
The Important Role of Ion Transport System in Cervical Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: An Overview of the Epidemiology and Pathogenesis of Cervical Cancer
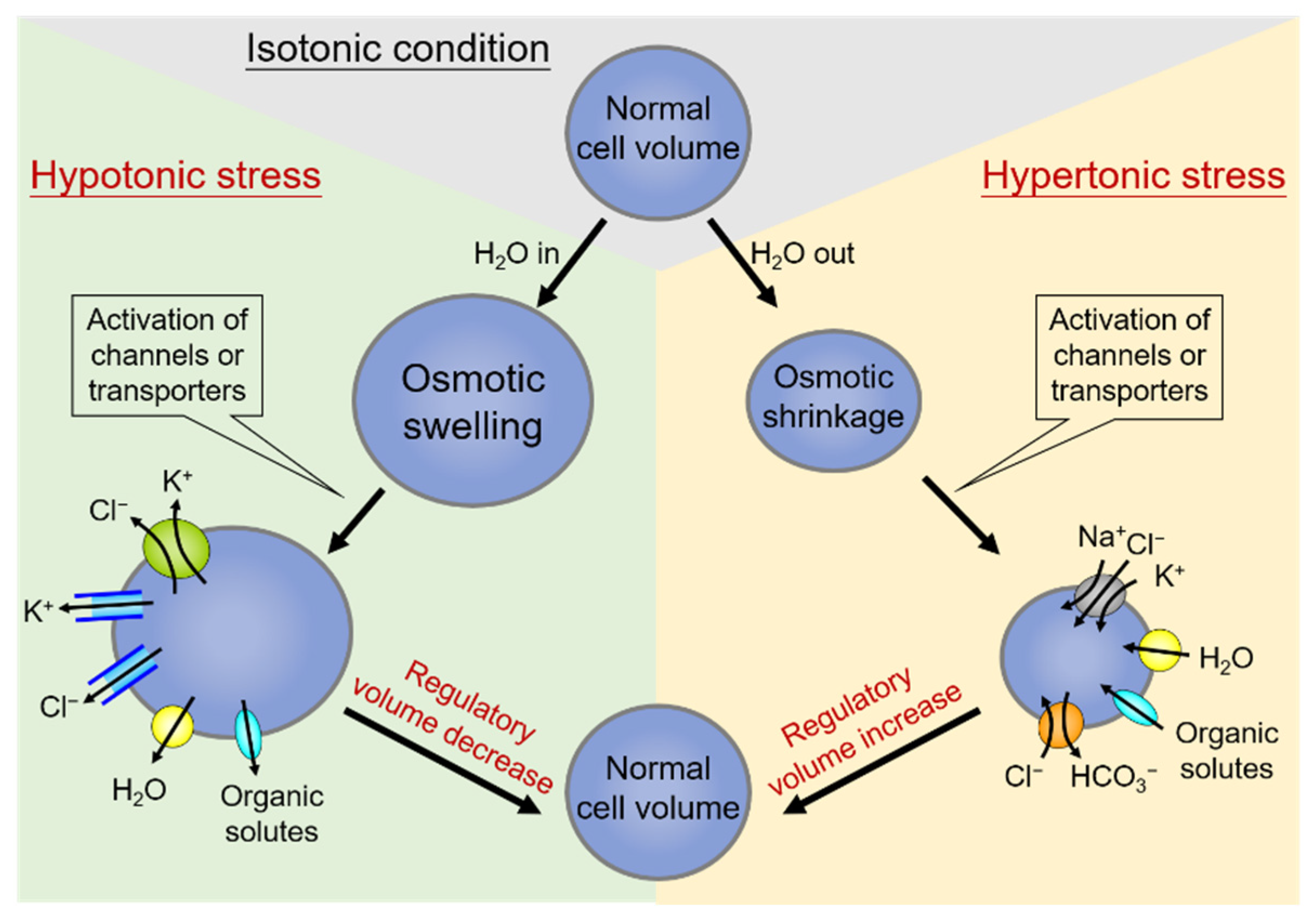
2. Cell Volume Regulation and Volume-Sensitive Cl− Channels
2.1. Volume-Sensitive Cl− Channels Associated with Human Cervical Carcinogenesis
2.2. Differential Osmosensing Signaling Pathways of Volume-Sensitive Cl− Channels Associated with Human Cervical Carcinogenesis
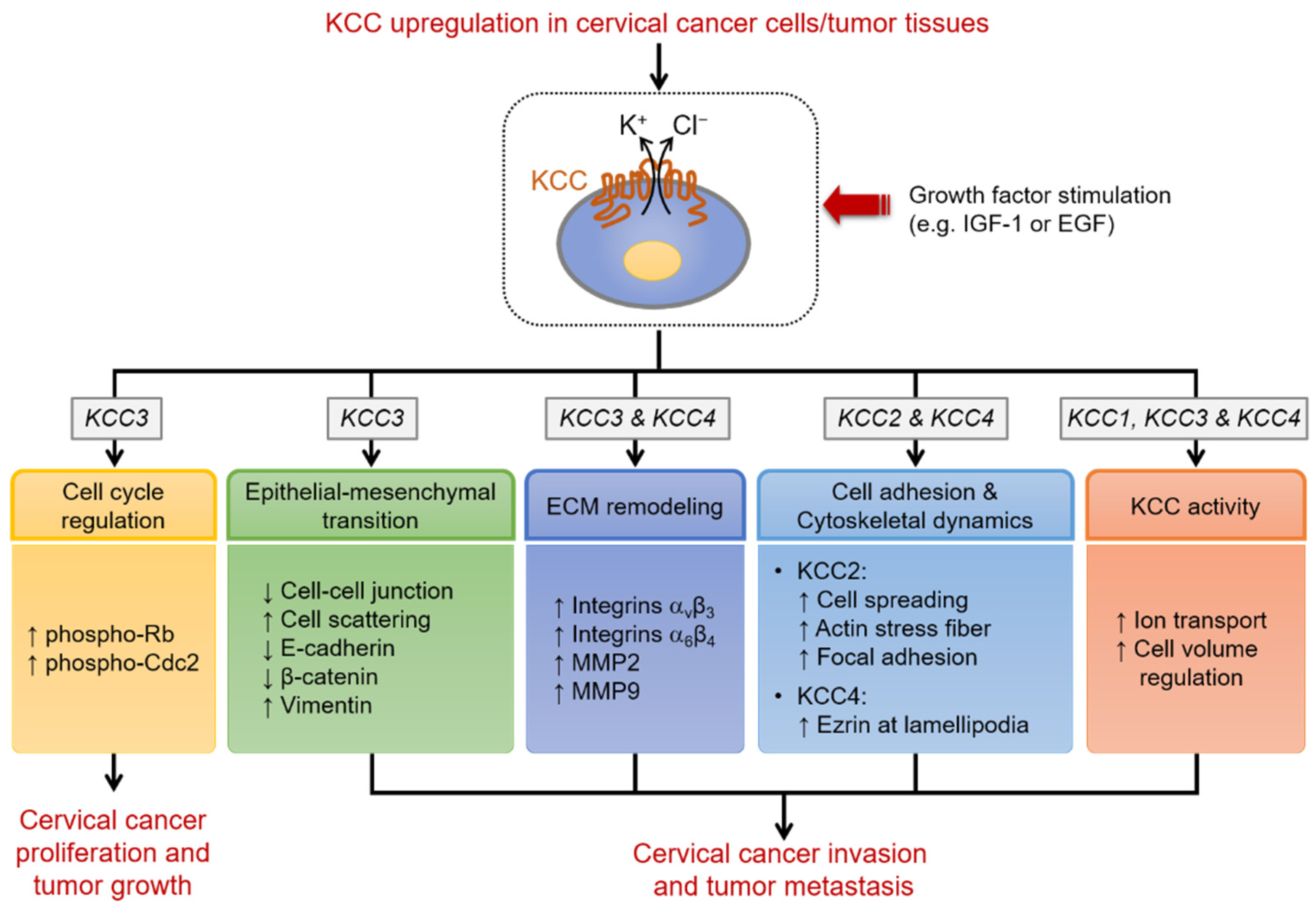
3. K+-Cl− Cotransport and K+-Cl− Cotransporter (KCC) Family
3.1. K+-Cl− Cotransport Activity Affects Malignant Transformation, Proliferation, and Invasion of Cervical Epithelial Cells
3.2. The Distinct Roles of KCC Isoforms in Cervical Carcinogenesis
3.3. Clinical Implications and Therapeutic Significance of KCC in Cervical Carcinogenesis
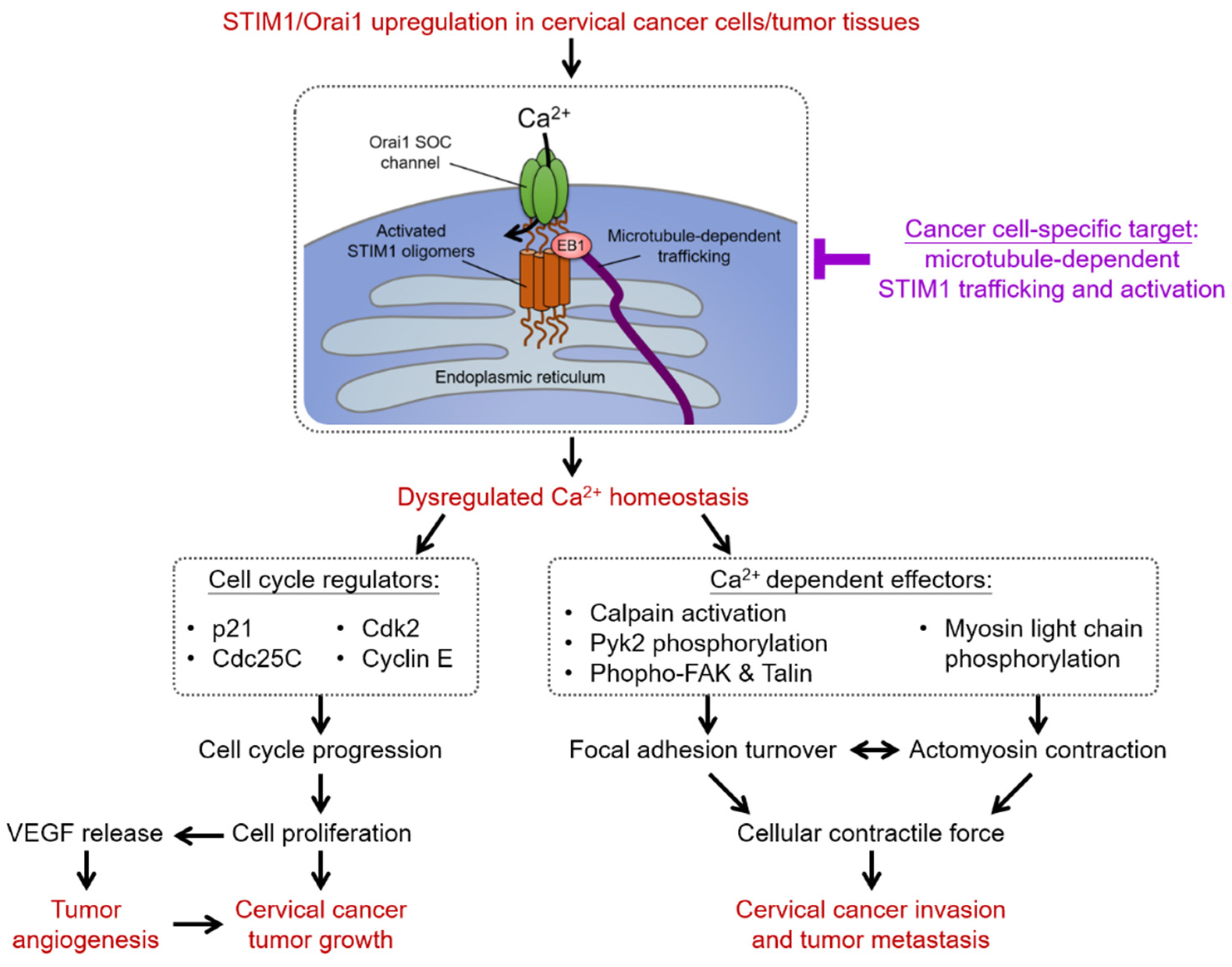
4. Intracellular Ca2+ Homeostasis and Store-Operated Ca2+ Entry (SOCE)
4.1. SOCE-Dependent Ca2+ Signaling Network in Cervical Carcinogenesis
4.1.1. Proliferation and Cell Cycle Regulation
4.1.2. Tumor Angiogenesis
4.1.3. Cell Migration
4.2. Diagnostic and Prognostic Values of SOCE in Cervical Carcinogenesis
4.3. Recent Development of Therapeutics Targeting SOCE in Cervical Carcinogenesis
5. Conclusions and Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef]
- Vu, M.; Yu, J.; Awolude, O.A.; Chuang, L. Cervical cancer worldwide. Curr. Probl. Cancer 2018, 42, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.D.; Balakrishnan, V.; Oon, C.E.; Kaur, G. Key Molecular Events in Cervical Cancer Development. Medicina 2019, 55, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadducci, A.; Guerrieri, M.E.; Greco, C. Tissue biomarkers as prognostic variables of cervical cancer. Crit. Rev. Oncol. Hematol. 2013, 86, 104–129. [Google Scholar] [CrossRef]
- Muñoz, N.; Bosch, F.X. The causal link between HPV and cervical cancer and its implications for prevention of cervical cancer. Bull. Pan Am. Health Organ. 1996, 30, 362–377. [Google Scholar] [PubMed]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Muoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J.L.M.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshmi, G.; Pillai, M.R. Beyond HPV: Oncomirs as new players in cervical cancer. FEBS Lett. 2008, 582, 4113–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łaniewski, P.; Cui, H.; Roe, D.J.; Barnes, D.; Goulder, A.; Monk, B.J.; Greenspan, D.L.; Chase, D.M.; Herbst-Kralovetz, M.M. Features of the cervicovaginal microenvironment drive cancer biomarker signatures in patients across cervical carcinogenesis. Sci. Rep. 2019, 9, 7333. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodat-Despoix, L.; Chamlali, M.; Ouadid-Ahidouch, H. Ion channels as key partners of cytoskeleton in cancer disease. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188627. [Google Scholar] [CrossRef]
- Stock, C. How Dysregulated Ion Channels and Transporters Take a Hand in Esophageal, Liver, and Colorectal Cancer. Rev. Physiol. Biochem. Pharmacol. 2020, 1–94. [Google Scholar] [CrossRef]
- Rashid, K.; Ahmad, A.; Liang, L.; Liu, M.; Cui, Y.; Liu, T. Solute carriers as potential oncodrivers or suppressors: Their key functions in malignant tumor formation. Drug Discov. Today 2021, 26, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Bentz, S.; Hediger, M.A.; Anderle, P. Solute carriers (SLCs) in cancer. Mol. Asp. Med. 2013, 34, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Fredriksson, R.; Schiöth, H.B. Solute carriers as drug targets: Current use, clinical trials and prospective. Mol. Asp. Med. 2013, 34, 702–710. [Google Scholar] [CrossRef]
- Davies, K.J. Adaptive homeostasis. Mol. Aspects Med. 2016, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Okada, T.; Sato-Numata, K.; Islam, M.R.; Ando-Akatsuka, Y.; Numata, T.; Kubo, M.; Shimizu, T.; Kurbannazarova, R.S.; Marunaka, Y.; et al. Cell Volume-Activated and Volume-Correlated Anion Channels in Mammalian Cells: Their Biophysical, Molecular, and Pharmacological Properties. Pharmacol. Rev. 2019, 71, 49–88. [Google Scholar] [CrossRef] [Green Version]
- Lambert, I.H.; Hoffmann, E.K.; Pedersen, S.F. Cell volume regulation: Physiology and pathophysiology. Acta Physiol. 2008, 194, 255–282. [Google Scholar] [CrossRef]
