
Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
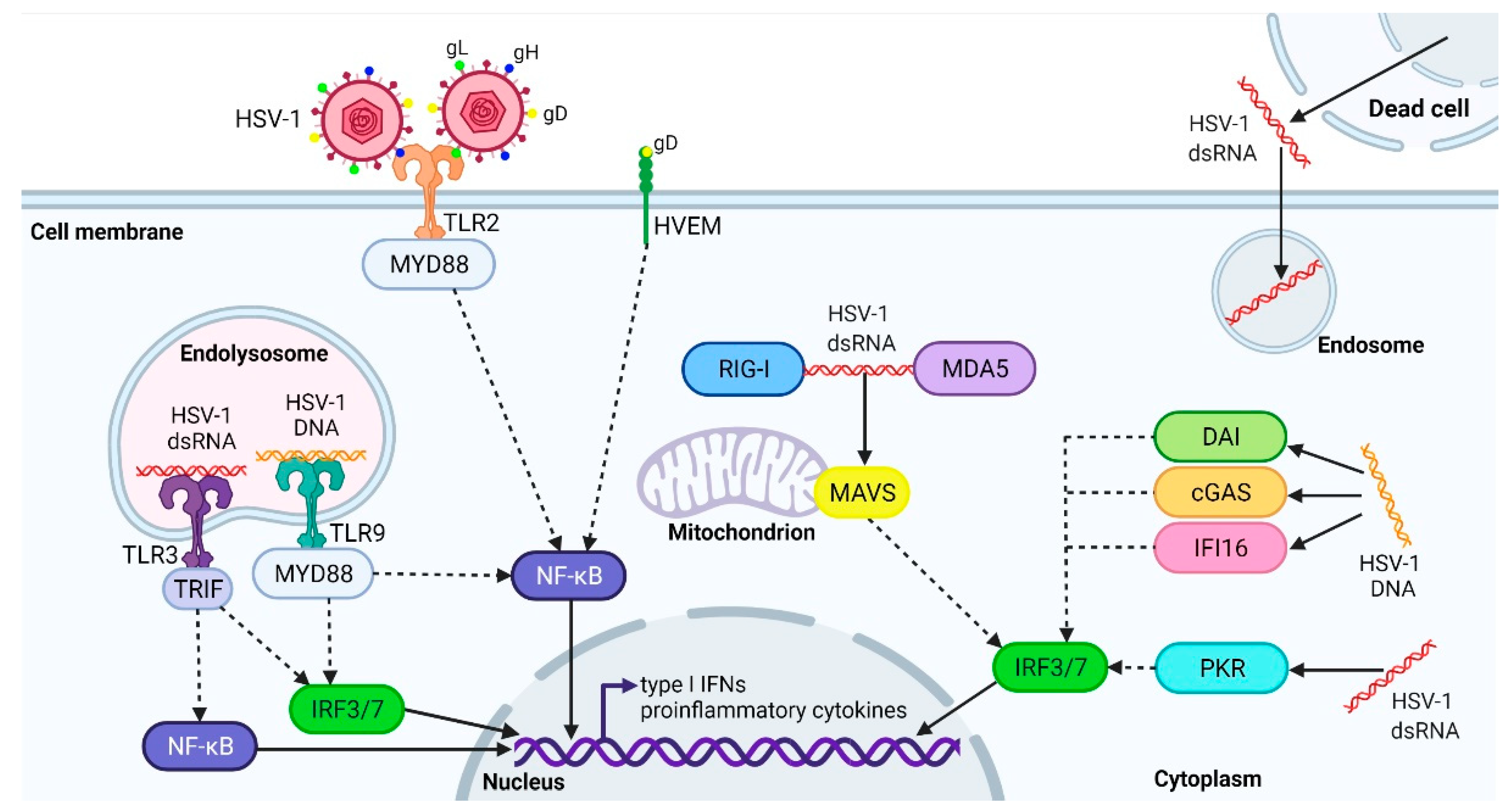
2. The Active and Latent HSV-1 Infection, and Antiviral Immune Response
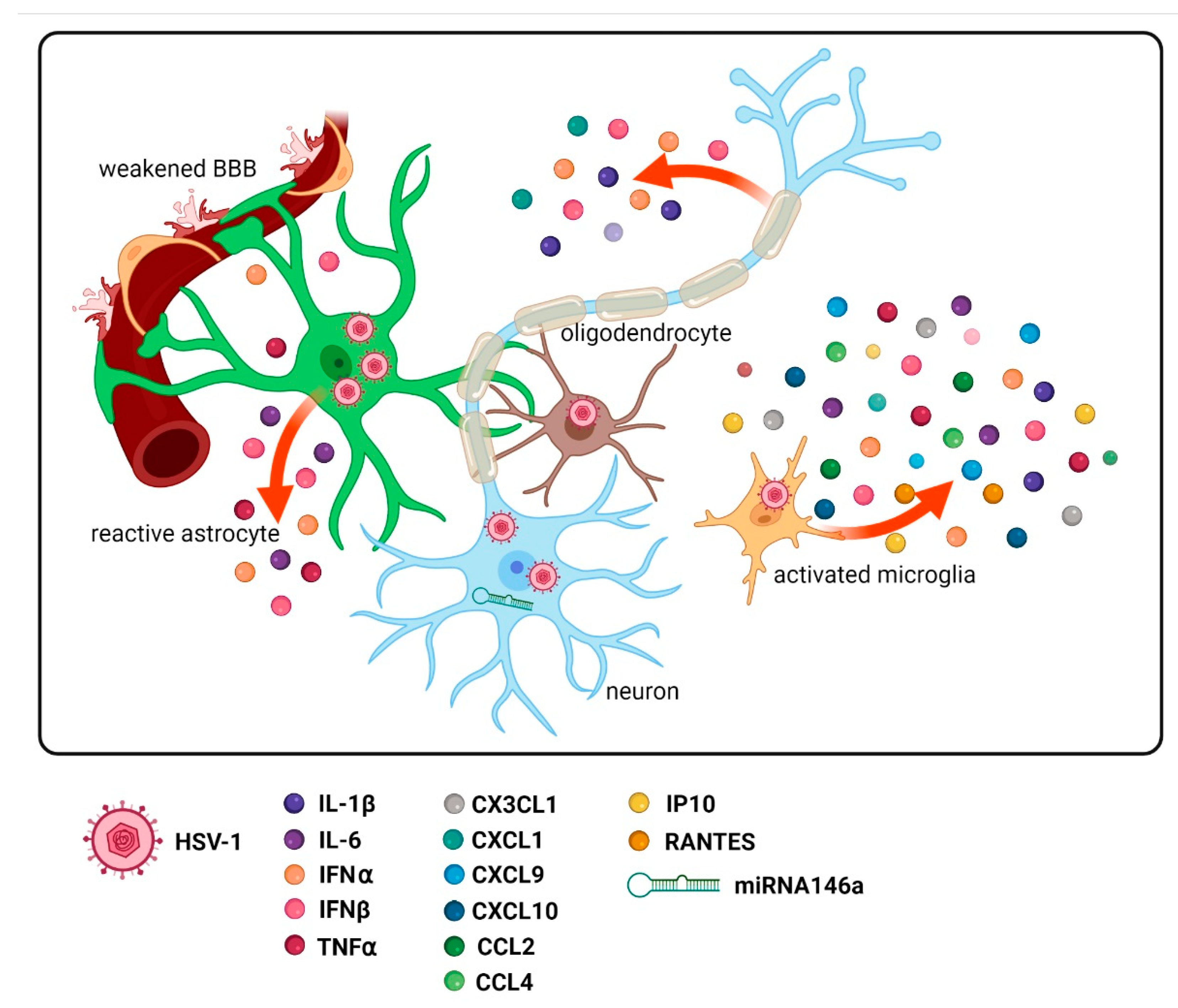
3. Amended Production of Various Inflammatory Factors
4. Functions of Astrocytes, Oligodendrocytes, Microglia, and Their Activation
4.1. Astrocytes
4.2. Microglia
4.3. Oligodendrocytes
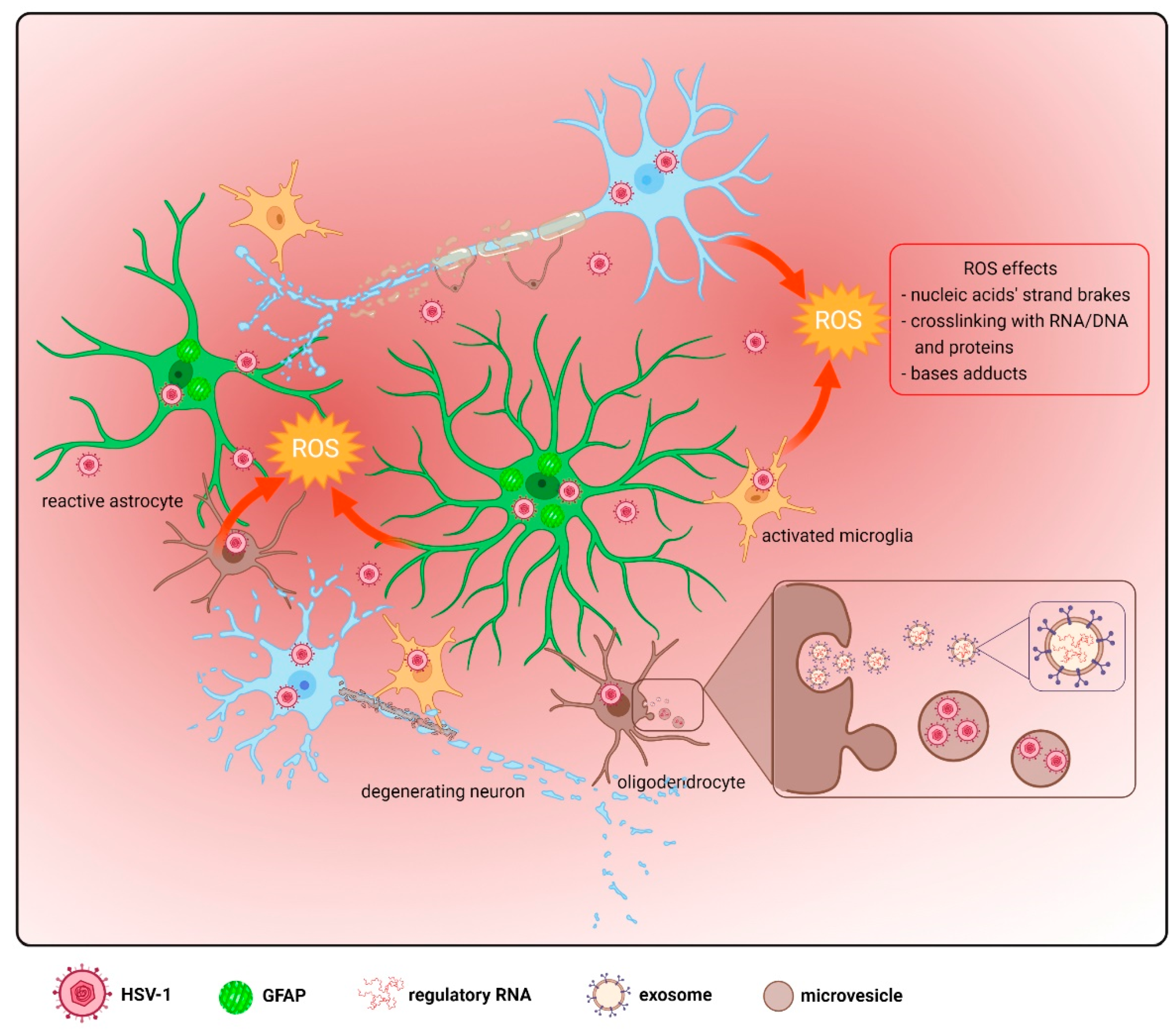
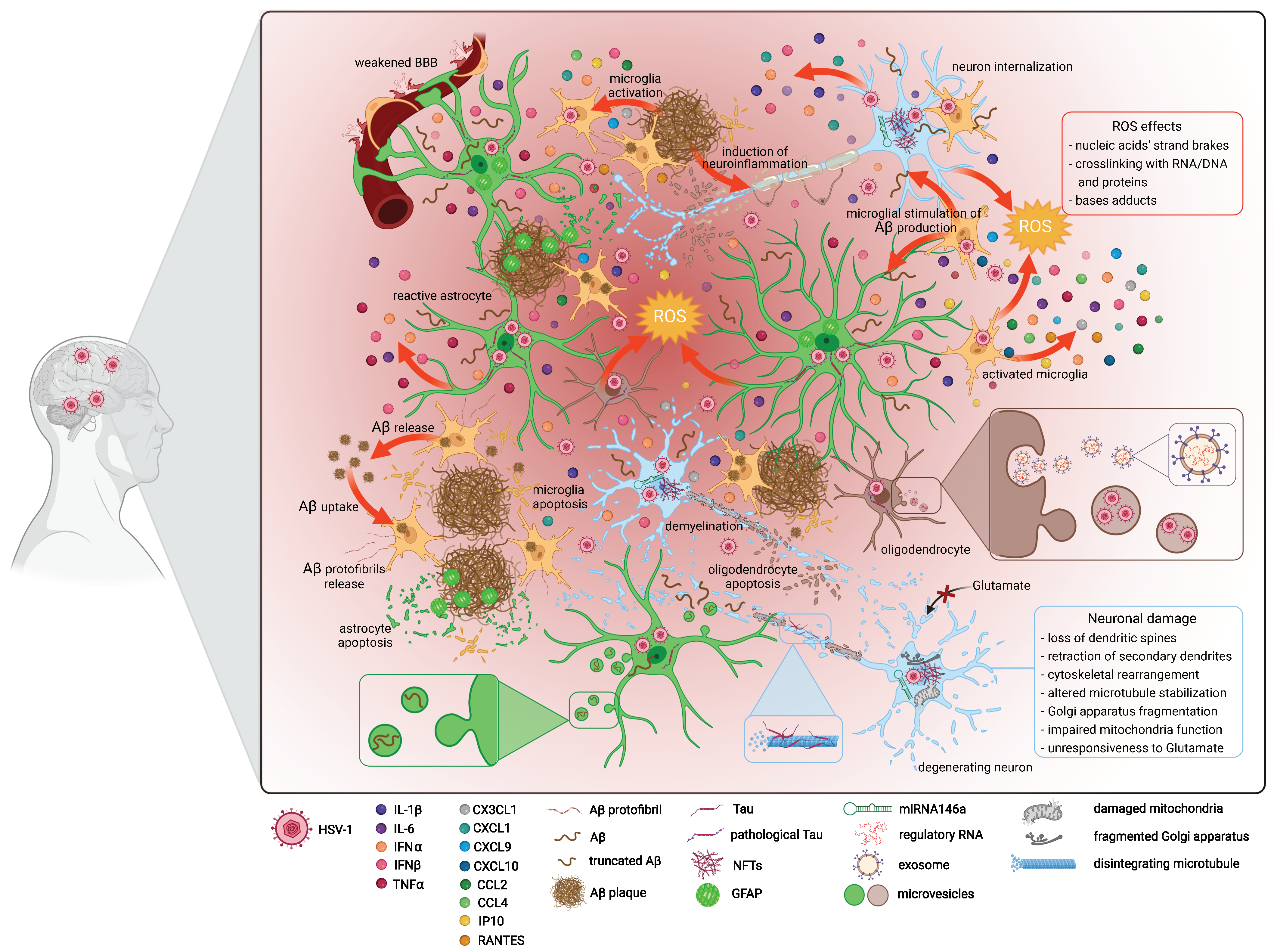
5. Oxidative Damage
6. Amyloid Beta Secretion
7. Tau Hyperphosphorylation
8. Apoptosis and Autophagy
9. Neuronal and Glial Cell Injury and Loss Combined with Advanced Age
10. Genetic Studies—Future Line of Investigation
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| APOE-ε4 | Apolipoprotein E ε4 |
| APP | Amyloid precursor protein |
| BACE1 | β-secretase |
| BBB | Blood–brain barrier |
| BECN1 | Beclin 1 |
| CCL | C–C motif chemokine ligand |
| cGAS | Cyclic guanosine monophosphate-adenosine monophosphate synthase |
| ChAc | Choline acetyltransferase |
| CNS | Central nervous system |
| CRMP | Collapsin response-mediated protein |
| CXCL | Chemokine (C–X–C motif) ligand |
| DAI | DNA-dependent activator of IFN-regulatory factor |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DRG | Dorsal root ganglia |
| ds | Double-stranded |
| FGFs | Fibroblast growth factors |
| g | Glycoprotein |
| GAD | Glutamine acid decarboxylase |
| GFAP | Glial fibrillary acidic protein |
| GRP78 | Glucose-regulated protein 78 |
| GSK-3β | Glycogen synthase kinase 3beta |
| HN | Human neural |
| HNE | 4-hydroxy trans-2-nonenal |
| HSE | Herpes simplex encephalitis |
| HSV-1 | Herpes simplex virus type 1 |
| HVEM | Herpes virus entry mediator |
| ICP0 | Infection cell protein 0 |
| IE | Immediate early |
| IFI16 | IFN-γ-inducible protein 16 |
| IFNs | Interferons |
| ILK | Integrin-linked kinase |
| IRF | Interferon regulatory factor |
| iNOS | Inducible nitric oxide synthase |
| IP-10 | IFN-γ-inducible protein 10 |
| ISGs | IFN-stimulated genes |
| LATs | Latency-associated transcripts |
| MAPs | Microtubule-associated proteins |
| MAVS | Mitochondrial antiviral-signaling protein |
| MDA5 | Melanoma differentiation-associated protein 5 |
| miRNAs | MicroRNAs |
| MMP | Matrix metalloprotease |
| MVs | Microvesicles |
| NF-κB | Nuclear factor kappa B |
| NFTs | Neurofibrillary tangles |
| NK | Natural killer |
| OBCs | Organotypic brain culture slices |
| OPCs | Oligodendrocyte progenitor cells |
| PAMPs | Pathogen-associated molecular patterns |
| PKA | Protein kinase A |
| PKR | Protein kinase R |
| PRRs | Pattern recognition receptors |
| PSD-95 | Postsynaptic density protein 95 |
| PUFA | Polyunsaturated fatty acid |
| RCS | Reactive carbonyl species |
| RIG-I | Retinoic acid-inducible gene I |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| sAβ | Soluble Aβ |
| snoRNAs | Small nucleolar RNAs |
| SOCS | Suppressor of cytokine signaling |
| STING | Stimulator of interferon gene |
| TG | Trigeminal ganglion |
| TH | Tyrosine hydroxylase |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor α |
| vhs | Virion host shutoff |
| 8-OHA | 8-hydroxyadenine |
| 8-OHG | 8-hydroxyguanine |
References
