
Cx43 Promotes Endothelial Cell Migration and Angiogenesis via the Tyrosine Phosphatase SHP-2

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
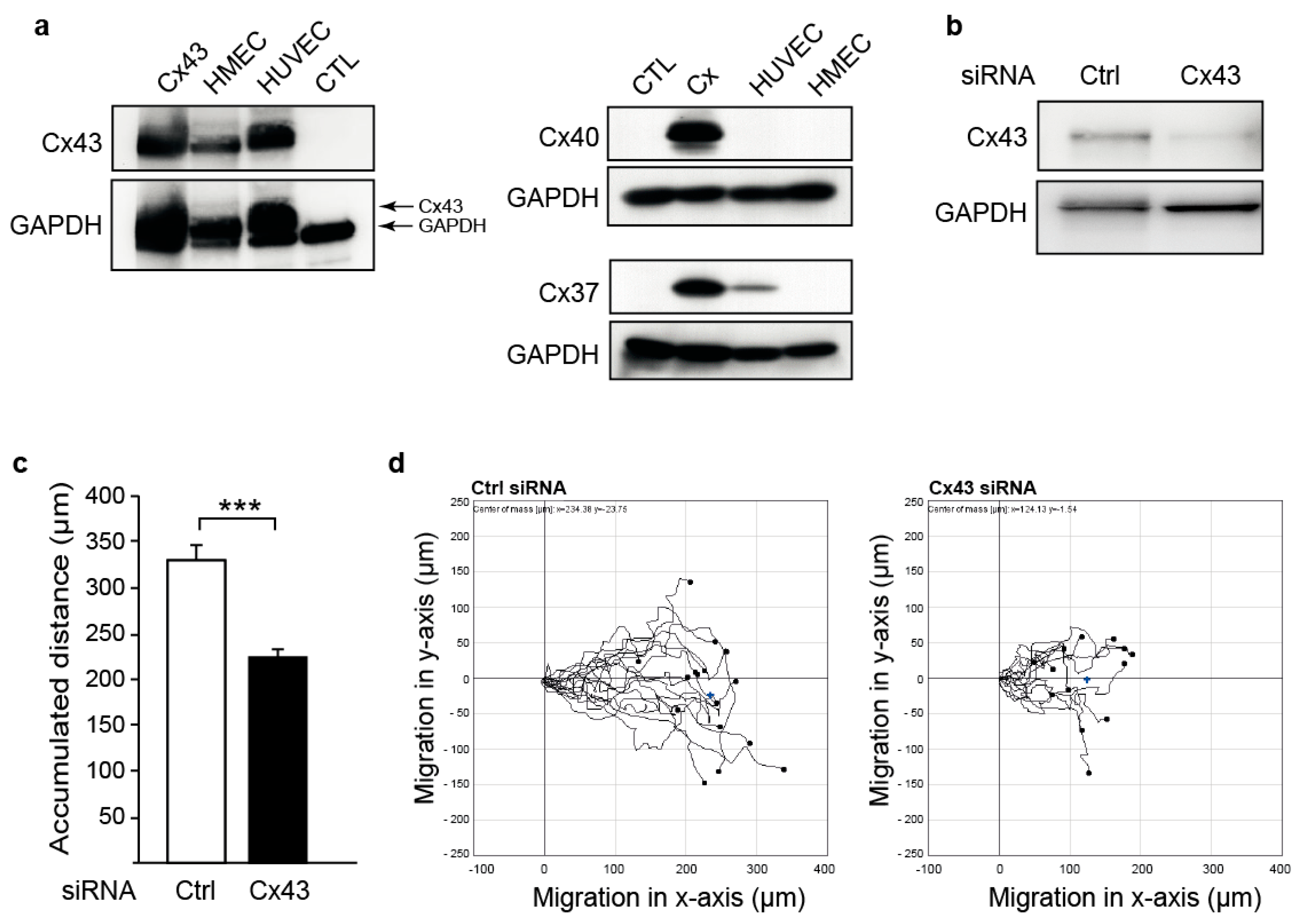
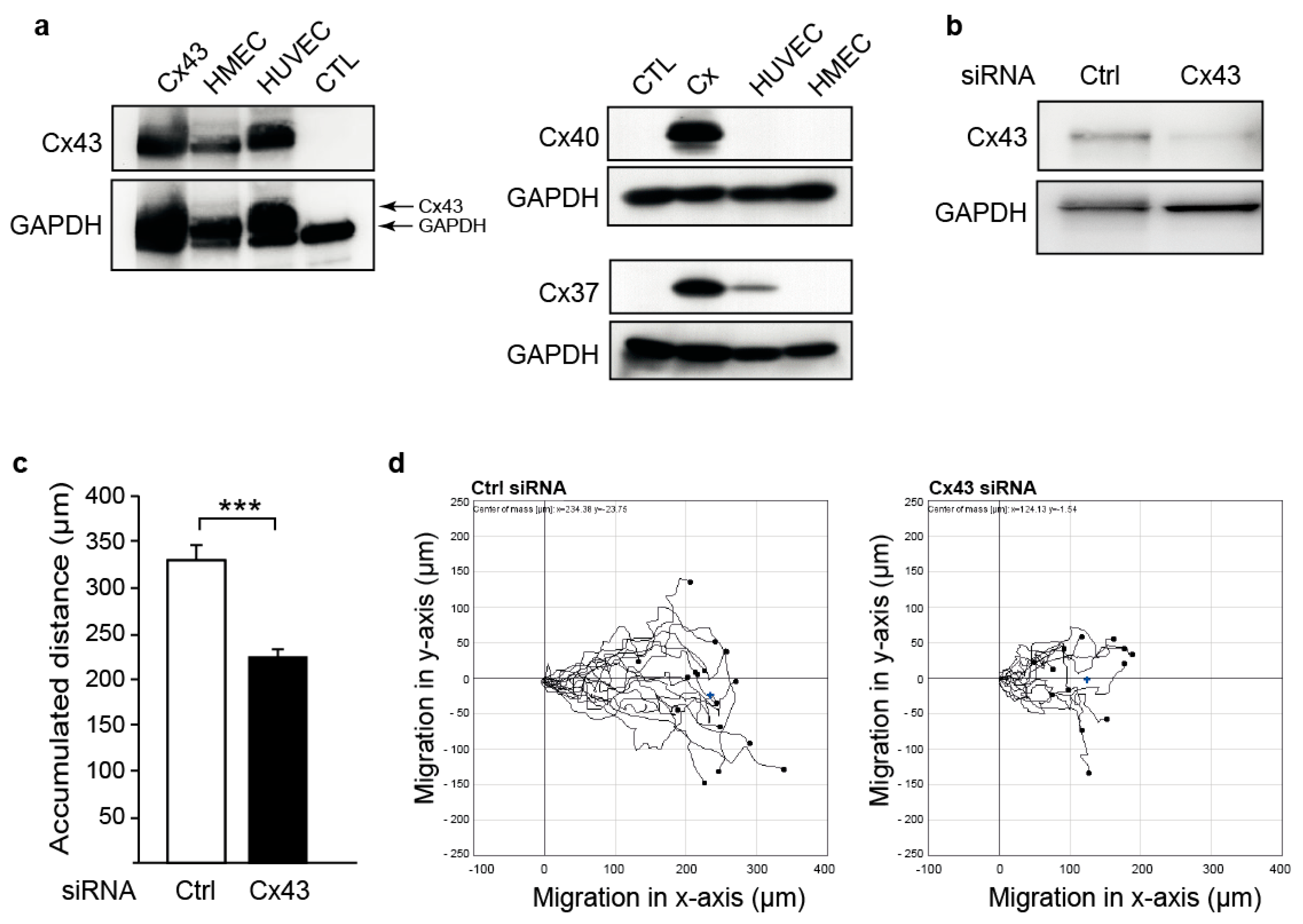
2.1. Downregulation of Cx43 Decreases Endothelial Cell Migration
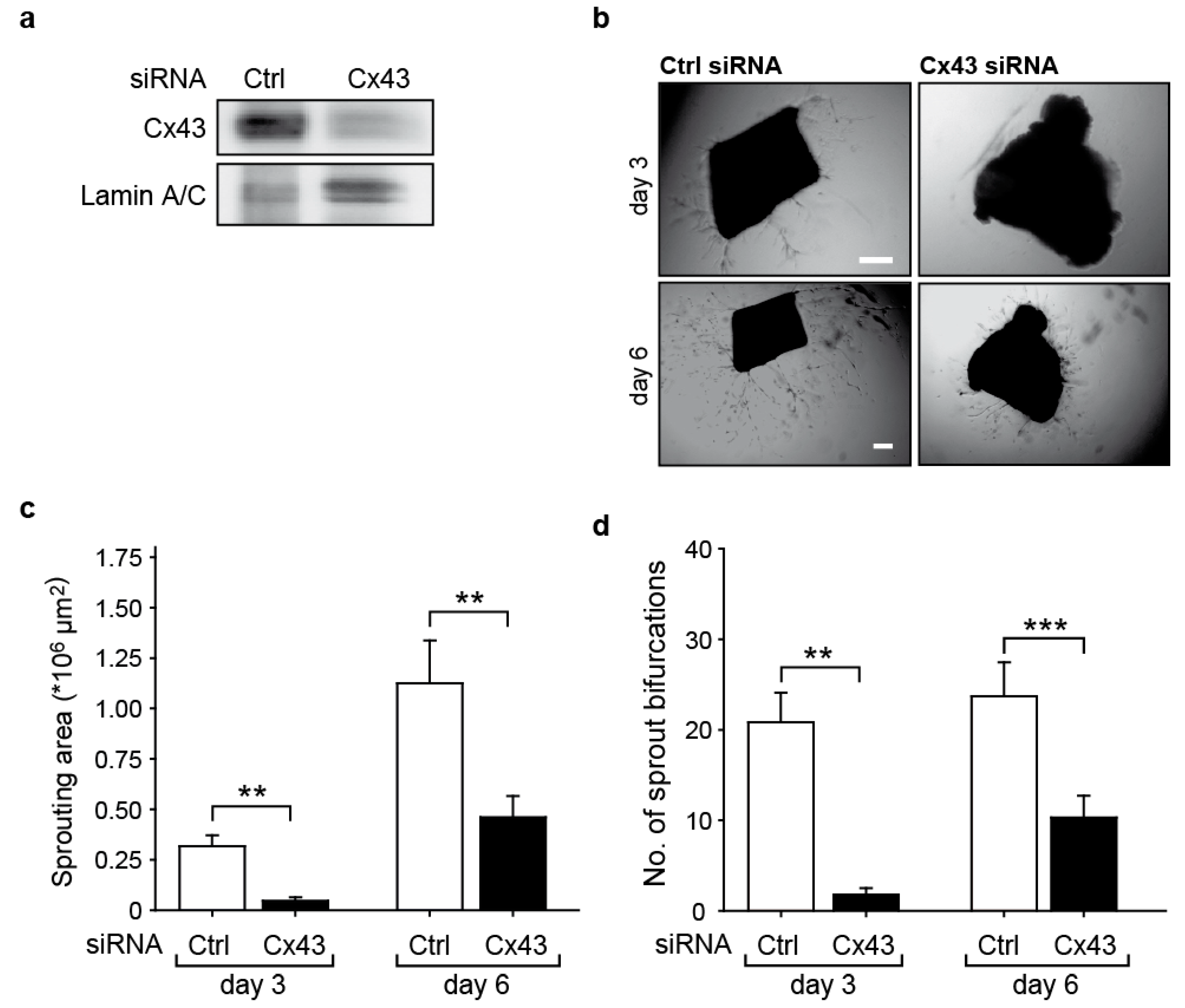
2.2. Downregulation of Cx43 Reduces Angiogenic Sprouting Ex Vivo
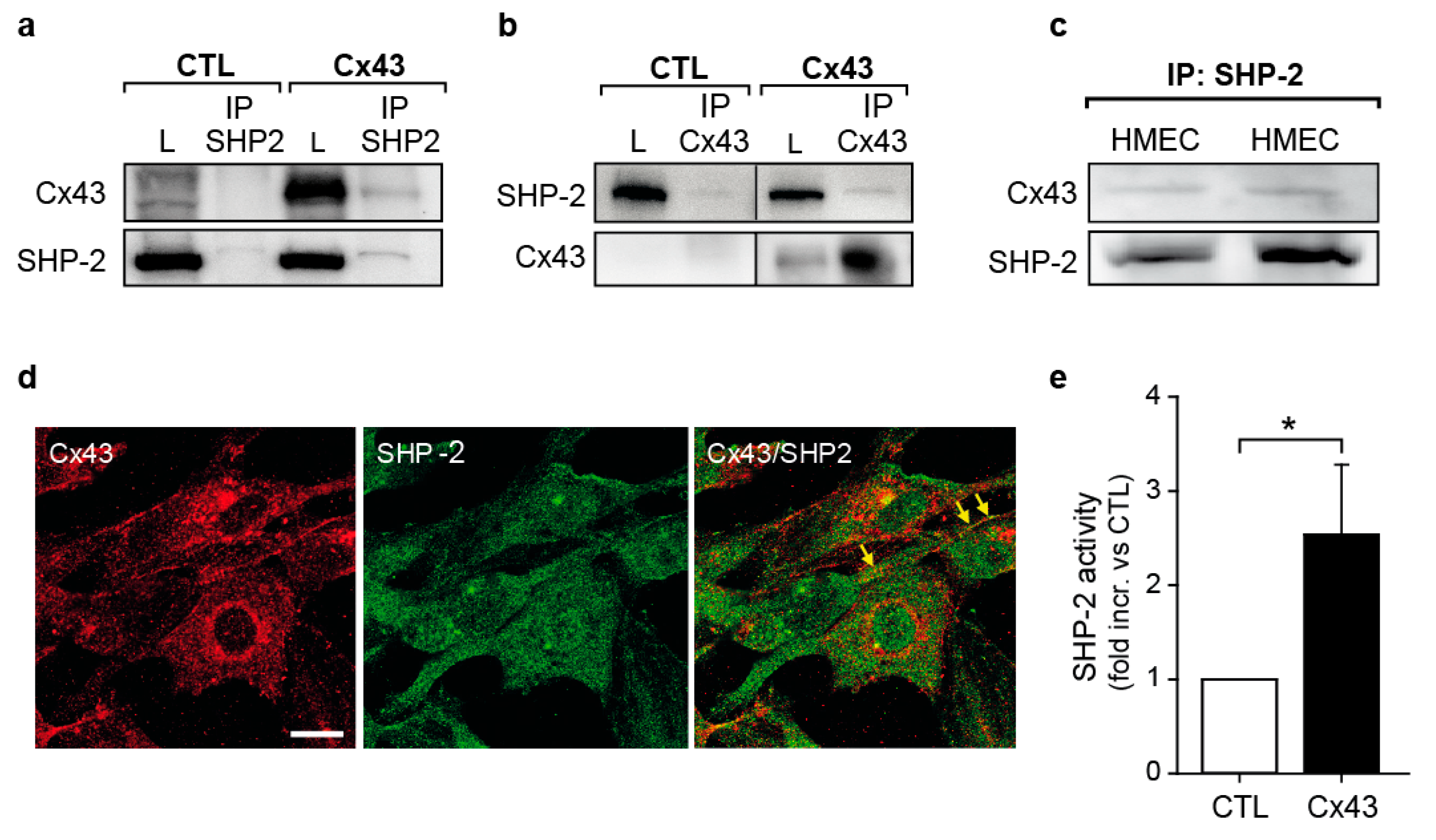
2.3. Cx43 Interacts with the Tyrosine Phosphatase SHP-2 and Induces Its Activity
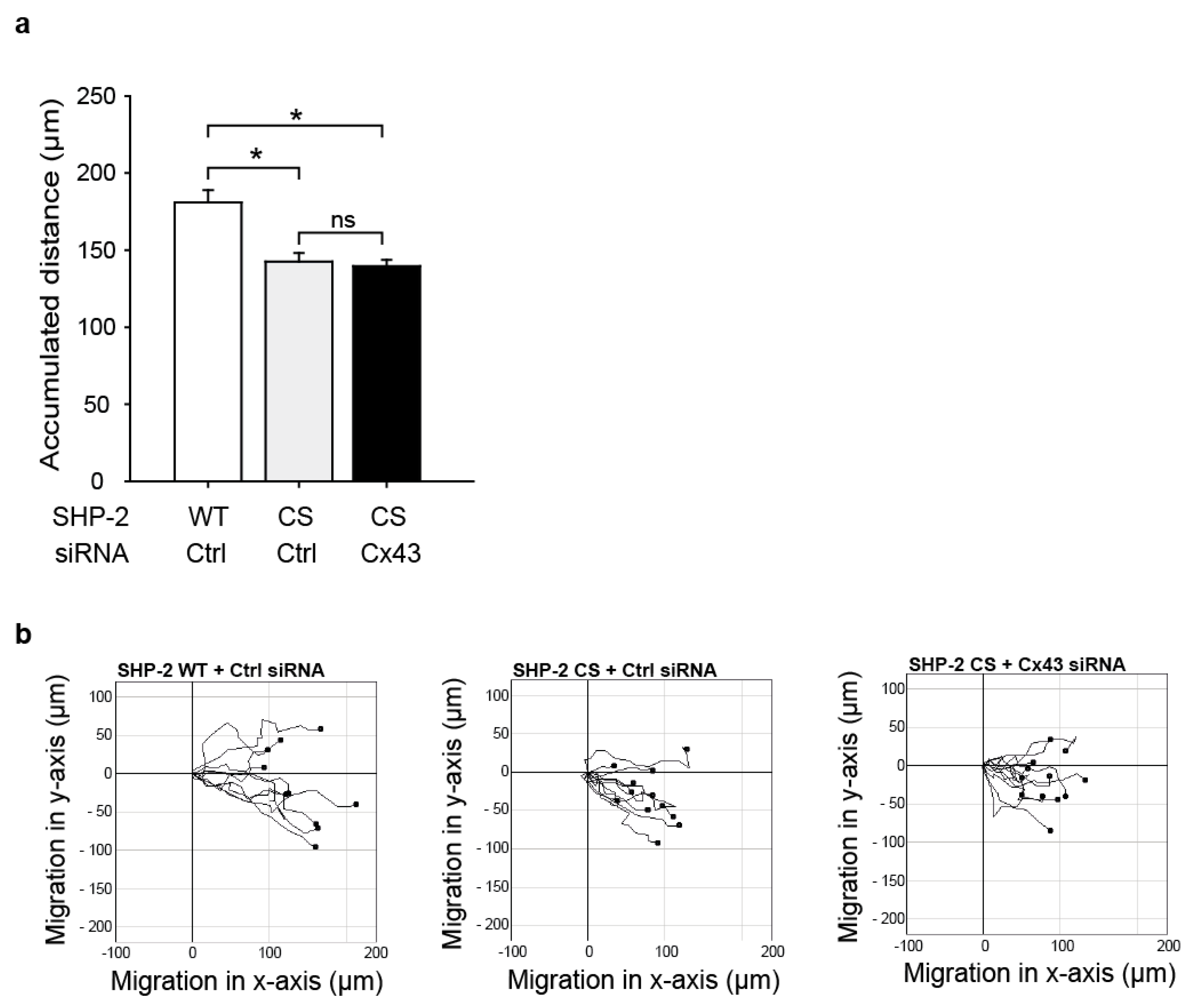
2.4. SHP-2 Inactivation Reduces Endothelial Migration
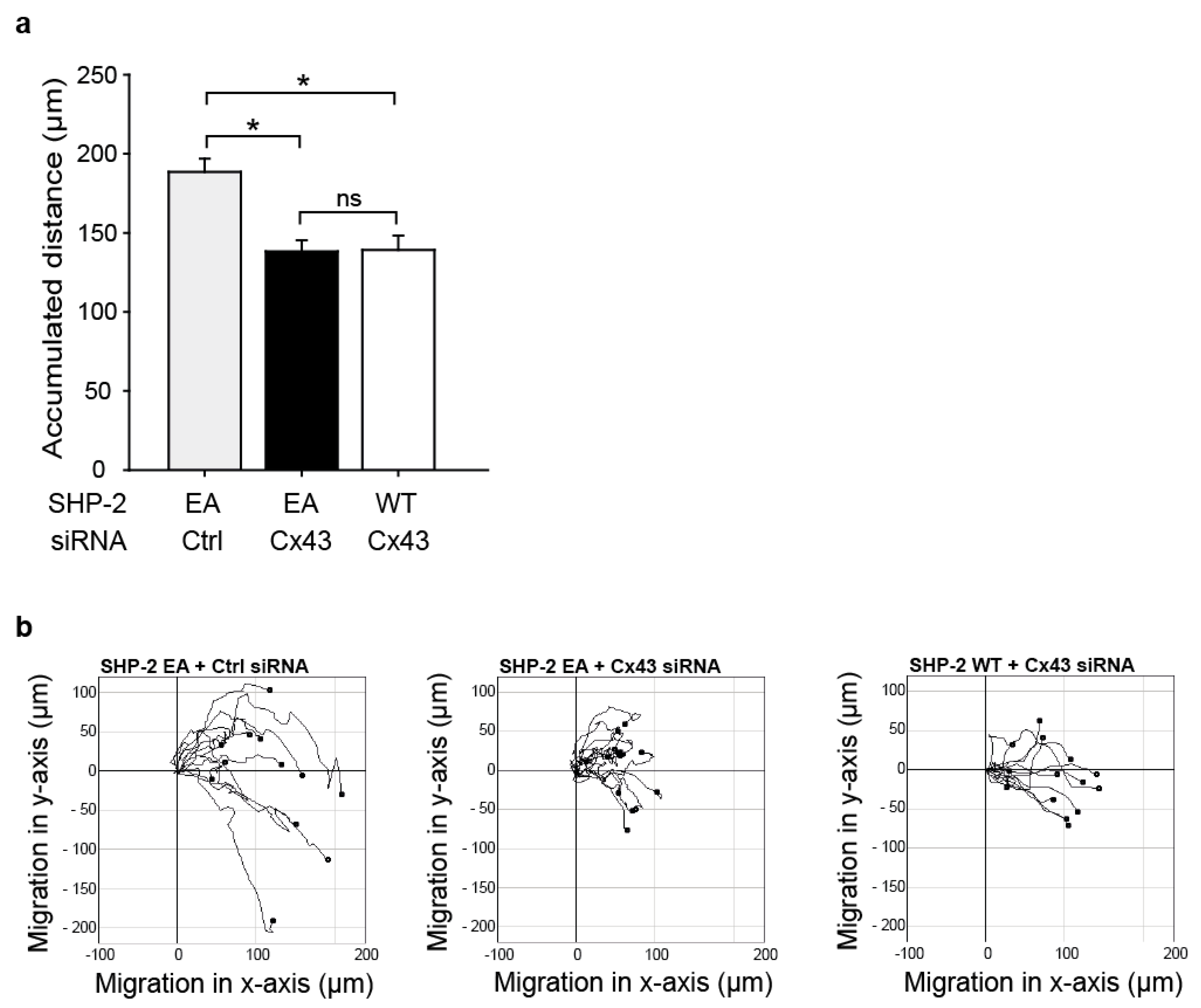
2.5. The Impaired Migration in Cx43 Knock-Out Cells Cannot Be Rescued by Expression of a Constitutively Active SHP-2
3. Discussion
4. Materials and Methods
4.1. Cells and Culture Conditions
4.2. Lentiviral Transduction of HMEC
4.3. siRNA-Mediated Knockdown
4.4. Cell Migration
4.5. Mouse Aortic Ring Assay
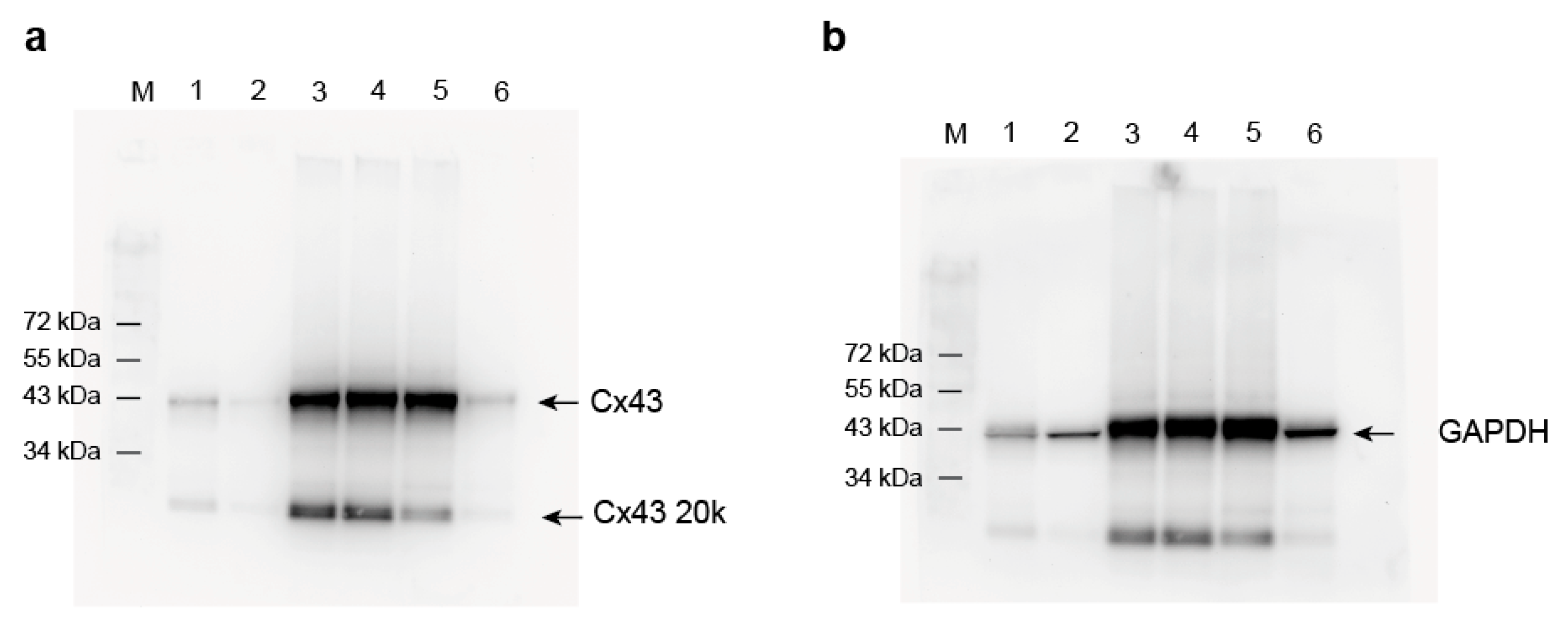
4.6. Western Blot Analysis
4.7. Lysis of Mouse Aortae
4.8. Immunoprecipitation
4.9. Immunofluorescent Staining
4.10. Measurement of SHP-2 Activity