- Guo, M.; Pegoraro, A.F.; Mao, A.; Zhou, E.H.; Arany, P.R.; Han, Y.; Burnette, D.T.; Jensen, M.H.; Kasza, K.E.; Moore, J.R.; et al. Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc. Natl. Acad. Sci. USA 2017, 114, E8618–E8627. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Häussinger, D.; Lang, F. Cell volume and hormone action. Trends Pharmacol. Sci. 1992, 13, 371–373. [Google Scholar] [CrossRef]
- Häussinger, D. The role of cellular hydration in the regulation of cell function. Biochem. J. 1996, 313 Pt 3, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Pasantes-Morales, H. Channels and Volume Changes in the Life and Death of the Cell. Mol. Pharmacol. 2016, 90, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Morishita, K.; Watanabe, K.; Ichijo, H. Cell volume regulation in cancer cell migration driven by osmotic water flow. Cancer Sci. 2019, 110, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yang, Y.; Han, L.; Han, S.; Liu, N.; Xu, F.; Li, F. Effect of three-dimensional ECM stiffness on cancer cell migration through regulating cell volume homeostasis. Biochem. Biophys. Res. Commun. 2020, 528, 459–465. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Jentsch, T. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef]
- Delpire, E.; Gagnon, K.B. Water Homeostasis and Cell Volume Maintenance and Regulation. Curr. Top Membr. 2018, 81, 3–52. [Google Scholar] [CrossRef]
- Okada, Y. Ion Channels and Transporters Involved in Cell Volume Regulation and Sensor Mechanisms. Cell Biochem. Biophys. 2004, 41, 233–258. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Pedersen, S.F. Cell volume homeostatic mechanisms: Effectors and signalling pathways. Acta Physiol. 2010, 202, 465–485. [Google Scholar] [CrossRef]
- Furst, J.; Gschwentner, M.; Ritter, M.; Botta, G.; Jakab, M.; Mayer, M.; Garavaglia, L.; Bazzini, C.; Rodighiero, S.; Meyer, G.; et al. Molecular and functional aspects of anionic channels activated during regulatory volume decrease in mammalian cells. Pflug. Arch. 2002, 444, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.; Rauh, O.; Fauth, T.; Buerger, C. Cell Volume Regulation in the Epidermis. Cell. Physiol. Biochem. 2021, 55, 57–70. [Google Scholar] [CrossRef]
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabo, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichl, M.; et al. O-GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to alpha-Integrin. Front Cell Dev. Biol. 2020, 8, 607080. [Google Scholar] [CrossRef]
- Bertelli, S.; Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Picco, C.; Gavazzo, P.; Pusch, M. Mechanisms of Activation of LRRC8 Volume Regulated Anion Channels. Cell. Physiol. Biochem. 2021, 55, 41–56. [Google Scholar] [CrossRef]
- Rust, M.B.; Alper, S.L.; Rudhard, Y.; Shmukler, B.E.; Vicente, R.; Brugnara, C.; Trudel, M.; Jentsch, T.J.; Hübner, C.A. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J. Clin. Investig. 2007, 117, 1708–1717. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflug. Arch. 2019, 471, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Ellory, J.C.; Gibson, J.S.; Stewart, G.W. Pathophysiology of Abnormal Cell Volume in Human Red Cells. Contrib. Nephrol. 1998, 123, 220–239. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Lauf, P.K.; Adragna, N.C. K-Cl Cotransport: Properties and Molecular Mechanism. Cell. Physiol. Biochem. 2000, 10, 341–354. [Google Scholar] [CrossRef]
- Wehner, F.; Tinel, H. Role of Na+ conductance, Na(+)-H+ exchange, and Na(+)-K(+)-2Cl- symport in the regulatory volume increase of rat hepatocytes. J. Physiol. 1998, 506 Pt 1, 127–142. [Google Scholar] [CrossRef]
- Alexander, R.T.; Grinstein, S. Na+/H+ exchangers and the regulation of volume. Acta Physiol. 2006, 187, 159–167. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-Potassium-Chloride Cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Counillon, L. The SLC9A-C Mammalian Na(+)/H(+) Exchanger Family: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Sørensen, B.H.; Sauter, D.P.R.; Lambert, I.H. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels 2015, 9, 380–396. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 587, 2141–2149. [Google Scholar]
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and Physiology of the Volume-Regulated Anion Channel (VRAC)/Volume-Sensitive Outwardly Rectifying Anion Channel (VSOR). Pflug. Arch. 2016, 468, 371–383. [Google Scholar] [CrossRef]