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Dosa, S.; Castellanos, K.; Bacsa, S.; Gagyi, E.; Kovacs, S.K.; Valyi-Nagy, K.; Shukla, D.; Dermody, T.S.; Valyi-Nagy, T. Chronic progressive deficits in neuron size, density, and number in the trigeminal ganglia of mice latently infected with herpes simplex virus. Brain Pathol. 2011, 21, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, E.M.N.; Rodrigues, L.D.; Oliveira, L.F.; Santos, J.C.C. dos Dementia and cognitive impairment in adults as sequels of HSV-1-related encephalitis: A review. Dement. Neuropsychol. 2021, 15, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tsao, J.W. Alzheimer Disease: REVUE; StatPearls Publishing: Treasure Island, CA, USA, 2018. [Google Scholar]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Wadhwa, P.; Jadhav, H. Reactive Astrogliosis: Role in Alzheimer’s Disease. CNS Neurol. Disord.-Drug Targets 2015, 14, 872–879. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Giovanni, G.D.; et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 2–28. [Google Scholar] [CrossRef] [Green Version]
- Ball, M.J. Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved? Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1982, 9, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Gannicliffe, A.; Sutton, P.R.N.; Itzhak, R.F. Viruses, brain and immunosuppression. Psychol. Med. 1986, 16, 247–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.J.; Mathews, R.; Steiner, I.; Hill, J.M.; Wisner, T.W.; Murdoch, G.H.; Kohama, S.; Sexton, G.; Nagalla, S. Latent HSV 1 virus in trigeminal ganglia: The optimal site for linking prevention of Alzheimer’s disease to vaccination. Neurobiol. Aging 2001, 22, 705–709. [Google Scholar] [CrossRef]
- Lewandowski, G.; Zimmerman, M.N.; Denk, L.L.; Porter, D.D.; Prince, G.A. Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch. Virol. 2002, 147, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Li Puma, D.D.; Piacentini, R.; Leone, L.; Gironi, K.; Marcocci, M.E.; De Chiara, G.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-β Protein Accumulation. Stem Cells 2019, 37, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.A.; Harris, E.A. Molecular Mechanisms for Herpes Simplex Virus Type 1 Pathogenesis in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes simplex virus type 1 infection of the central nervous system: Insights into proposed interrelationships with neurodegenerative disorders. Front. Cell. Neurosci. 2019, 13, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laval, K.; Enquist, L.W. The Potential Role of Herpes Simplex Virus Type 1 and Neuroinflammation in the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2021, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Overwhelming evidence for a major role for herpes simplex virus type 1 (Hsv1) in alzheimer’s disease (ad); underwhelming evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef] [PubMed]
- Hemmat, N.; Asadzadeh, H.; Asadzadeh, Z.; Shadbad, M.A.; Baradaran, B. The Analysis of Herpes Simplex Virus Type 1 (HSV-1)-Encoded MicroRNAs Targets: A Likely Relationship of Alzheimer’s Disease and HSV-1 Infection. Cell. Mol. Neurobiol. 2021. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lellouch-Tubiana, A.; Fohlen, M.; Robain, O.; Rozenberg, F. Immunocytochemical characterization of long-term persistent immune activation in human brain after herpes simplex encephalitis. Neuropathol. Appl. Neurobiol. 2000, 26, 285–294. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Wozniak, M.A. Could antivirals be used to treat Alzheimer’s disease? Future Microbiol. 2012, 7, 307–309. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.A.; Itzhaki, R.F. Antiviral agents in Alzheimer-s disease: Hope for the future? Ther. Adv. Neurol. Disord. 2010, 3, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F.; Lin, W.