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Fraisl, P.; Mazzone, M.; Schmidt, T.; Carmeliet, P. Regulation of angiogenesis by oxygen and metabolism. Dev. Cell 2009, 16, 167–179. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Koepple, C.; Zhou, Z.; Huber, L.; Schulte, M.; Schmidt, K.; Gloe, T.; Kneser, U.; Schmidt, V.J.; de Wit, C. Expression of Connexin43 Stimulates Endothelial Angiogenesis Independently of Gap Junctional Communication In Vitro. Int. J. Mol. Sci. 2021, 22, 7400. [Google Scholar] [CrossRef]
- Wang, H.H.; Su, C.H.; Wu, Y.J.; Li, J.Y.; Tseng, Y.M.; Lin, Y.C.; Hsieh, C.L.; Tsai, C.H.; Yeh, H.I. Reduction of connexin43 in human endothelial progenitor cells impairs the angiogenic potential. Angiogenesis 2013, 16, 553–560. [Google Scholar] [CrossRef]
- Yu, W.; Jin, H.; Sun, W.; Nan, D.; Deng, J.; Jia, J.; Yu, Z.; Huang, Y. Connexin43 promotes angiogenesis through activating the HIF-1α/VEGF signaling pathway under chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 2021, 41, 2656–2675. [Google Scholar] [CrossRef]
- Wang, D.G.; Zhang, F.X.; Chen, M.L.; Zhu, H.J.; Yang, B.; Cao, K.J. Cx43 in mesenchymal stem cells promotes angiogenesis of the infarcted heart independent of gap junctions. Mol. Med. Rep. 2014, 9, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Kung, C.I.; Tseng, Y.Y.; Lin, Y.C.; Chen, C.H.; Tsai, C.H.; Yeh, H.I. Activation of endothelial cells to pathological status by down-regulation of connexin43. Cardiovasc. Res. 2008, 79, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, U. Connexins: Key Players in the Control of Vascular Plasticity and Function. Physiol. Rev. 2020, 100, 525–572. [Google Scholar] [CrossRef]
