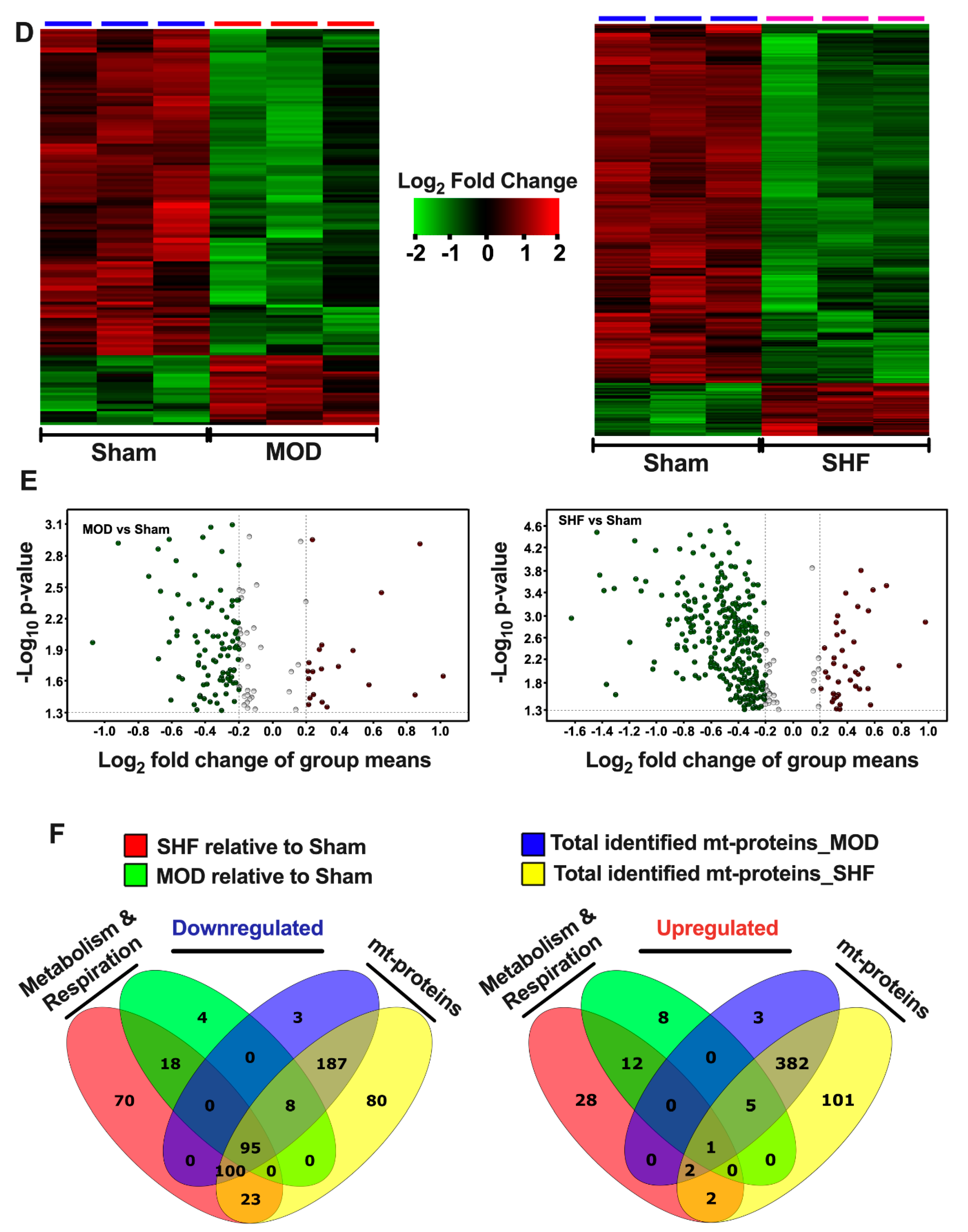
2.7. Mitochondrial Proteome and Oxidative Capacity
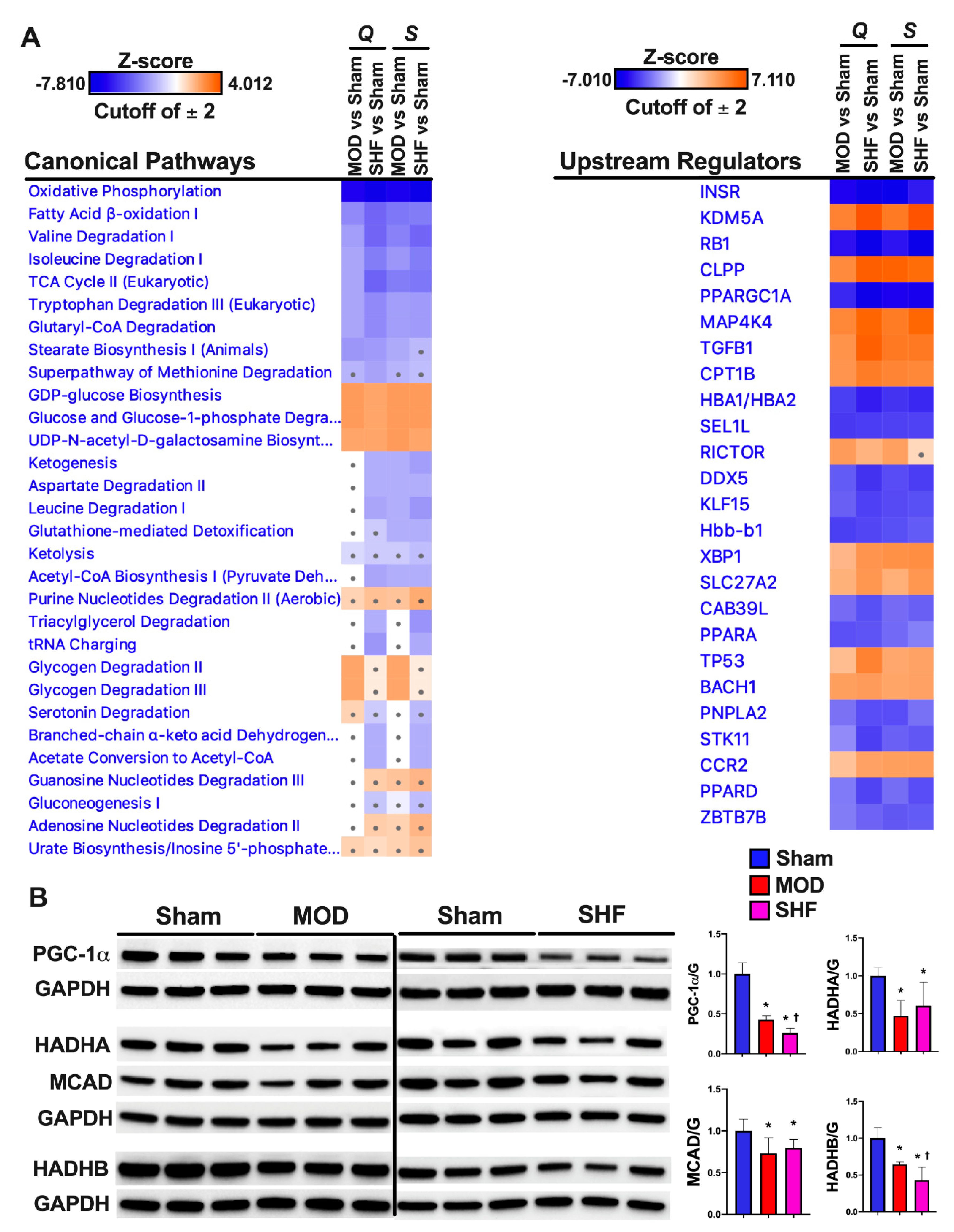
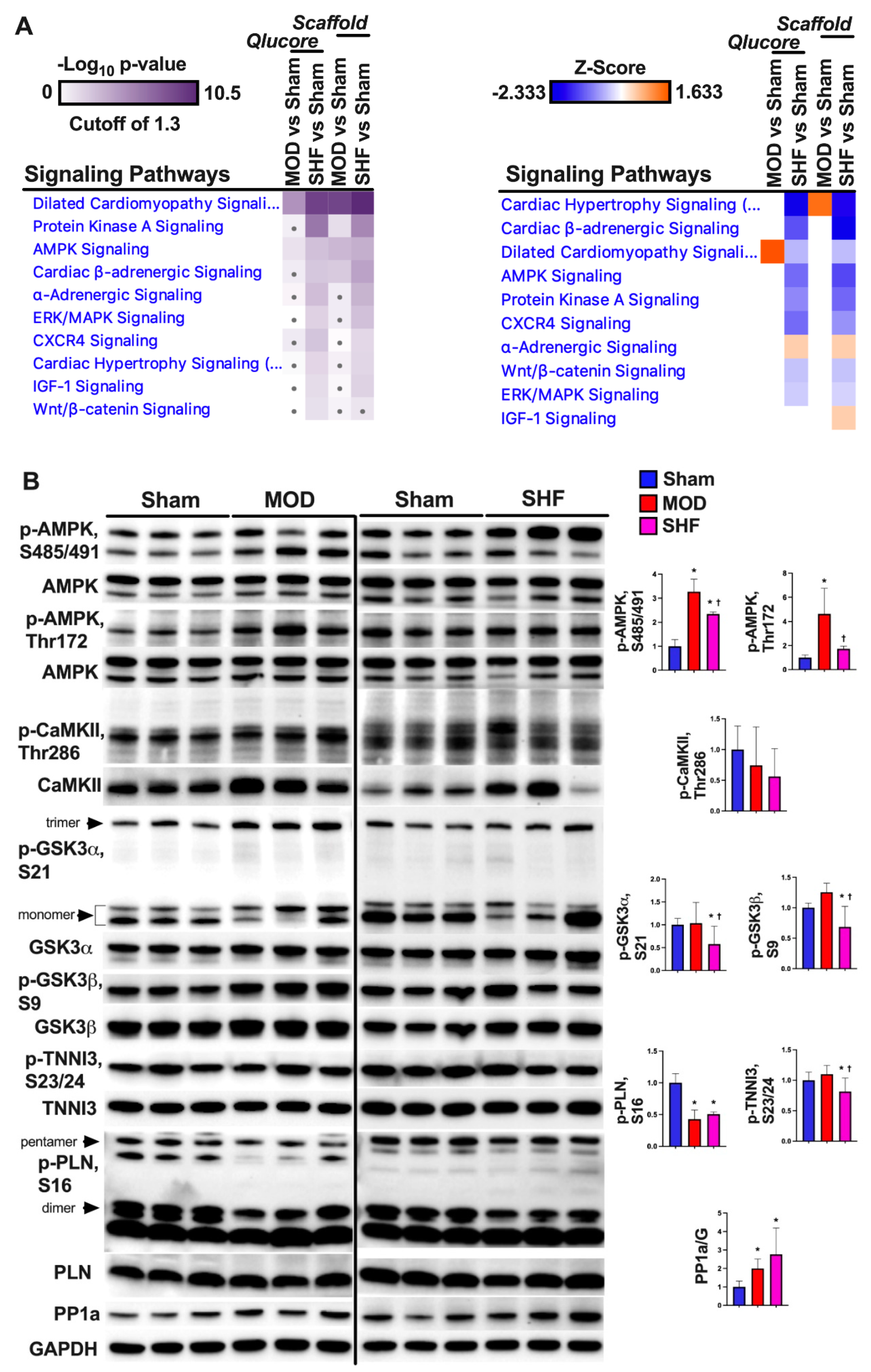
2.8. PKA and AMPK Signaling and their Implication in Metabolic Remodeling
2.9. Signaling Implicated in Mitochondrial Function other than PKA and AMPK
3. Discussion
4. Materials and Methods
4.1. Experimental Model of Ascending Aortic Banding for the Creation of the MOD and SHF Phenotypes
4.2. Discovery-Based Proteomic and p-Proteomic Analysis Using Tandem Mass Tags and Liquid Chromatography–Mass Spectrometry
4.3. Criteria for Protein Odentification
4.4. Protein Quantification in Scaffold
4.5. Bioinformatic Analyses
4.6. Criteria for Protein Identification
4.7. Protein Quantification in Scaffold
4.8. Phosphopeptide Quantification in Scaffold PTM
4.9. Bioinformatic Analyses
4.10. Validation by Immunoblotting
4.11. Discovery-Based Non-Targeted Central Carbon Metabolism and Acyl-Carnitine Analysis by LC–MS
4.11.1. Central Carbon Metabolism Data Identification and Normalization
4.11.2. Acyl Carnitine Data Identification
4.11.3. Statistical Analysis of the Metabolomics MS Raw Data
4.11.4. Bioinformatic Analyses
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W., II; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure—A Report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell Cardiol. 2013, 55, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Sreekumaran Nair, K.; Bergen, R.H., 3rd; Klaus, K.; Guenzel, A.J.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Integrity and Function in the Progression of Early Pressure Overload-Induced Left Ventricular Remodeling. J. Am. Heart Assoc. 2017, 6, e005869. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [Green Version]
- Kreusser, M.M.; Lehmann, L.H.; Keranov, S.; Hoting, M.O.; Oehl, U.; Kohlhaas, M.; Reil, J.C.; Neumann, K.; Schneider, M.D.; Hill, J.A.; et al. Cardiac CaM Kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 2014, 130, 1262–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tandan, S.; Hill, J.A. Calcineurin-dependent ion channel regulation in heart. Trends Cardiovasc. Med. 2014, 24, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdyga, A.; Surdo, N.C.; Monterisi, S.; Di Benedetto, G.; Grisan, F.; Penna, E.; Pellegrini, L.; Zaccolo, M.; Bortolozzi, M.; Swietach, P.; et