- Stutzin, A.; Hoffmann, E.K. Swelling-activated ion channels: Functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol. 2006, 187, 27–42. [Google Scholar] [CrossRef]
- Tseng, G.N. Cell swelling increases membrane conductance of canine cardiac cells: Evidence for a volume-sensitive Cl channel. Am. J. Physiol. 1992, 262, C1056–C1068. [Google Scholar] [CrossRef]
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive Chloride Channels Involved in Apoptotic Volume Decrease and Cell Death. J. Membr. Biol. 2006, 209, 21–29. [Google Scholar] [CrossRef]
- Chou, C.Y.; Shen, M.R.; Wu, S.N. Volume-sensitive chloride channels associated with human cervical carcinogenesis. Cancer Res. 1995, 55, 6077–6083. [Google Scholar]
- Shen, M.-R.; Wu, S.-N.; Chou, C.-Y. Volume-sensitive chloride channels in the primary culture cells of human cervical carcinoma. Biochim. Biophys. Acta 1996, 1315, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Tzur, A.; Kafri, R.; LeBleu, V.S.; Lahav, G.; Kirschner, M.W. Cell Growth and Size Homeostasis in Proliferating Animal Cells. Science 2009, 325, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.C. The regulation of cell size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Bryan, A.K.; Goranov, A.; Amon, A.; Manalis, S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Cikes, M. Relationship Between Growth Rate, Cell Volume, Cell Cycle Kinetics, and Antigenic Properties of Cultured Murine Lymphoma Cells23. J. Natl. Cancer Inst. 1970, 45, 978–988. [Google Scholar] [CrossRef]
- Lang, F.; Föller, M.; Lang, K.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion Channels in Cell Proliferation and Apoptotic Cell Death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Shen, M.R.; Droogmans, G.; Eggermont, J.; Voets, T.; Ellory, J.C.; Nilius, B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 2000, 529 Pt 2, 385–394. [Google Scholar] [CrossRef]
- Schumacher, P.A.; Sakellaropoulos, G.; Phipps, D.J.; Schlichter, L.C. Small-conductance chloride channels in human peripheral T lymphocytes. J. Membr. Biol. 1995, 145, 217–232. [Google Scholar] [CrossRef]
- Voets, T.; Szucs, G.; Droogmans, G.; Nilius, B. Blockers of volume-activated Cl? currents inhibit endothelial cell proliferation. Pflug. Arch. 1995, 431, 132–134. [Google Scholar] [CrossRef]
- Schlichter, L.C.; Sakellaropoulos, G.; Ballyk, B.; Pennefather, P.S.; Phipps, D.; Schlichter, L.; Sakellaropoulos, G.; Ballyk, B.; Pennefather, P.; Phipps, D. Properties of K+ and Cl–? channels and their involvement in proliferation of rat microglial cells. Glia 1996, 17, 225–236. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Shen, M.-R.; Chen, T.-M.; Huang, K.-E. Volume-activated taurine transport is differentially activated in human cervical cancer ht-3 cells but not in human papillomavirus-immortalized z183a and normal cervical epithelial cells. Clin. Exp. Pharmacol. Physiol. 1997, 24, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Pasantes-Morales, H.; Cardin, V.; Tuz, K. Signaling events during swelling and regulatory volume decrease. Neurochem. Res. 2000, 25, 1301–1314. [Google Scholar] [CrossRef]
- Vázquez-Juárez, E.; Ramos-Mandujano, G.; Hernández-Benítez, R.; Pasantes-Morales, H. On the Role of G-Protein Coupled Receptors in Cell Volume Regulation. Cell. Physiol. Biochem. 2008, 21, 001–014. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Panayiotidis, M.I.; de la Paz, L.D. Autocrine signaling involved in cell volume regulation: The role of released transmitters and plasma membrane receptors. J. Cell. Physiol. 2008, 216, 14–28. [Google Scholar] [CrossRef] [PubMed]
- van der Wijk, T.; Tomassen, S.F.; de Jonge, H.R.; Tilly, B.C. Signalling mechanisms involved in volume regulation of intestinal epithelial cells. Cell Physiol. Biochem. 2000, 10, 289–296. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Shen, M.-R.; Hsu, K.-S.; Huang, H.-Y.; Lin, H.-C. Involvement of PKC-α in regulatory volume decrease responses and activation of volume-sensitive chloride channels in human cervical cancer HT-3 cells. J. Physiol. 1998, 512 Pt 2, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzochi, C.; Benos, D.J.; Smith, P.R. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am. J. Physiol. Renal. Physiol. 2006, 291, F1113–F1122. [Google Scholar] [CrossRef] [Green Version]