-R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.S.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Alzheimer Disease Overview; GeneReviews®: Bethesda, MD, USA, 2018. [Google Scholar]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, D.; Liang, C. Autophagy interaction with herpes simplex virus type-1 infection. Autophagy 2016, 12, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Stoeger, T.; Adler, H. “Novel” triggers of herpesvirus reactivation and their potential health relevance. Front. Microbiol. 2019, 10, 3207. [Google Scholar] [CrossRef]
- Knezevic, A.; Martic, J.; Stanojevic, M.; Jankovic, S.; Nedeljkovic, J.; Nikolic, L.; Pasic, S.; Jankovic, B.; Jovanovic, T. Disseminated Neonatal Herpes Caused by Herpes Simplex Virus Types 1 and 2. Emerg. Infect. Dis. 2007, 13, 302. [Google Scholar] [CrossRef] [PubMed]
- Saleh, D.; Yarrarapu, S.N.S.; Sharma, S. Herpes Simplex Type 1; StatPearls Publishing: Treasure Island, CA, USA, 2021. [Google Scholar]
- Rajasagi, N.K.; Rouse, B.T. Application of our understanding of pathogenesis of herpetic stromal keratitis for novel therapy. Microbes Infect. 2018, 20, 526–530. [Google Scholar] [CrossRef]
- Niemialtowski, M.G.; Rouse, B.T. Phenotypic and functional studies on ocular T cells during herpetic infections of the eye. J. Immunol. 1992, 148, 1864–1870. [Google Scholar] [PubMed]
- Zhang, J.; Liu, H.; Wei, B. Immune response of T cells during herpes simplex virus type 1 (HSV-1) infection. J. Zhejiang Univ. Sci. B 2017, 18, 277–288. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Orzalli, M.H.; Knipe, D.M. Innate Immune Mechanisms and Herpes Simplex Virus Infection and Disease. Adv. Anat. Embryol. Cell Biol. 2017, 223, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2020, 11, 613799. [Google Scholar] [CrossRef] [PubMed]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res. 2020, 179, 104811. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and Adaptive Immune Responses to Herpes Simplex Virus. Viruses 2009, 1, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. Fems Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef]
- Stevens, J.G.; Haarr, L.; Porter, D.D.; Cook, M.L.; Wagner, E.K. Prominence of the herpes simplex virus latency-associated transcript in trigeminal ganglia from seropositive humans. J. Infect. Dis. 1988, 158, 117–123. [Google Scholar] [CrossRef]
- Brdovčak, M.C.; Zubković, A.; Jurak, I. Herpes Simplex Virus 1 Deregulation of Host MicroRNAs. Non-Coding RNA 2018, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.F.; Coen, D.M. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1995, 69, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The Latency-Associated Transcript Inhibits Apoptosis via Downregulation of Components of the Type I Interferon Pathway during Latent Herpes Simplex Virus 1 Ocular Infection. J. Virol. 2019, 93, e00103-19. [Google Scholar] [CrossRef] [Green Version]
- Theil, D.; Derfuss, T.; Paripovic, I.; Herberger, S.; Meinl, E.; Schueler, O.; Strupp, M.; Arbusow, V.; Brandt, T. Latent Herpesvirus Infection in Human Trigeminal Ganglia Causes Chronic Immune Response. Am. J. Pathol. 2003, 163, 2179–2184. [Google Scholar] [CrossRef] [Green Version]
- Valyi-Nagy, T.; Olson, S.J.; Valyi-Nagy, K.; Montine, T.J.; Dermody, T.S. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology 2000, 278, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Fields virology-NLM Catalog-NCBI. Available online: https://www.ncbi.nlm.nih.gov/nlmcatalog/101601028 (accessed on 5 November 2021).