al. Phosphatases control PKA-dependent functional microdomains at the outer mitochondrial membrane. Proc. Natl. Acad. Sci. USA 2018, 115, E6497–E6506. [Google Scholar] [CrossRef] [Green Version]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Luczak, E.D.; Wu, Y.; Granger, J.M.; Joiner, M.A.; Wilson, N.R.; Gupta, A.; Umapathi, P.; Murphy, K.R.; Reyes Gaido, O.E.; Sabet, A.; et al. Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nat. Commun. 2020, 11, 4416. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Kabra, D.G.; Aichler, M.; Schriever, S.C.; Pfuhlmann, K.; García, V.C.; Lehti, M.; Weber, J.; Kutschke, M.; Rozman, J.; et al. Calcineurin Links Mitochondrial Elongation with Energy Metabolism. Cell Metab. 2015, 22, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Morgan, H.E.; Parmeggiani, A. Regulation of glycogenolysis in muscle. II. control of glycogen phosphorylase reaction in isolated perfused heart. J. Biol. Chem. 1964, 239, 2435–2439. [Google Scholar] [CrossRef]
- Kumar, G.S.; Choy, M.S.; Koveal, D.M.; Lorinsky, M.K.; Lyons, S.P.; Kettenbach, A.N.; Page, R.; Peti, W. Identification of the substrate recruitment mechanism of the muscle glycogen protein phosphatase 1 holoenzyme. Sci. Adv. 2018, 4, eaau6044. [Google Scholar] [CrossRef] [Green Version]
- Joseph, B.K.; Liu, H.Y.; Francisco, J.; Pandya, D.; Donigan, M.; Gallo-Ebert, C.; Giordano, C.; Bata, A.; Nickels, J.T., Jr. Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d. J. Biol. Chem. 2015, 290, 10588–10598. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; Dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 8144–8149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 53, 465–479. [Google Scholar]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 2, e84897. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y. Branched chain amino acid metabolic reprogramming in heart failure. Biochim. Biophys. Acta 2016, 1862, 2270–2275. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Kolwicz, S.C., Jr.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective Branched-Chain Amino Acid Catabolism Disrupts Glucose Metabolism and Sensitizes the Heart to Ischemia-Reperfusion Injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Depre, C.; Vanoverschelde, J.L.; Taegtmeyer, H. Glucose for the heart. Circulation 1999, 99, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Nguyêñ, V.T.; Taegtmeyer, H. Feeding and fasting determine postischemic glucose utilization in isolated working rat hearts. Am. J. Physiol. 1991, 260 Pt 2, H542–H548. [Google Scholar] [CrossRef]