- Di Ciano-Oliveira, C.; Thirone, A.C.; Szaszi, K.; Kapus, A. Osmotic stress and the cytoskeleton: The R(h)ole of Rho GTPases. Acta Physiol. 2006, 187, 257–272. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Hoffmann, E.K.; Mills, J.W. The cytoskeleton and cell volume regulation. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 385–399. [Google Scholar] [CrossRef]
- Shen, M.R.; Chou, C.Y.; Hsu, K.F.; Hsu, K.S.; Wu, M.L. Modulation of volume-sensitive Cl -channels and cell volume by actin filaments and microtubules in human cervical cancer HT-3 cells. Acta Physiol. Scand. 1999, 167, 215–225. [Google Scholar] [CrossRef]
- Ando-Akatsuka, Y.; Shimizu, T.; Numata, T.; Okada, Y. Involvements of the ABC protein ABCF2 and α-actinin-4 in regulation of cell volume and anion channels in human epithelial cells. J. Cell. Physiol. 2012, 227, 3498–3510. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Almonte, C.; Mollard, P.; Garber, S.S. Modulation of a volume-regulated chloride current by F-actin. J. Membr. Biol. 1995, 147, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Larsen, T.H.; Lieberman, M. F-actin modulates swelling-activated chloride current in cultured chick cardiac myocytes. Am. J. Physiol. 1997, 273, C1215–C1224. [Google Scholar] [CrossRef]
- Shimizu, T.; Fujii, T.; Ohtake, H.; Tomii, T.; Takahashi, R.; Kawashima, K.; Sakai, H. Impaired actin filaments decrease cisplatin sensitivity via dysfunction of volume-sensitive Cl—Channels in human epidermoid carcinoma cells. J. Cell. Physiol. 2020, 235, 9589–9600. [Google Scholar] [CrossRef] [PubMed]
- Burow, P.; Klapperstück, M.; Markwardt, F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflug. Arch. 2015, 467, 1215–1226. [Google Scholar] [CrossRef]
- Wei, H.; Mei, Y.A.; Sun, J.T.; Zhou, H.Q.; Zhang, Z.H. Regulation of swelling-activated chloride channels in embryonic chick heart cells. Cell Res. 2003, 13, 21–28. [Google Scholar] [CrossRef]
- Xu, R.; Wang, X.; Shi, C. Volume-regulated anion channel as a novel cancer therapeutic target. Int. J. Biol. Macromol. 2020, 159, 570–576. [Google Scholar] [CrossRef]
- Lauf, P.K.; Theg, B.E. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem. Biophys. Res. Commun. 1980, 92, 1422–1428. [Google Scholar] [CrossRef]
- Dunham, P.B.; Stewart, G.W.; Ellory, J.C. Chloride-activated passive potassium transport in human erythrocytes. Proc. Natl. Acad. Sci. USA 1980, 77, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adragna, N.C.; Ferrell, C.M.; Zhang, J.; Di Fulvio, M.; Temprana, C.F.; Sharma, A.; Fyffe, R.E.; Cool, D.R.; Lauf, P.K. Signal transduction mechanisms of K+-Cl- cotransport regulation and relationship to disease. Acta Physiol. 2006, 187, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Conway, L.C.; Cardarelli, R.A.; Moore, Y.E.; Jones, K.; McWilliams, L.J.; Baker, D.J.; Burnham, M.P.; Bürli, R.W.; Wang, Q.; Brandon, N.J.; et al. N-Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem. 2017, 292, 21253–21263. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784. [Google Scholar] [CrossRef] [Green Version]
- Melo, Z.; Heros, P.D.L.; Cruz-Rangel, S.; Vázquez, N.; Bobadilla, N.A.; Pasantes-Morales, H.; Alessi, D.R.; Mercado, A.; Gamba, G. N-terminal Serine Dephosphorylation Is Required for KCC3 Cotransporter Full Activation by Cell Swelling. J. Biol. Chem. 2013, 288, 31468–31476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adragna, N.C.; Ravilla, N.B.; Lauf, P.K.; Begum, G.; Khanna, A.R.; Sun, D.; Kahle, K.T. Regulated phosphorylation of the K-Cl cotransporter KCC3 is a molecular switch of intracellular potassium content and cell volume homeostasis. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, C.A.; Barros, P.; Matos, P.; Jordan, P. Tyrosine phosphorylation modulates cell surface expression of chloride cotransporters NKCC2 and KCC3. Arch. Biochem. Biophys. 2019, 669, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Wake, H.; Moorhouse, A.J.; Nabekura, J. Clustering of Neuronal K+-Cl− Cotransporters in Lipid Rafts by Tyrosine Phosphorylation. J. Biol. Chem. 2009, 284, 27980–27988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strange, K.; Singer, T.D.; Morrison, R.; Delpire, E. Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am. J. Physiol. Cell Physiol. 