- Braun, E.; Zimmerman, T.; Hur, T.B.; Reinhartz, E.; Fellig, Y.; Panet, A.; Steiner, I. Neurotropism of herpes simplex virus type 1 in brain organ cultures. J. Gen. Virol. 2006, 87, 2827–2837. [Google Scholar] [CrossRef]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cymerys, J.; Kowalczyk, A.; Mikołajewicz, K.; Słońska, A.; Krzyżowska, M. Nitric Oxide Influences HSV-1-Induced Neuroinflammation. Oxid. Med. Cell. Longev. 2019, 2019, 2302835. [Google Scholar] [CrossRef]
- Villalba, M.; Hott, M.; Martin, C.; Aguila, B.; Valdivia, S.; Quezada, C.; Zambrano, Á.; Concha, M.I.; Otth, C. Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med. Microbiol. Immunol. 2012, 201, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galán-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; López-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, A.M.; Truman, A.W.; Marriott, I. The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells. J. Neurovirol. 2020, 26, 544–555. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Vahlne, A.; Nystrom, B.; Sandberg, M.; Hamberger, A.; Lycke, E. Attachment of herpes simplex virus to neurons and glial cells. J. Gen. Virol. 1978, 40, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Vahlne, A.; Svennerholm, B.; Sandberg, M.; Hamberger, A.; Lycke, E. Differences in attachment between herpes simplex type 1 and type 2 viruses to neurons and glial cells. Infect. Immun. 1980, 28, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Gekker, G.; Lokensgard, J.R. Differential apoptotic signaling in primary glial cells infected with herpes simplex virus 1. J. Neurovirol. 2006, 12, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Raker, V.; Förthmann, B.; Detering, N.T.; Kubinski, S.; Buch, A.; Katzilieris-Petras, G.; Spanier, J.; Gudi, V.; Wagenknecht, S.; et al. HSV-1 triggers paracrine fibroblast growth factor response from cortical brain cells via immediate-early protein ICP0. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Puma, D.D.; Marcocci, M.E.; Lazzarino, G.; De Chiara, G.; Tavazzi, B.; Palamara, A.T.; Piacentini, R.; Grassi, C. Ca2+-dependent release of ATP from astrocytes affects herpes simplex virus type 1 infection of neurons. Glia 2021, 69, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Charpin, C.; Gambarelli, D.; Lavaut, M.N.; Seigneurin, J.M.; Raphael, M.; Berard, M.; Toga, M. Herpes simplex virus antigen detection in human acute encephalitis:-An immunohistochemical study using avidin-biotin-peroxidase complex method. Acta Neuropathol. 1985, 68, 245–252. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.S.; Kim, S.U. Multifocal CNS demyelination following peripheral inoculation with herpes simplex virus type 1. Ann. Neurol. 1987, 22, 52–59. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.S.; Kim, S.U. Herpes Simplex Virus Type 1 Induced Multifocal Demyelination of the Central Nervous System in Mice. Ann. N. Y. Acad. Sci. 1988, 540, 654–656. [Google Scholar] [CrossRef]
- Sköldenberg, B. Herpes simplex encephalitis. Scand. J. Infect. Dis. Suppl. 1996, 100, 8–13. [Google Scholar]
- Bello-Morales, R.; Fedetz, M.; Alcina, A.; Tabarés, E.; López-Guerrero, J.A. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J. NeuroVirology 2005, 11, 190–198. [Google Scholar] [CrossRef]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [Green Version]
- Denver, P.; McClean, P.L. Distinguishing normal brain aging from the development of Alzheimer’s disease: Inflammation, insulin signaling and cognition. Neural Regen. Res. 2018, 13, 1719. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Goshima, F.; Ito, H.; Koide, N.; Yoshida, T.; Yokochi, T.; Kimura, Y.; Nishiyama, Y. The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virology 2005, 334, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangold, C.A.; Szpara, M.L. Persistent infection with herpes simplex virus 1 and Alzheimer’s disease-a call to study how variability in both virus and host may impact disease. Viruses 2019, 11, 966. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C. Inflammaging as a Major Characteristic of Old People: Can It Be Prevented or Cured? Nutr. Rev. 2007, 65, S173–S176. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508. [Google Scholar] [CrossRef]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, M.; Bergmann, C.C. Alpha/Beta Interferon (IFN-α/β) Signaling in Astrocytes Mediates Protection against Viral Encephalomyelitis and Regulates IFN-γ-Dependent Responses. J. Virol. 2018, 92, e01901-17. [Google Scholar] [CrossRef] [Green Version]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef]
- Apelt, J.; Schliebs, R. β-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001, 894, 21–30. [Google Scholar] [CrossRef]
- Abbas, N.; Bednar, I.; Mix, E.; Marie, S.; Paterson, D.; Ljungberg, A.; Morris, C.; Winblad, B.; Nordberg, A.; Zhu, J. Up-regulation of the inflammatory cytokines IFN-γ and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APPSWE transgenic mice. J. Neuroimmunol. 2002, 126, 50–57. [Google Scholar] [CrossRef]
- Minami, M.; Kita, M.; Yan, X.Q.; Yamamoto, T.; Iida, T.; Sekikawa, K.; Iwakura, Y.; Imanishi, J. Role of IFN-γ and tumor necrosis factor-α in herpes simplex virus type 1 infection. J. Interf. Cytokine Res. 2002, 22, 671–676. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Insights into the pathogenesis of herpes simplex encephalitis from mouse models. Mamm. Genome 2018, 29, 425–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Liu, H.; Huang, C.; Wang, R.; Luo, M.; Lu, W. Herpes Simplex Virus 1-Induced Blood-Brain Barrier Damage Involves Apoptosis Associated with GM130-Mediated Golgi Stress. Front. Mol. Neurosci. 2020, 13, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furr, S.R.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Marriott, I. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflamm. 2011, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.J.; Beltran-Lobo, P.; Johnson, L.; Perez-Nievas, B.G.; Noble, W. Astrocytes in Tauopathies. Front. Neurol. 2020, 11, 572850. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Hu, S.; Sheng, W.; VanOijen, M.; Cox, D.; Cheeran, M.C.J.; Peterson, P.K. Robust expression of TNF-α, IL-1β, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 2001, 7, 208–219. [Google Scholar] [CrossRef]
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell Rep. 2020, 32, 108150. [Google Scholar] [CrossRef]
- Rosato, P.C.; Katzenell, S.; Pesola, J.M.; North, B.; Coen, D.M.; Leib, D.A. Neuronal IFN signaling is dispensable for the establishment of HSV-1 latency. Virology 2016, 497, 323–327. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500. [Google Scholar] [CrossRef] [Green Version]
- Leonoudakis, D.; Rane, A.; Angeli, S.; Lithgow, G.J.; Andersen, J.K.; Chinta, S.J. Anti-Inflammatory and Neuroprotective Role of Natural Product Securinine in Activated Glial Cells: Implications for Parkinson’s Disease. Mediators Inflamm. 2017, 2017, 8302636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease-A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The Herpes Simplex Virus ICP0 RING Finger Domain Inhibits IRF3- and IRF7-Mediated Activation of Interferon-Stimulated Genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhvalova, M.S.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes Simplex Virus 1 Induces Brain Inflammation and Multifocal Demyelination in the Cotton Rat Sigmodon hispidus. J. Virol. 2020, 94, e01161-19. [Google Scholar] [CrossRef] [Green Version]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions. Front. Cell. Neurosci. 2020, 14, 79. [Google Scholar] [CrossRef] [Green Version]
- Papuć, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345. [Google Scholar] [CrossRef]
- AK, A.K.; Mendez, M.D. Herpes Simplex Encephalitis; StatPearls Publishing: Treasure Island, CA, USA, 2021. [Google Scholar]
- Klapper, P.E.; Cleator, G.M.; Longson, M. Mild forms of herpes encephalitis. J. Neurol. Neurosurg. Psychiatry 1984, 47, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.; Lovheim, H.; Honkala, E.; Karhunen, P.J.; Elgh, F.; Kok, E.H. HSV presence in brains of individuals without dementia: The TASTY brain series. DMM Dis. Model. Mech. 2016, 9, 1349–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.-B.E.; Vestergaard, H.T.; Dessau, R.B.; Bodilsen, J.; Andersen, N.S.; Omland, L.H.; Christiansen, C.B.; Ellermann-Eriksen, S.; Nielsen, L.; Benfield, T.; et al. Long-Term Survival, Morbidity, Social Functioning and Risk of Disability in Patients with a Herpes Simplex Virus Type 1 or Type 2 Central Nervous System Infection, Denmark, 2000–2016. Clin. Epidemiol. 2020, 12, 745. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.; Kimberlin, D.W.; Prober, C.G. Pathogenesis and disease. Rhizoctonia Solani, Biol. Pathol. 2007, 15, 161–171. [Google Scholar]