- Kameritsch, P.; Khandoga, N.; Pohl, U.; Pogoda, K. Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 2013, 4, e584. [Google Scholar] [CrossRef]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys. Acta 2012, 1818, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [Green Version]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. Biophys. Acta 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameritsch, P.; Kiemer, F.; Mannell, H.; Beck, H.; Pohl, U.; Pogoda, K. PKA negatively modulates the migration enhancing effect of Connexin 43. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 828–838. [Google Scholar] [CrossRef]
- Van Campenhout, R.; Cooreman, A.; Leroy, K.; Rusiecka, O.M.; Van Brantegem, P.; Annaert, P.; Muyldermans, S.; Devoogdt, N.; Cogliati, B.; Kwak, B.R.; et al. Non-Canonical roles of connexins. Prog. Biophys. Mol. Biol. 2020, 153, 35–41. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. 1), 11. [Google Scholar] [CrossRef] [Green Version]
- Pogoda, K.; Kameritsch, P.; Mannell, H.; Pohl, U. Connexins in the control of vasomotor function. Acta Physiol. 2019, 225, e13108. [Google Scholar] [CrossRef]
- Atkinson, M.M.; Lampe, P.D.; Lin, H.H.; Kollander, R.; Li, X.R.; Kiang, D.T. Cyclic AMP modifies the cellular distribution of connexin43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J. Cell Sci. 1995, 108 Pt 9, 3079–3090. [Google Scholar] [CrossRef]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Li, W.E.; Nagy, J.I. Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur. J. Neurosci. 2000, 12, 2644–2650. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Spagnol, G.; Naslavsky, N.; Caplan, S.; Sorgen, P.L. TC-PTP directly interacts with connexin43 to regulate gap junction intercellular communication. J. Cell Sci. 2014, 127, 3269–3279. [Google Scholar] [PubMed] [Green Version]
- Feng, G.S. Shp-2 tyrosine phosphatase: Signaling one cell or many. Exp. Cell Res. 1999, 253, 47–54. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Kamiya, N.; Kim, H.K.; King, P.D. Regulation of bone and skeletal development by the SHP-2 protein tyrosine phosphatase. Bone 2014, 69, 55–60. [Google Scholar] [CrossRef]
- Geraldes, P. Protein phosphatases and podocyte function. Curr. Opin. Nephrol. Hypertens. 2018, 27, 49–55. [Google Scholar] [CrossRef]
- Coulombe, G.; Rivard, N. New and Unexpected Biological Functions for the Src-Homology 2 Domain-Containing Phosphatase SHP-2 in the Gastrointestinal Tract. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Heun, Y.; Pircher, J.; Czermak, T.; Bluem, P.; Hupel, G.; Bohmer, M.; Kraemer, B.F.; Pogoda, K.; Pfeifer, A.; Woernle, M.; et al. Inactivation of the tyrosine phosphatase SHP-2 drives vascular dysfunction in Sepsis. EBioMedicine 2019, 42, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. Role of protein phosphatases in the cancer microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Mannell, H.; Krotz, F. SHP-2 regulates growth factor dependent vascular signalling and function. Mini Rev. Med. Chem. 2014, 14, 471–483. [Google Scholar] [CrossRef]
- Mannell, H.; Hellwig, N.; Gloe, T.; Plank, C.; Sohn, H.Y.; Groesser, L.; Walzog, B.; Pohl, U.; Krotz, F. Inhibition of the tyrosine phosphatase SHP-2 suppresses angiogenesis in vitro and in vivo. J. Vasc. Res. 2008, 45, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Heun, Y.; Pogoda, K.; Anton, M.; Pircher, J.; Pfeifer, A.; Woernle, M.; Ribeiro, A.; Kameritsch, P.; Mykhaylyk, O.; Plank, C.; et al. HIF-1α Dependent Wound Healing Angiogenesis In Vivo Can Be Controlled by Site-Specific Lentiviral Magnetic Targeting of SHP-2. Mol. Ther. 2017, 25, 1616–1627. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, J.; Qi, T.; Huang, Y.; Lu, Y.; Zhan, T.; Gong, H.; Zhu, Z.; Shi, Y.; Zhou, J.; et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2019, 33, 1124–1137. [Google Scholar] [CrossRef]