2000, 279, C860–C867. [Google Scholar] [CrossRef]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early Changes in KCC2 Phosphorylation in Response to Neuronal Stress Result in Functional Downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [Green Version]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M.; et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-De-Ozores, A.R.; Chávez-Canales, M.; Heros, P.D.L.; Gamba, G.; Castañeda-Bueno, M. Physiological Processes Modulated by the Chloride-Sensitive WNK-SPAK/OSR1 Kinase Signaling Pathway and the Cation-Coupled Chloride Cotransporters. Front. Physiol. 2020, 11, 585907. [Google Scholar] [CrossRef]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdörfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef] [PubMed]
- Frenette-Cotton, R.; Marcoux, A.-A.; Garneau, A.P.; Noel, M.; Isenring, P. Phosphoregulation of K+-Cl−cotransporters during cell swelling: Novel insights. J. Cell. Physiol. 2018, 233, 396–408. [Google Scholar] [CrossRef]
- Bize, I.; Güvenç, B.; Buchbinder, G.; Brugnara, C. Stimulation of Human Erythrocyte K-Cl Cotransport and Protein Phosphatase Type 2A by n-Ethylmaleimide: Role of Intracellular Mg++. J. Membr. Biol. 2000, 177, 159–168. [Google Scholar] [CrossRef]
- Hebert, S.C.; Mount, D.B.; Gamba, G. Molecular physiology of cation-coupled Cl? cotransport: The SLC12 family. Pflüg. Arch. 2004, 447, 580–593. [Google Scholar] [CrossRef]
- Gamba, G. Molecular Physiology and Pathophysiology of Electroneutral Cation-Chloride Cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [PubMed]
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, B., 3rd. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 1996, 271, 16237–16244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.A.; Stevenson, T.J.; Donaldson, L.F. Molecular Characterization of a Putative K-Cl Cotransporter in Rat Brain. A neuronal-specific isoform. J. Biol. Chem. 1996, 271, 16245–16252. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Mercado, A.; Vázquez, N.; Xie, Q.; Desai, R.; George, A.L.; Gamba, G.; Mount, D.B. Molecular, functional, and genomic characterization of human KCC2, the neuronal K–Cl cotransporter. Mol. Brain Res. 2002, 103, 91–105. [Google Scholar] [CrossRef]
- Race, J.E.; Makhlouf, F.N.; Logue, P.J.; Wilson, F.H.; Dunham, P.B.; Holtzman, E.J. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am. J. Physiol. 1999, 277, C1210–C1219. [Google Scholar] [CrossRef] [PubMed]
- Hiki, K.; D’Andrea, R.J.; Furze, J.; Crawford, J.; Woollatt, E.; Sutherland, G.R.; Vadas, M.A.; Gamble, J.R. Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J. Biol. Chem. 1999, 274, 10661–10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.A. Molecular operation of the cation chloride cotransporters: Ion binding and inhibitor interaction. Curr. Top Membr. 2012, 70, 215–237. [Google Scholar] [PubMed]
- Hartmann, A.-M.; Nothwang, H.G. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front. Cell. Neurosci. 2014, 8, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Chang, S.; Zhao, C.; Wang, F.; Liu, S.; Wang, J.; Delpire, E.; Ye, S.; Guo, J. Structures and an activation mechanism of human potassium-chloride cotransporters. Sci. Adv. 2020, 6, eabc5883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Zhang, Y.; Liu, T.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.; Liu, Y.; et al. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Li, X.; Chen, Y.; Zhang, Y.; Su, Q.; Zhou, Q. Cryo-EM structures of the full-length human KCC2 and KCC3 cation-chloride cotransporters. Cell Res. 2021, 31, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.S.; Kern, D.M.; Brohawn, S.G. Cryo-EM structure of the potassium-chloride cotransporter KCC4 in lipid nanodiscs. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chang, S.; Han, B.; Xu, L.; Zhang, M.; Zhao, C.; Yang, W.; Wang, F.; Li, J.; Delpire, E.; et al. Cryo-EM structures of the human cation-chloride cotransporter KCC1. Science 2019, 366, 505–508. [Google Scholar] [CrossRef]
- Garneau, A.P.; Slimani, S.; Tremblay, L.E.; Fiola, M.J.; Marcoux, A.A.; Isenring, P. K+-Cl− cotransporter 1 (KCC1): A housekeeping membrane protein that plays key supplemental roles in hematopoietic and cancer cells. J. Hematol. Oncol. 2019, 12, 74. [Google Scholar] [CrossRef]