- Yang, Q.; Zhou, J. wei Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J. Astrocytes: News about brain health and diseases. Biomedicines 2020, 8, 394. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Gumenyuk, A.V.; Tykhomyrov, A.A.; Savosko, S.I.; Guzyk, M.M.; Rybalko, S.L.; Ryzha, A.O.; Chaikovsky, Y.B. State of Astrocytes in the Mice Brain under Conditions of Herpes Viral Infection and Modeled Stroke. Neurophysiol. 2019, 50, 326–331. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive Oxygen Species from Human Astrocytes Induced Functional Impairment and Oxidative Damage. Neurochem. Res. 2013, 38, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Tretyakova, N.Y.; Groehler, A.; Ji, S. DNA-Protein Cross-links: Formation, Structural Identities, and Biological Outcomes. Acc. Chem. Res. 2015, 48, 1631–1644. [Google Scholar] [CrossRef] [Green Version]
- Hwa Yun, B.; Guo, J.; Bellamri, M.; Turesky, R.J. DNA Adducts: Formation, biological effects, and new biospecimens for mass spectrometric measurements in humans. Mass Spectrom. Rev. 2020, 39, 55–82. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, G.K.; Fu, M.M.; Docherty, J.J. Innate and adaptive host response during the initial phase of herpes simplex virus encephalitis in the neonatal mouse. J. Neurovirol. 2006, 12, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Bello-Morales, R.; Andreu, S.; López-Guerrero, J.A. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1–19. [Google Scholar] [CrossRef]
- Armien, A.G.; Hu, S.; Little, M.R.; Robinson, N.; Lokensgard, J.R.; Low, W.C.; Cheeran, M.C. Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol. 2010, 20, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Wnęk, M.; Ressel, L.; Ricci, E.; Rodriguez-Martinez, C.; Guerrero, J.C.V.; Ismail, Z.; Smith, C.; Kipar, A.; Sodeik, B.; Chinnery, P.F.; et al. Herpes simplex encephalitis is linked with selective mitochondrial damage; a post-mortem and in vitro study. Acta Neuropathol. 2016, 132, 433–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Guan, Y.; Sun, X.; Shi, L.; Liang, R.; Lv, X.; Xin, W. HSV-1 activates NF-kappaB in mouse astrocytes and increases TNF-alpha and IL-6 expression via Toll-like receptor 3. Neurol. Res. 2013, 35, 755–762. [Google Scholar] [CrossRef]
- Reinert, L.S.; Harder, L.; Holm, C.K.; Iversen, M.B.; Horan, K.A.; Dagnæs-Hansen, F.; Ulhøi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J. Clin. Investig. 2012, 122, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Bansode, Y.D.; Chattopadhyay, D.; Saha, B. Innate immune response in astrocytes infected with herpes simplex virus 1. Arch. Virol. 2019, 164, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Tykhomyrov, A.A.; Pavlova, A.S.; Nedzvetsky, V.S. Glial Fibrillary Acidic Protein (GFAP): On the 45th Anniversary of Its Discovery. Neurophysiology 2016, 48, 54–71. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yuan, Y.; Chen, J.; Zhu, Y.; Qiu, Y.; Zhu, F.; Huang, A.; He, C. Reactive astrocytes inhibit the survival and differentiation of oligodendrocyte precursor cells by secreted TNF-α. J. Neurotrauma 2011, 28, 1089–1100. [Google Scholar] [CrossRef]
- Konat, G.W.; Kielian, T.; Marriott, I. The role of Toll-like receptors in CNS response to microbial challenge. J. Neurochem. 2006, 99, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, P.A.; Duncan, D.S.; Miller, S.D. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain. Behav. Immun. 2008, 22, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Falsig, J.; Van Beek, J.; Hermann, C.; Leist, M. Molecular basis for detection of invading pathogens in the brain. J. Neurosci. Res. 2008, 86, 1434–1447. [Google Scholar] [CrossRef]
- Lannes, N.; Eppler, E.; Etemad, S.; Yotovski, P.; Filgueira, L. Microglia at center stage: A comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget 2017, 8, 114393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Urban, S.L.; Lokensgard, J.R. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J. Immunol. 2008, 181, 6417–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Lokensgard, J.R. Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis. J. Neurovirol. 2008, 14, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, O.; Pandit, N.P.; Molesworth, K.; Fisher, A.; Mychko, O.; Makarava, N.; Baskakov, I.V. Alzheimer’s disease-associated β-amyloid does not protect against herpes simplex virus 1 infection in the mouse brain. J. Biol. Chem. 2021, 297, 100845. [Google Scholar] [CrossRef]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; De La Rocha, I.C.; Rivera, A. Oligodendroglial cells in alzheimer’s disease. Adv. Exp. Med. Biol. 2019, 1175, 325–333. [Google Scholar]
- McKenzie, A.T.; Moyon, S.; Wang, M.; Katsyv, I.; Song, W.-M.; Zhou, X.; Dammer, E.B.; Duong, D.M.; Aaker, J.; Zhao, Y.; et al. Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-X.; Zhang, H.-Y.; Li, H.-Y.; Liu, P.-H.; Sui, Y.; Sun, X.-H. Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction. Neural Regen. Res. 2018, 13, 908. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiao, M. Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 2016, 126, 97–104. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early Oligodendrocyte/Myelin Pathology in Alzheimer’s Disease Mice Constitutes a Novel Therapeutic Target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef]
- Beckhauser, T.F.; Francis-Oliveira, J.; Pasquale, R. De Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23. [Google Scholar] [CrossRef]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paternò, R.; Avvedimento, E.V.; Santillo, M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front. Cell. Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, Y.; Takemoto, T.; Itoh, K.; Ishida, A.; Yamazaki, T. Dual Role of Superoxide Dismutase 2 Induced in Activated Microglia: Oxidative stress tolerance and convergence of inflammatory responses*. J. Biol. Chem. 2015, 290, 22805–22817. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.D.; Núñez, M.T. Oligodendrocytes: Functioning in a delicate balance between high metabolic requirements and oxidative damage. Adv. Exp. Med. Biol. 2016, 949, 167–181. [Google Scholar] [CrossRef]
- Valyi-Nagy, T.; Dermody, T.S. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol. Histopathol. 2005, 20, 957–967. [Google Scholar] [PubMed]
- Marino-Merlo, F.; Papaianni, E.; Frezza, C.; Pedatella, S.; Nisco, M.; Macchi, B.; Grelli, S.; Mastino, A. NF-κB-Dependent Production of ROS and Restriction of HSV-1 Infection in U937 Monocytic Cells. Viruses 2019, 11, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiano, M.; Chastel, O.; Thoisy, B.; Eens, M.; Costantini, D. Oxidative stress favours herpes virus infection in vertebrates: Ameta-analysis. Curr. Zool. 2016, 62, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavouras, J.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 2007, 13, 416–425. [Google Scholar] [CrossRef]
- Elias, T.; Lee, L.H.; Rossi, M.; Caruso, F.; Adams, S.D. In vitro analysis of the antioxidant and antiviral activity of embelin against herpes simplex virus-1. Microorganisms 2021, 9, 1–19. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem. Biol. Interact. 2015, 234, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Schachtele, S.J.; Hu, S.; Little, M.R.; Lokensgard, J.R. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflamm. 2010, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [Green Version]
- Weidner, A.M.; Bradley, M.A.; Beckett, T.L.; Niedowicz, D.M.; Dowling, A.L.S.; Matveev, S.V.; LeVine, H., III; Lovell, M.A.; Murphy, M.P. RNA Oxidation Adducts 8-OHG and 8-OHA Change with Aβ42 Levels in Late-Stage Alzheimer’s Disease. PLoS ONE 2011, 6, e24930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protto, V.; Tramutola, A.; Fabiani, M.; Marcocci, M.E.; Napoletani, G.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Perluigi, M.; Domenico, F.D.; et al. Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms 2020, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kristen, H.; Sastre, I.; Muñoz-Galdeano, T.; Recuero, M.; Aldudo, J.; Bullido, M.J. The lysosome system is severely impaired in a cellular model of neurodegeneration induced by HSV-1 and oxidative stress. Neurobiol. Aging 2018, 68, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana, S.; Sastre, I.; Recuero, M.; Bullido, M.J.; Aldudo, J. Oxidative Stress Enhances Neurodegeneration Markers Induced by Herpes Simplex Virus Type 1 Infection in Human Neuroblastoma Cells. PLoS ONE 2013, 8, e75842. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.A.; Itzhaki, R.F.; Shipley, S.J.; Dobson, C.B. Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci. Lett. 2007, 429, 95–100. [Google Scholar] [CrossRef]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 1–12. [Google Scholar] [CrossRef]