- Cross, H.R.; Opie, L.H.; Radda, G.K.; Clarke, K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ. Res. 1996, 78, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.L.; Proia, A.D.; Spencer-Manzon, M.J.; Butany, J.; Wechsler, S.B.; Kishnani, P.S. Cardiac Pathology in Glycogen Storage Disease Type III. JIMD Rep. 2012, 6, 65–72. [Google Scholar] [PubMed] [Green Version]
- Opie, L.H.; Knuuti, J. The adrenergic-fatty acid load in heart failure. J. Am. Coll. Cardiol. 2009, 54, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wende, A.R.; Abel, E.D. Lipotoxicity in the heart. Biochim. Biophys. Acta 2010, 1801, 311–319. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.M.; Fields, A.D.; Sorokina, N.; Lewandowski, E.D. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J. Mol. Cell Cardiol. 2008, 44, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Melenovsky, V.; Benes, J.; Skaroupkova, P.; Sedmera, D.; Strnad, H.; Kolar, M.; Vlcek, C.; Petrak, J.; Benes, J., Jr.; Papousek, F.; et al. Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol. Cell. Biochem. 2011, 354, 83–96. [Google Scholar] [CrossRef]
- Ito, T.; Kimura, Y.; Uozumi, Y.; Takai, M.; Muraoka, S.; Matsuda, T.; Ueki, K.; Yoshiyama, M.; Ikawa, M.; Okabe, M.; et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J. Mol. Cell Cardiol. 2008, 44, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Shimada-Takaura, K.; Jong, C.J.; Ito, T.; Takahashi, K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2016, 48, 549–558. [Google Scholar] [CrossRef]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism underlying the antioxidant activity of taurine: Prevention of mitochondrial oxidant production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Ramila, K.C.; Jong, C.J.; Pastukh, V.; Ito, T.; Azuma, J.; Schaffer, S.W. Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts. American journal of physiology. Heart Circ. Physiol. 