- Garneau, A.P.; Marcoux, A.A.; Frenette-Cotton, R.; Mac-Way, F.; Lavoie, J.L.; Isenring, P. Molecular insights into the normal operation, regulation, and multisystemic roles of K(+)-Cl(-) cotransporter 3 (KCC3). Am. J. Physiol. Cell Physiol. 2017, 313, C516–C532. [Google Scholar] [CrossRef]
- Marcoux, A.; Garneau, A.; Frenette-Cotton, R.; Slimani, S.; Mac-Way, F.; Isenring, P. Molecular features and physiological roles of K+-Cl− cotransporter 4 (KCC4). Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 3154–3166. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. The Expanding Therapeutic Potential of Neuronal KCC2. Cells 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Côme, E.; Heubl, M.; Schwartz, E.J.; Poncer, J.C.; Lévi, S. Reciprocal Regulation of KCC2 Trafficking and Synaptic Activity. Front. Cell. Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef]
- Howard, H.C.; Mount, D.B.; Rochefort, D.; Byun, N.; Dupré, N.; Lu, J.; Fan, X.; Song, L.; Rivière, J.-B.; Prévost, C.; et al. The K–Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 2002, 32, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Boettger, T.; Rust, M.B.; Maier, H.; Seidenbecher, T.; Schweizer, M.; Keating, D.J.; Faulhaber, J.; Ehmke, H.; Pfeffer, C.; Scheel, O.; et al. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003, 22, 5422–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-T.; Tzeng, S.-F.; Lin, T.-S.; Hsu, K.-S.; Delpire, E.; Shen, M.-R. KCC3 deficiency-induced disruption of paranodal loops and impairment of axonal excitability in the peripheral nervous system. Neuroscience 2016, 335, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.B.; Faulhaber, J.; Budack, M.K.; Pfeffer, C.; Maritzen, T.; Didié, M.; Beck, F.-X.; Boettger, T.; Schubert, R.; Ehmke, H.; et al. Neurogenic Mechanisms Contribute to Hypertension in Mice With Disruption of the K-Cl Cotransporter KCC3. Circ. Res. 2006, 98, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.-R.; Chou, C.-Y.; Hsu, K.-F.; Liu, H.-S.; Dunham, P.B.; Holtzman, E.J.; Ellory, J.C. The KCl cotransporter isoform KCC3 can play an important role in cell growth regulation. Proc. Natl. Acad. Sci. USA 2001, 98, 14714–14719. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.C.; Furey, C.G.; Zeng, X.; Allocco, A.; Nelson-Williams, C.; Dong, W.; Karimy, J.K.; Wang, K.; Ma, S.; Delpire, E.; et al. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Mol. Genet. Genom. Med. 2019, 7, e892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettger, T.; Hübner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Portioli, C.; Munevar, M.J.R.; De Vivo, M.; Cancedda, L. Cation-coupled chloride cotransporters: Chemical insights and disease implications. Trends Chem. 2021, 3, 832–849. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.R.; Chou, C.Y.; Ellory, J.C. Volume-sensitive KCI cotransport associated with human cervical carcinogenesis. Pflug. Arch. 2000, 440, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-R.; Chou, C.-Y.; Hsu, K.-F.; Hsu, Y.-M.; Chiu, W.-T.; Tang, M.-J.; Alper, S.L.; Ellory, J.C. KCl Cotransport Is an Important Modulator of Human Cervical Cancer Growth and Invasion. J. Biol. Chem. 2003, 278, 39941–39950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Marshall, J.F.; Hart, I.R. Integrin trafficking and its role in cancer metastasis. Cancer Metastasis Rev. 2007, 26, 567–578. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, Insulin, and Cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.-R.; Lin, A.-C.; Hsu, Y.-M.; Chang, T.-J.; Tang, M.-J.; Alper, S.L.; Ellory, J.C.; Chou, C.-Y. Insulin-like Growth Factor 1 Stimulates KCl Cotransport, Which Is Necessary for Invasion and Proliferation of Cervical Cancer and Ovarian Cancer Cells. J. Biol. Chem. 2004, 279, 40017–40025. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-M.; Chou, C.-Y.; Chen, H.H.; Lee, W.-Y.; Chen, Y.-F.; Lin, P.-W.; Alper, S.L.; Ellory, J.C.; Shen, M.-R. IGF-1 upregulates electroneutral K-Cl cotransporter KCC3 and KCC4 which are differentially required for breast cancer cell proliferation and invasiveness. J. Cell. Physiol. 2006, 210, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.M.; Chen, Y.F.; Chou, C.Y.; Tang, M.J.; Chen, J.H.; Wilkins, R.J.; Ellory, J.C.; Shen, M.R. KCl cotransporter-3 down-regulates E-cadherin/beta-catenin complex to promote epithelial-mesenchymal transition. Cancer Res. 2007, 67, 11064–11073. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Chou, C.