- Katsouri, L.; Birch, A.M.; Renziehausen, A.W.J.; Zach, C.; Aman, Y.; Steeds, H.; Bonsu, A.; Palmer, E.O.C.; Mirzaei, N.; Ries, M.; et al. Ablation of reactive astrocytes exacerbates disease pathology in a model of Alzheimer’s disease. Glia 2020, 68, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Lee, M.; McGeer, E.; McGeer, P.L. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: Implications for Alzheimer’s disease pathogenesis. Neurobiol. Aging 2015, 36, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Chen, B.; Yang, L.; Qi, K.; Lu, J.; Zhao, D. Amyloid-β Activates Microglia and Regulates Protein Expression in a Manner Similar to Prions. J. Mol. Neurosci. 2015, 56, 509–518. [Google Scholar] [CrossRef]
- Saucken, V.E.; Jay, T.R.; Landreth, G.E. The effect of amyloid on microglia-neuron interactions before plaque onset occurs independently of TREM2 in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2020, 145, 105072. [Google Scholar] [CrossRef]
- Floden, A.M.; Li, S.; Combs, C.K. β-Amyloid-Stimulated Microglia Induce Neuron Death via Synergistic Stimulation of Tumor Necrosis Factor α and NMDA Receptors. J. Neurosci. 2005, 25, 2566–2575. [Google Scholar] [CrossRef]
- Powell-Doherty, R.D.; Abbott, A.R.N.; Nelson, L.A.; Bertke, A.S. Amyloid-β and p-Tau Anti-Threat Response to Herpes Simplex Virus 1 Infection in Primary Adult Murine Hippocampal Neurons. J. Virol. 2020, 94, e01874-19. [Google Scholar] [CrossRef]
- Álvarez, G.; Aldudo, J.; Alonso, M.; Santana, S.; Valdivieso, F. Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J. Neurosci. Res. 2012, 90, 1020–1029. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Evans, N.A.; Soden, P.E.; Rosin, C.; Facci, L.; Richardson, J.C. Oligodendrocytes are a novel source of amyloid peptide generation. Neurochem. Res. 2009, 34, 2243–2250. [Google Scholar] [CrossRef]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-beta peptides are cytotoxic to oligodendrocytes. J. Neurosci. 2001, 21, 2001. [Google Scholar] [CrossRef]
- Lee, J.-T.; Xu, J.; Lee, J.-M.; Ku, G.; Han, X.; Yang, D.-I.; Chen, S.; Hsu, C.Y. Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase–ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.D.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble aβ through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouwens, L.K.; Makoni, N.J.; Rogers, V.A.; Nichols, M.R. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. 2016, 1648, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef] [Green Version]
- ILL-Raga, G.; Palomer, E.; Wozniak, M.A.; Ramos-Fernández, E.; Bosch-Morató, M.; Tajes, M.; Guix, F.X.; Galán, J.J.; Clarimón, J.; Antúnez, C.; et al. Activation of PKR causes amyloid ß-peptide accumulation via De-Repression of Bace1 expression. PLoS ONE 2011, 6, e21456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Ferland, P.; Webster, P.; Bearer, E.L. Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS ONE 2011, 6, e17966. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.; Mee, A.; Itzhaki, R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Vissel, B. Amyloid β: One of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br. J. Pharmacol. 2015, 172, 3714–3727. [Google Scholar] [CrossRef] [Green Version]
- Kosik, K.S. The molecular and cellular biology of tau. Brain Pathol. 1993, 3, 39–43. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiberlich, V.; Bauer, N.G.; Schwarz, L.; Ffrench-Constant, C.; Goldbaum, O.; Richter-Landsberg, C. Downregulation of the microtubule associated protein tau impairs process outgrowth and myelin basic protein mRNA transport in oligodendrocytes. Glia 2015, 63, 1621–1635. [Google Scholar] [CrossRef]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Stieler, J.; Strijkstra, A.M.; Hut, R.A.; Rüdiger, J.; Van der Zee, E.A.; Harkany, T.; Holzer, M.; Härtig, W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 2003, 23, 6972–6981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenessey, A.; Yen, S.H.C. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993, 629, 40–46. [Google Scholar] [CrossRef]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M.Y. Abnormal tau phosphorylation at Ser396 in alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Brion, J.-P.; Smith, C.; Couck, A. -M.; Gallo, J. -M.; Anderton, B.H. Developmental Changes in τ Phosphorylation: Fetal τ Is Transiently Phosphorylated in a Manner Similar to Paired Helical Filament-τ Characteristic of Alzheimer’s Disease. J. Neurochem. 1993, 61, 2071–2080. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Six, J.; Lubke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.Q.; Lee, V.M.Y. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. USA 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [Green Version]
- Seubert, P.; Mawal-Dewan, M.; Barbour, R.; Jakes, R.; Goedert, M.; Johnson, G.V.W.; Litersky, J.M.; Schenk, D.; Lieberburg, I.; Trojanowski, J.Q.; et al. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J. Biol. Chem. 1995, 270, 18917–18922. [Google Scholar] [CrossRef] [Green Version]
- Hefti, M.M.; Kim, S.; Bell, A.J.; Betters, R.K.; Fiock, K.L.; Iida, M.A.; Smalley, M.E.; Farrell, K.; Fowkes, M.E.; Crary, J.F. Tau Phosphorylation and Aggregation in the Developing Human Brain. J. Neuropathol. Exp. Neurol. 2019, 78, 930–938. [Google Scholar] [CrossRef]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Takeda, S. Tau Propagation as a Diagnostic and Therapeutic Target for Dementia: Potentials and Unanswered Questions. Front. Neurosci. 2019, 13, 1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217, e20190783. [Google Scholar] [CrossRef] [PubMed]
- Shimohama, S. Apoptosis in Alzheimer’s disease—An update. Apoptosis 2000, 5, 9–16. [Google Scholar] [CrossRef]
- Dickson, D.W. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: Cause or effect? J. Clin. Investig. 2004, 114, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, M.A.; Frost, A.L.; Itzhaki, R.F. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimer’s Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef]
- Benetti, L.; Roizman, B. Herpes simplex virus protein kinase Us3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 9411–9416. [Google Scholar] [CrossRef] [Green Version]
- Lerchundi, R.; Neira, R.; Valdivia, S.; Vio, K.; Concha, M.I.; Zambrano, A.; Otth, C. Tau cleavage at D421 by caspase-3 is induced in neurons and astrocytes infected with Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2011, 23, 513–520. [Google Scholar] [CrossRef]
- Bandea, C.I. Aβ, tau, α-synuclein, huntingtin, TDP-43, PrP and AA are members of the innate immune system: A unifying hypothesis on the etiology of AD, PD, HD, ALS, CJD and RSA as innate immunity disorders. bioRxiv 2013, 000604. [Google Scholar] [CrossRef] [Green Version]
- Calissano, P.; Matrone, C.; Amadoro, G. Apoptosis and in vitro Alzheimer disease neuronal models. Commun. Integr. Biol. 2009, 2, 163–169. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus infection and death receptor-mediated apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, V.; Roizman, B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 3931–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBiasi, R.L.; Kleinschmidt-DeMasters, B.K.; Richardson-Burns, S.; Tyler, K.L. Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J. Infect. Dis. 2002, 186, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Scanlan, P.M.; Tiwari, V.; Sheth, V.; Clement, C.; Guzman-Hartman, G.; Dermody, T.S.; Valyi-Nagy, T. Expression of nectin-1 in normal and herpes simplex virus type 1-infected murine brain. Appl. Immunohistochem. Mol. Morphol. 2006, 14, 341–347. [Google Scholar] [CrossRef]
- Marques, C.P.; Hu, S.; Sheng, W.; Lokensgard, J.R. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. 2006, 121, 1–10. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Guégan, C.; Przedborski, S. Programmed cell death in amyotrophic lateral sclerosis. J. Clin. Investig. 2003, 111, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.L.; Blaho, J.A. Apoptosis During Herpes Simplex Virus Infection. Adv. Virus Res. 2006, 69, 67–97. [Google Scholar]
- Zambrano, Á.; Solis, L.; Salvadores, N.; Cortés, M.; Lerchundi, R.; Otth, C. Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J. Alzheimer’s Dis. 2008, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Neuronal Apoptosis in Herpes Simplex Virus-1 Encephalitis (HSE)-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/17664814/ (accessed on 15 October 2021).