2015, 308, H232–H239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, J.; Sawamura, A.; Awata, N. Usefulness of taurine in chronic congestive heart failure and its prospective application. Jpn. Circ. J. 1992, 56, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Beyranvand, M.R.; Khalafi, M.K.; Roshan, V.D.; Choobineh, S.; Parsa, S.A.; Piranfar, M.A. Effect of taurine supplementation on exercise capacity of patients with heart failure. J. Cardiol. 2011, 57, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2, ecosalplus.3.6.3.4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit-Jacques, J.; Hartzell, H.C. Effect of arachidonic acid on the L-type calcium current in frog cardiac myocytes. J. Physiol. 1996, 493 Pt 1, 67–81. [Google Scholar] [CrossRef]
- Damron, D.S.; Darvish, A.; Murphy, L.; Sweet, W.; Moravec, C.S.; Bond, M. Arachidonic acid-dependent phosphorylation of troponin I and myosin light chain 2 in cardiac myocytes. Circ. Res. 1995, 76, 1011–1019. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Leopold, J.A. Xanthine oxidase inhibition and heart failure: Novel therapeutic strategy for ventricular dysfunction? Circ. Res. 2006, 98, 169–171. [Google Scholar] [CrossRef] [Green Version]
- Minhas, K.M.; Saraiva, R.M.; Schuleri, K.H.; Lehrke, S.; Zheng, M.; Saliaris, A.P.; Berry, C.E.; Barouch, L.A.; Vandegaer, K.M.; Li, D.; et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ. Res. 2006, 98, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshwani, M.M.; Klammt, C.; von Daake, S.; Ma, Y.; Kornev, A.P.; Choe, S.; Insel, P.A.; Taylor, S.S. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2012, 109, E1221–E1229. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Barré, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, S.; Soltys, C.L.; Barr, A.J.; Shiojima, I.; Walsh, K.; Dyck, J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003, 278, 39422–39427. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884s–890s. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Haj Slimane, Z.; Bedioune, I.; Lechêne, P.; Varin, A.; Lefebvre, F.; Mateo, P.; Domergue-Dupont, V.; Dewenter, M.; Richter, W.; Conti, M.; et al. Control of cytoplasmic and nuclear protein kinase A by phosphodiesterases and phosphatases in cardiac myocytes. Cardiovasc. Res. 2014, 102, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Hinke, S.A.; Navedo, M.F.; Ulman, A.; Whiting, J.L.; Nygren, P.J.; Tian, G.; Jimenez-Caliani, A.J.; Langeberg, L.K.; Cirulli, V.; Tengholm, A.; et al. Anchored phosphatases modulate glucose homeostasis. EMBO J. 2012, 31, 3991–4004. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Le, A.V.; Tavalin, S.J.; Dodge-Kafka, K.L. Identification of AKAP79 as a protein phosphatase 1 catalytic binding protein. Biochemistry 2011, 50, 5279–5291. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, M.D.; Feliciello, A.; Jones, J.K.; Avvedimento, E.V.; Gottesman, M.E. PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. J. Mol. Biol. 2003, 327, 885–897. [Google Scholar] [CrossRef]
- Opalińska, M.; Meisinger, C. Mitochondrial protein import under kinase surveillance. Microb. Cell 2014, 1, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.; Harbauer, A.B.; Rao, S.; Eyrich, B.; Zahedi, R.P.; Stojanovski, D.; Schönfisch, B.; Guiard, B.; Sickmann, A.; Pfanner, N.; et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell 2011, 144, 227–239. [Google Scholar] [CrossRef]
- Carlucci, A.; Lignitto, L.; Feliciello, A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008, 18, 604–613. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation pattern of the NDUFS4 subunit of complex I of the mammalian respiratory chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell. Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucero, M.; Suarez, A.E.; Chambers, J.W. Phosphoregulation on mitochondria: Integration of cell and organelle responses. CNS Neurosci. Ther. 2019, 25, 837–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, R.; Zheng, Y.M.; Li, X.Q.; Wang, Q.S.; Liu, Q.H.; Ginnan, R.; Singer, H.A.; Ho, Y.S.; Wang, Y.X. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, A.; Kang, B.P.; Opawumi, D.; Belizaire, W.; Meggs, L.G. Molecular biology of protein kinase C signaling in cardiac myocytes. Mol. Cell. Biochem. 2001, 225, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Nowak, G.; Bakajsova, D.; Clifton, G.L. Protein kinase C-epsilon modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. American journal of physiology. Ren. Physiol. 2004, 286, F307–F316. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Hajjar, R.J. Characterization of the Differential Progression of Left Ventricular Remodeling in a Rat Model of Pressure Overload Induced Heart Failure. Does Clip Size Matter? Methods Mol. Biol. 2018, 1816, 195–206. [Google Scholar] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Shadforth, I.P.; Dunkley, T.P.; Lilley, K.S.; Bessant, C. i-Tracker: For quantitative proteomics using iTRAQ. BMC Genom. 