-Y.; Wilkins, R.J.; Ellory, J.C.; Mount, D.B.; Shen, M.-R. Motor Protein-Dependent Membrane Trafficking of KCl Cotransporter-4 Is Important for Cancer Cell Invasion. Cancer Res. 2009, 69, 8585–8593. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Shen, M.-R. Epithelial-mesenchymal transition in cervical carcinoma. Am. J. Transl. Res. 2012, 4, 1–13. [Google Scholar] [PubMed]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.R.; Hsu, Y.M.; Hsu, K.F.; Chen, Y.F.; Tang, M.J.; Chou, C.Y. Insulin-like growth factor 1 is a potent stimulator of cervical cancer cell invasiveness and proliferation that is modulated by alphavbeta3 integrin signaling. Carcinogenesis 2006, 27, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.-C.; Akerman, C.J.; Newey, S.E.; Pan, J.; Clinch, N.W.; Jacob, Y.; Shen, M.-R.; Wilkins, R.J.; Ellory, J.C. The potassium-chloride cotransporter 2 promotes cervical cancer cell migration and invasion by an ion transport-independent mechanism. J. Physiol. 2011, 589, 5349–5359. [Google Scholar] [CrossRef]
- Zdebik, A.A. Beyond ion transport: KCC2 makes cells walk and talk. J. Physiol. 2011, 589, 5903. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I. Solute Carrier Transporters as Targets for Drug Delivery and Pharmacological Intervention for Chemotherapy. J. Pharm. Sci. 2011, 100, 3731–3750. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.-H.; Liu, H.-S.; Wu, Y.-H.; Shen, M.-R.; Chou, C.-Y. SPAK mediates KCC3-enhanced cervical cancer tumorigenesis. FEBS J. 2014, 281, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2016, 1864, 900–906. [Google Scholar] [CrossRef]
- Bakowski, D.; Murray, F.; Parekh, A.B. Store-Operated Ca(2+) Channels: Mechanism, Function, Pharmacology, and Therapeutic Targets. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 629–654. [Google Scholar] [CrossRef]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2020, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.; Gouveia, S.; Van Der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.; Putney, J.; Hoogenraad, C.; et al. STIM1 Is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.; Buey, R.M.; Lawera, A.; et al. An EB1-Binding Motif Acts as a Microtubule Tip Localization Signal. Cell 2009, 138, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chen, L.-H.; Shen, M.-R. The distinct role of STIM1 and STIM2 in the regulation of store-operated Ca2+ entry and cellular function. J. Cell. Physiol. 2019, 234, 8727–8739. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Chen, Y.-F.; Chiu, W.-T.; Liu, K.-Y.; Liu, Y.-L.; Chang, J.-Y.; Chang, H.-C.; Shen, M.-R. Microtubule-Associated Histone Deacetylase 6 Supports the Calcium Store Sensor STIM1 in Mediating Malignant Cell Behaviors. Cancer Res. 2013, 73, 4500–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.-C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-T.; Hsu, Y.-T.; Chen, Y.-F.; Shen, M.-R. Super-Resolution Microscopy Reveals That Stromal Interaction Molecule 1 Trafficking Depends on Microtubule Dynamics. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef]
- Yeung, P.S.; Yamashita, M.; Prakriya, M. Molecular basis of allosteric Orai1 channel activation by STIM1. J. Physiol. 2020, 598, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [Green Version]
- Qiu, R.; Lewis, R.S. Structural features of STIM and Orai underlying store-operated calcium entry. Curr. Opin. Cell Biol. 2019, 57, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Hogan, P.G. STIM calcium sensing and conformational change. J. Physiol. 2020, 598, 1695–1705. [Google Scholar] [CrossRef]
- Trebak, M.; Putney, J.W., Jr. ORAI Calcium Channels. Physiology 2017, 32, 332–342. [Google Scholar] [CrossRef]
- Prakriya, M.; Yeung, P.S.; Yamashita, M. An open pore structure of the Orai channel, finally. Cell Calcium 2021, 94, 102366. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Borgström, A.; Stokłosa, P.; Dörr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Woodard, G.E.; Rosado, J.A. Orais and STIMs: Physiological mechanisms and disease. J. Cell. Mol. Med. 2012, 16, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channelopathies. Pflug. Arch. 2010, 460, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Lin, P.-C.; Yeh, Y.-M.; Chen, L.-H.; Shen, M.-R. Store-Operated Ca2+ Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Hsu, K.-F.; Shen, M.