- Rathnakumar, K.; Savant, S.; Giri, H.; Ghosh, A.; Fisslthaler, B.; Fleming, I.; Ram, U.; Bera, A.K.; Augustin, H.G.; Dixit, M. Angiopoietin-2 mediates thrombin-induced monocyte adhesion and endothelial permeability. J. Thromb. Haemost. 2016, 14, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Heun, Y.; Grundler Groterhorst, K.; Pogoda, K.; Kraemer, B.F.; Pfeifer, A.; Pohl, U.; Mannell, H. The Phosphatase SHP-2 Activates HIF-1α in Wounds In Vivo by Inhibition of 26S Proteasome Activity. Int. J. Mol. Sci. 2019, 20, 4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.Z.; Jiang, J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions—An update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef] [Green Version]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Mannell, H.; Blodow, S.; Schneider, H.; Schubert, K.M.; Qiu, J.; Schmidt, A.; Imhof, A.; Beck, H.; Tanase, L.I.; et al. NO Augments Endothelial Reactivity by Reducing Myoendothelial Calcium Signal Spreading: A Novel Role for Cx37 (Connexin 37) and the Protein Tyrosine Phosphatase SHP-2. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Plate, K.H.; Breier, G.; Risau, W. Molecular mechanisms of developmental and tumor angiogenesis. Brain Pathol. 1994, 4, 207–218. [Google Scholar] [CrossRef]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [Green Version]