- He, S.; Han, J. Manipulation of Host Cell Death Pathways by Herpes Simplex Virus. Curr Top Microbiol Immunol. 2020, 6, 15. [Google Scholar]
- Martin, C.; Leyton, L.; Hott, M.; Arancibia, Y.; Spichiger, C.; McNiven, M.A.; Court, F.A.; Concha, M.I.; Burgos, P.V.; Otth, C. Herpes simplex virus type 1 neuronal infection perturbs golgi apparatus integrity through activation of src tyrosine kinase and Dyn-2 GTPase. Front. Cell. Infect. Microbiol. 2017, 7, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toscano, E.C.B.; Sousa, L.F. da C.; Lima, G.K.; Mesquita, L.A.; Vilela, M.C.; Rodrigues, D.H.; Ferreira, R.N.; Soriani, F.M.; Campos, M.A.; Kroon, E.G.; et al. Neuroinflammation is associated with reduced SOCS2 and SOCS3 expression during intracranial HSV-1 infection. Neurosci. Lett. 2020, 736, 135295. [Google Scholar] [CrossRef]
- Acuña-Hinrichsen, F.; Covarrubias-Pinto, A.; Ishizuka, Y.; Stolzenbach, M.F.; Martin, C.; Salazar, P.; Castro, M.A.; Bramham, C.R.; Otth, C. Herpes Simplex Virus Type 1 Neuronal Infection Triggers the Disassembly of Key Structural Components of Dendritic Spines. Front. Cell. Neurosci. 2021, 15, 580717. [Google Scholar] [CrossRef]
- Doll, J.R.; Hoebe, K.; Thompson, R.L.; Sawtell, N.M. Resolution of herpes simplex virus reactivation in vivo results in neuronal destruction. PLoS Pathog. 2020, 16, e1008296. [Google Scholar] [CrossRef] [Green Version]
- Aravalli, R.N.; Hu, S.; Lokensgard, J.R. Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia. J. Neuroinflamm. 2007, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sanfilippo, C.M.; Blaho, J.A. ICP0 Gene Expression Is a Herpes Simplex Virus Type 1 Apoptotic Trigger. J. Virol. 2006, 80, 6810–6821. [Google Scholar] [CrossRef] [Green Version]
- Mangold, C.A.; Rathbun, M.M.; Renner, D.W.; Kuny, C.V.; Szpara, M.L. Viral infection of human neurons triggers strain-specific differences in host neuronal and viral transcriptomes. PLoS Pathog. 2021, 17, e1009441. [Google Scholar] [CrossRef]
- Lussignol, M.; Esclatine, A. Herpesvirus and autophagy: “All right, everybody be cool, this is a robbery! ” Viruses 2017, 9, 372. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.C.C.; Suen, K.C.; Ma, C.H.; Elyaman, W.; Ng, H.K.; Hugon, J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2α in neuronal degeneration. J. Neurochem. 2002, 83, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Tallóczy, Z.; Jiang, W.; Virgin IV, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the elF2α kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.P.; Ha, T.; et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.B.; Horan, K.A.; Holm, C.K.; Stranks, A.J.; Mettenleiter, T.C.; Simon, A.K.; Jensen, S.B.; Rixon, F.J.; He, B.; Paludan, S.R. Activation of autophagy by alpha-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on STING. J. Immunol. 2011, 187, 5268–5276. [Google Scholar] [CrossRef] [Green Version]
- Katzenell, S.; Leib, D.A. Herpes Simplex Virus and Interferon Signaling Induce Novel Autophagic Clusters in Sensory Neurons. J. Virol. 2016, 90, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.-F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The Herpes Simplex Virus 1 Us11 Protein Inhibits Autophagy through Its Interaction with the Protein Kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Siracusano, G.; Venuti, A.; Lombardo, D.; Mastino, A.; Esclatine, A.; Sciortino, M.T. Early activation of MyD88-mediated autophagy sustains HSV-1 replication in human monocytic THP-1 cells. Sci. Rep. 2016, 6, 31302. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the Role of Autophagy in Replication of Herpes Simplex Virus in Cell Culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [Green Version]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Santana, S.; Recuero, M.; Bullido, M.J.; Valdivieso, F.; Aldudo, J. Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol. Aging 2012, 33, e19–e430. [Google Scholar] [CrossRef]
- Šimić, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; Silva, R.; Giovanni, G.D.; et al. Monoaminergic Neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimić, G.; Kostović, I.; Winblad, B.; Bogdanović, N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol. 1997, 379, 482–494. [Google Scholar] [CrossRef]
- de Flores, R.; La Joie, R.; Chételat, G. Structural imaging of hippocampal subfields in healthy aging and Alzheimer’s disease. Neuroscience 2015, 309, 29–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.H.; Hof, P.R. Life and death of neurons in the aging brain. Science 1997, 278, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Avramopoulos, D.; Szymanski, M.; Wang, R.; Bassett, S. Gene expression reveals overlap between normal aging and Alzheimer’s disease genes. Neurobiol. Aging 2011, 32, 2319.e27. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.A.; Shipley, S.J.; Combrinck, M.; Wilcock, G.K.; Itzhaki, R.F. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J. Med. Virol. 2005, 75, 300–306. [Google Scholar] [CrossRef]
- Baringer, J.R.; Pisani, P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann. Neurol. 1994, 36, 823–829. [Google Scholar] [CrossRef]
- Fraser, N.W.; Lawrence, W.C.; Wroblewska, Z.; Gilden, D.H.; Koprowski, H. Herpes simplex type 1 DNA in human brain tissue. Proc. Natl. Acad. Sci. USA 1981, 78, 6461. [Google Scholar] [CrossRef] [Green Version]
- Bearer, E.L. HSV, axonal transport and Alzheimer’s disease: In vitro and in vivo evidence for causal relationships. Future Virol. 2012, 7, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Studahl, M.; Rosengren, L.; Günther, G.; Hagberg, L. Difference in pathogenesis between herpes simplex virus type 1 encephalitis and tick-borne encephalitis demonstrated by means of cerebrospinal fluid markers of glial and neuronal destruction. J. Neurol. 2000, 247, 636–642. [Google Scholar] [CrossRef]
- Itoyama, Y.; Sekizawa, T.; Openshaw, H.; Kogure, K.; Goto, I. Early loss of astrocytes in herpes simplex virus–induced central nervous system demyelination. Ann. Neurol. 1991, 29, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Bak, I.J.; Markham, C.H. Alteration in tyrosine hydroxylase, glutamic acid decarylase and choline acetyltransferase in basal ganglia following herpes simplex virus inoculation in rat neostriatum. Brain Res. 1979, 169, 401–405. [Google Scholar] [CrossRef]
- Päivärinta, M.A.; Marttila, R.J.; Rinne, J.O.; Rinne, U.K. Dopaminergic neurotransmission in chronic herpes simplex virus brain infection in rabbits. J. Neural Transm. 1993, 93, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8, 14727. [Google Scholar] [CrossRef]