2005, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Oberg, A.L.; Mahoney, D.W.; Eckel-Passow, J.E.; Malone, C.J.; Wolfinger, R.D.; Hill, E.G.; Cooper, L.T.; Onuma, O.K.; Spiro, C.; Therneau, T.M.; et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008, 7, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villén, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Schwartz, D.; Gygi, S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005, 23, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Thonusin, C.; IglayReger, H.B.; Soni, T.; Rothberg, A.E.; Burant, C.F.; Evans, C.R. Evaluation of intensity drift correction strategies using MetaboDrift, a normalization tool for multi-batch metabolomics data. J. Chromatogr. A 2017, 1523, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kachman, M.; Habra, H.; Duren, W.; Wigginton, J.; Sajjakulnukit, P.; Michailidis, G.; Burant, C.; Karnovsky, A. Deep annotation of untargeted LC-MS metabolomics data with Binner. Bioinformatics 2020, 36, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metab. Off. J. Metab. Soc. 2007, 3, 211–221. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaanine, A.H.; Higgins, L.; Markowski, T.; Harman, J.; Kachman, M.; Burant, C.; Navar, L.G.; Busija, D.; Delafontaine, P. Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure. Int. J. Mol. Sci. 2022, 23, 235. https://doi.org/10.3390/ijms23010235
Chaanine AH, Higgins L, Markowski T, Harman J, Kachman M, Burant C, Navar LG, Busija D, Delafontaine P. Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure. International Journal of Molecular Sciences. 2022; 23(1):235. https://doi.org/10.3390/ijms23010235
Chicago/Turabian StyleChaanine, Antoine H., LeeAnn Higgins, Todd Markowski, Jarrod Harman, Maureen Kachman, Charles Burant, L. Gabriel Navar, David Busija, and Patrice Delafontaine. 2022. "Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure" International Journal of Molecular Sciences 23, no. 1: 235. https://doi.org/10.3390/ijms23010235
APA StyleChaanine, A. H., Higgins, L., Markowski, T., Harman, J., Kachman, M., Burant, C., Navar, L. G., Busija, D., & Delafontaine, P. (2022). Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure. International Journal of Molecular Sciences, 23(1), 235. https://doi.org/10.3390/ijms23010235