- Kanemitsu, M.Y.; Loo, L.W.; Simon, S.; Lau, A.F.; Eckhart, W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J. Biol. Chem. 1997, 272, 22824–22831. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Xie, J.; Zhong, Q.; Wang, Y.; Zhang, S.; Luo, F.; Wen, F.; Xie, J.; Zhao, J.; Sun, X.; et al. A novel partially open state of SHP2 points to a “multiple gear” regulation mechanism. J. Biol. Chem. 2021, 296, 100538. [Google Scholar] [CrossRef]
- Tabernero, A.; Gangoso, E.; Jaraíz-Rodríguez, M.; Medina, J.M. The role of connexin43-Src interaction in astrocytomas: A molecular puzzle. Neuroscience 2016, 323, 183–194. [Google Scholar] [CrossRef]
- Salat-Canela, C.; Sesé, M.; Peula, C.; Ramón y Cajal, S.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an immortalized human microvascular endothelial cell line. J. Investig. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.; Wenzel, D.; Becher, U.M.; Freitag, D.F.; Klein, A.M.; Eberbeck, D.; Schulte, M.; Zimmermann, K.; Bergemann, C.; Gleich, B.; et al. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc. Natl. Acad. Sci. USA 2009, 106, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Krötz, F.; Sohn, H.Y.; Gloe, T.; Plank, C.; Pohl, U. Magnetofection potentiates gene delivery to cultured endothelial cells. J. Vasc. Res. 2003, 40, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Krötz, F.; Engelbrecht, B.; Buerkle, M.A.; Bassermann, F.; Bridell, H.; Gloe, T.; Duyster, J.; Pohl, U.; Sohn, H.Y. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. J. Am. Coll. Cardiol. 2005, 45, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannell, H.; Kameritsch, P.; Beck, H.; Pfeifer, A.; Pohl, U.; Pogoda, K. Cx43 Promotes Endothelial Cell Migration and Angiogenesis via the Tyrosine Phosphatase SHP-2. Int. J. Mol. Sci. 2022, 23, 294. https://doi.org/10.3390/ijms23010294
Mannell H, Kameritsch P, Beck H, Pfeifer A, Pohl U, Pogoda K. Cx43 Promotes Endothelial Cell Migration and Angiogenesis via the Tyrosine Phosphatase SHP-2. International Journal of Molecular Sciences. 2022; 23(1):294. https://doi.org/10.3390/ijms23010294
Chicago/Turabian StyleMannell, Hanna, Petra Kameritsch, Heike Beck, Alexander Pfeifer, Ulrich Pohl, and Kristin Pogoda. 2022. "Cx43 Promotes Endothelial Cell Migration and Angiogenesis via the Tyrosine Phosphatase SHP-2" International Journal of Molecular Sciences 23, no. 1: 294. https://doi.org/10.3390/ijms23010294
APA StyleMannell, H., Kameritsch, P., Beck, H., Pfeifer, A., Pohl, U., & Pogoda, K. (2022). Cx43 Promotes Endothelial Cell Migration and Angiogenesis via the Tyrosine Phosphatase SHP-2. International Journal of Molecular Sciences, 23(1), 294. https://doi.org/10.3390/ijms23010294