-R. The store-operated Ca2+ entry-mediated signaling is important for cancer spread. Biochim. Biophys. Acta. 2016, 1863, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 2013, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Hussain, Y.; Luqman, S.; Meena, A. Targeting Ca2+ signalling through phytomolecules to combat cancer. Pharmacol. Res. 2019, 146, 104282. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Deng, Y.; Ye, J.; Luo, Y.; Weng, J.; He, Q.; Liu, F.; Li, M.; Liang, R.; Lin, Y.; et al. Store-operated Ca2+ entry as a key oncogenic Ca2+ signaling driving tumor invasion-metastasis cascade and its translational potential. Cancer Lett. 2021, 516, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, A.; Zhang, Y.; Parrington, J. Deciphering the Role of Ca2+ Signalling in Cancer Metastasis: From the Bench to the Bedside. Cancers 2021, 13, 179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Chiu, W.T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Lindemann, O.; Schwab, A. TRP channels and STIM/ORAI proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Li, Y.; Lin, S.; Cheng, H.; Yang, S. Spatiotemporal regulation of store-operated calcium entry in cancer metastasis. Biochem. Soc. Trans. 2021, 49, 2581–2589. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Chen, Y.-F.; Chiu, W.-T.; Wang, Y.-K.; Chang, H.-C.; Shen, M.-R. The ER Ca2+ sensor STIM1 regulates actomyosin contractility of migratory cells. J. Cell Sci. 2013, 126, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-W.; Lai, C.-S.; Chen, Y.-F.; Chiu, W.-T.; Chen, H.-C.; Shen, M.-R. STIM1-dependent Ca2+ signaling regulates podosome formation to facilitate cancer cell invasion. Sci. Rep. 2017, 7, 11523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Zhang, N.; Pan, H.; Xie, J.; Han, W. Development of Store-Operated Calcium Entry-Targeted Compounds in Cancer. Front. Pharmacol. 2021, 12, 1349. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Roy, S.; Pan, Z. Store-Operated Calcium Channels as Drug Target in Gastroesophageal Cancers. Front. Pharmacol. 2021, 12, 944. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, A.; Derler, I. Molecular Choreography and Structure of Ca(2+) Release-Activated Ca(2+) (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Mallu, P.; Joshi, H.; Schultz, B.; Go, C.; Soboloff, J. Ca2+ as a therapeutic target in cancer. Adv. Cancer Res. 2020, 148, 233–317. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.I.E.; James, A.D. Targeting the Calcium Signalling Machinery in Cancer. Cancers 2020, 12, 2351. [Google Scholar] [CrossRef] [PubMed]
- Malliri, A.; Caswell, P.; Ballestrem, C.; Hurlstone, A.; Garcin, C.; Straube, A. Microtubules in cell migration. Essays Biochem. 2019, 63, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Gadadhar, S.; Bodakuntla, S.; Natarajan, K.; Janke, C. The tubulin code at a glance. J. Cell Sci. 2017, 130, 1347–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2020, 163, 105274. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-H.; Chen, S.-J.; Shen, M.-R.; Huang, Y.-T.; Hsieh, H.-P.; Lin, S.-Y.; Lin, C.-C.; Chang, W.W.; Chang, J.-Y. Lysosomal cysteine protease cathepsin S is involved in cancer cell motility by regulating store-operated Ca2+ entry. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2019, 1866, 118517. [Google Scholar] [CrossRef]
- McDowell, S.H.; Gallaher, S.A.; Burden, R.E.; Scott, C.J. Leading the invasion: The role of Cathepsin S in the tumour microenvironment. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118781. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-F.; Shen, M.-R. The Important Role of Ion Transport System in Cervical Cancer. Int. J. Mol. Sci. 2022, 23, 333. https://doi.org/10.3390/ijms23010333
Chen Y-F, Shen M-R. The Important Role of Ion Transport System in Cervical Cancer. International Journal of Molecular Sciences. 2022; 23(1):333. https://doi.org/10.3390/ijms23010333
Chicago/Turabian StyleChen, Yih-Fung, and Meng-Ru Shen. 2022. "The Important Role of Ion Transport System in Cervical Cancer" International Journal of Molecular Sciences 23, no. 1: 333. https://doi.org/10.3390/ijms23010333
APA StyleChen, Y.-F., & Shen, M.-R. (2022). The Important Role of Ion Transport System in Cervical Cancer. International Journal of Molecular Sciences, 23(1), 333. https://doi.org/10.3390/ijms23010333