- D’Amelio, M.; Puglisi-Allegra, S.; Mercuri, N. The role of dopaminergic midbrain in Alzheimer’s disease: Translating basic science into clinical practice. Pharmacol. Res. 2018, 130, 414–419. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Prasad, K.M.; Upton, C.H.; Viggiano, L.; Milosevic, J.; Raimondi, G.; McClain, L.; Chowdari, K.; Tischfield, J.; Sheldon, M.; et al. Persistent Infection by HSV-1 Is Associated with Changes in Functional Architecture of iPSC-Derived Neurons and Brain Activation Patterns Underlying Working Memory Performance. Schizophr. Bull. 2015, 41, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña-Hinrichsen, F.; Muñoz, M.; Hott, M.; Martin, C.; Mancilla, E.; Salazar, P.; Leyton, L.; Zambrano, A.; Concha, M.I.; Burgos, P.V.; et al. Herpes Simplex Virus Type 1 Enhances Expression of the Synaptic Protein Arc for Its Own Benefit. Front. Cell. Neurosci. 2018, 12, 505. [Google Scholar] [CrossRef]
- Hensel, N.; Raker, V.; Förthmann, B.; Buch, A.; Sodeik, B.; Pich, A.; Claus, P. The Proteome and Secretome of Cortical Brain Cells Infected with Herpes Simplex Virus. Front. Neurol. 2020, 11, 844. [Google Scholar] [CrossRef]
- Lathe, R.; Haas, J.G. Distribution of cellular HSV-1 receptor expression in human brain. J. Neurovirol. 2017, 23, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Casrouge, A.; Zhang, S.-Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes Simplex Virus Encephalitis in Human UNC-93B Deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Bastard, P.; Manry, J.; Chen, J.; Rosain, J.; Seeleuthner, Y.; AbuZaitun, O.; Lorenzo, L.; Khan, T.; Hasek, M.; Hernandez, N.; et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J. Clin. Investig. 2021, 131, e139980. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Shimizu, V.; Pérez De Diego, R.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.L.; Mørk, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jørgensen, S.E.; Skipper, K.A.; Höning, K.; Gad, H.H.; Østergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Zaid Al-Mohsen, I.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Audry, M.; Ciancanelli, M.; Yang, K.; Cobat, A.; Chang, H.H.; Sancho-Shimizu, V.; Lorenzo, L.; Niehues, T.; Reichenbach, J.; Li, X.X.; et al. NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 2011, 128, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef]
- Mørk, N.; Kofod-Olsen, E.; Sørensen, K.B.; Bach, E.; Ørntoft, T.F.; Østergaard, L.; Paludan, S.R.; Christiansen, M.; Mogensen, T.H. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015, 16, 552–566. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.; Ciancanelli, M.; Ou, Y.H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; Pérez de Diego, R.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.K.; Seppanen, M.; Hautala, T.; Ciancanelli, M.J.; Itan, Y.; Lafaille, F.G.; Dell, W.; Lorenzo, L.; Byun, M.; Pauwels, E.; et al. TLR3 deficiency in herpes simplex encephalitis: High allelic heterogeneity and recurrence risk. Neurology 2014, 83, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.-Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.-W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Baucells, B.J.; Vlagea, A.; Petit-Pedrol, M.; Esteve-Solé, A.; Deyà-Martínez, A.; Ruiz-García, R.; Juan, M.; Pérez de Diego, R.; Dalmau, J.; et al. Toll-like receptor 3 deficiency in autoimmune encephalitis post–herpes simplex encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitturi, B.K.; Rosemberg, S.; Arita, F.N.; da Rocha, A.J.; Forte, W.C.N.; Tilbery, C.P. Multiphasic disseminated encephalomyelitis associated with herpes virus infection in a patient with TLR3 deficiency. Mult. Scler. Relat. Disord. 2019, 36, 101379. [Google Scholar] [CrossRef] [PubMed]
- Bravo García-Morato, M.; Calvo Apalategi, A.; Bravo-Gallego, L.Y.; Blázquez Moreno, A.; Simón-Fuentes, M.; Garmendia, J.V.; Méndez Echevarría, A.; del Rosal Rabes, T.; Domínguez-Soto, Á.; López-Granados, E.; et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J. Allergy Clin. Immunol. 2019, 144, 309–312.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Y.; Clark, N.E.; Freije, C.A.; Pauwels, E.; Taggart, A.J.; Okada, S.; Mandel, H.; Garcia, P.; Ciancanelli, M.J.; Biran, A.; et al. Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell 2018, 172, 952–965.e18. [Google Scholar] [CrossRef] [Green Version]
- Lafaille, F.G.; Harschnitz, O.; Lee, Y.S.; Zhang, P.; Hasek, M.L.; Kerner, G.; Itan, Y.; Ewaleifoh, O.; Rapaport, F.; Carlile, T.M.; et al. Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat. Med. 2019, 25, 1873–1884. [Google Scholar] [CrossRef]
- Sato, R.; Kato, A.; Chimura, T.; Saitoh, S.I.; Shibata, T.; Murakami, Y.; Fukui, R.; Liu, K.; Zhang, Y.; Arii, J.; et al. Combating herpesvirus encephalitis by potentiating a TLR3–mTORC2 axis. Nat. Immunol. 2018, 19, 1071–1082. [Google Scholar] [CrossRef]
- Mielcarska, M.B.; Bossowska-Nowicka, M.; Toka, F.N. Functional failure of TLR3 and its signaling components contribute to herpes simplex encephalitis. J. Neuroimmunol. 2018, 316, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Jouanguy, E.; Béziat, V.; Mogensen, T.H.; Casanova, J.L.; Tangye, S.G.; Zhang, S.Y. Human inborn errors of immunity to herpes viruses. Curr. Opin. Immunol. 2020, 62, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.; Yadavalli, T.; Suryawanshi, R.; Hopkins, J.; Agelidis, A.; Patil, C.; Fredericks, B.; Tseng, H.; Valyi-Nagy, T.; Shukla, D. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Ciancanelli, M.J.; Zhang, P.; Harschnitz, O.; Bondet, V.; Hasek, M.; Chen, J.; Mu, X.; Itan, Y.; Cobat, A.; et al. TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Investig. 2021, 131, e134529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Harschnitz, O.; Studer, L.; Casanova, J.L. Neuron-intrinsic immunity to viruses in mice and humans. Curr. Opin. Immunol. 2021, 72, 309–317. [Google Scholar] [CrossRef]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 2006, 53, 484–490. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Krämer-Albers, E.M. Emerging Roles of Exosomes in Neuron–Glia Communication. Front. Physiol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- De Chiara, G.; Marcocci, M.E.; Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. Biomed Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mielcarska, M.B.; Skowrońska, K.; Wyżewski, Z.; Toka, F.N. Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 242. https://doi.org/10.3390/ijms23010242
Mielcarska MB, Skowrońska K, Wyżewski Z, Toka FN. Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(1):242. https://doi.org/10.3390/ijms23010242
Chicago/Turabian StyleMielcarska, Matylda Barbara, Katarzyna Skowrońska, Zbigniew Wyżewski, and Felix Ngosa Toka. 2022. "Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 1: 242. https://doi.org/10.3390/ijms23010242
APA StyleMielcarska, M. B., Skowrońska, K., Wyżewski, Z., & Toka, F. N. (2022). Disrupting Neurons and Glial Cells Oneness in the Brain—The Possible Causal Role of Herpes Simplex Virus Type 1 (HSV-1) in Alzheimer’s Disease. International Journal of Molecular Sciences, 23(1), 242. https://doi.org/10.3390/ijms23010242