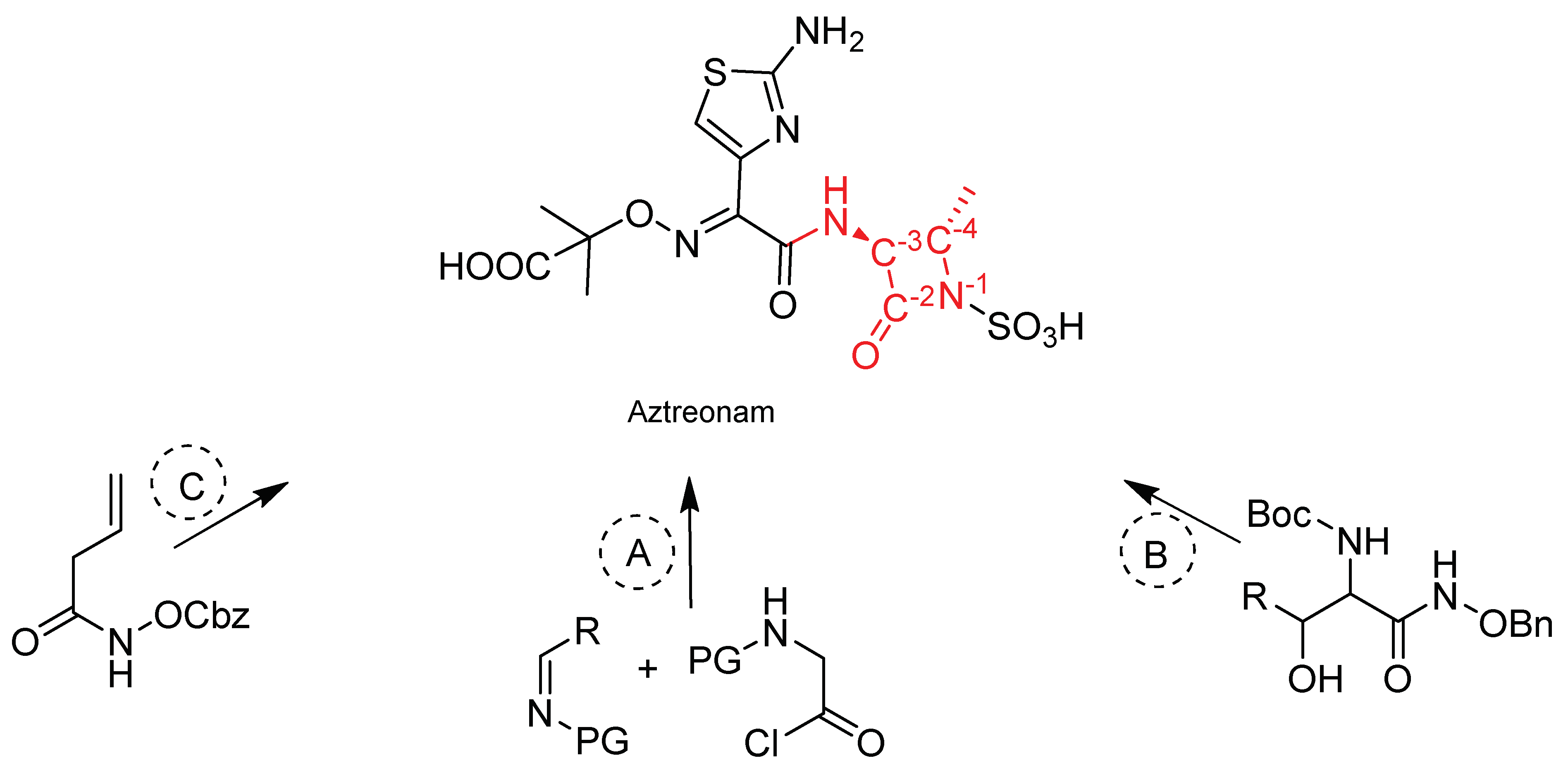
4. Materials and Methods
4.1. Chemistry and Chemical Characterization of Compounds
Unless otherwise stated, all reactions were carried out under argon atmosphere in flame-dried glassware. Chemicals and solvents were obtained from commercial sources (Sigma-Aldrich, Acros Organics, TCI Europe, fluorochem, and Apollo Sci) and were used as supplied. Dry solvents were prepared by distillation from CaH2 (CH2Cl2) or from a mixture of sodium and benzophenone (tetrahydrofuran). Other solvents (dimethylformamide, toluene, methanol, and CH3CN) were used directly from anhydrous Aldrich Sure/Seal bottles. Evaporation of the solvent was carried out under reduced pressure. Reactions were monitored by thin-layer chromatography (TLC) on silica gel aluminum plates (Merck DC Fertigplatten Kieselgel 60 GF254), visualized under UV light (254 nm), and stained with appropriate TLC stains for detection (ninhydrin, dinitrophenylhydrazine, and phospho-molybdic acid). The products were purified by flash column chromatography performed on Merck silica gel 60 (mesh size, 70–230) using the indicated solvents. Yields are reported for the purified products. 1H NMR and 13C NMR spectra were recorded at 295 K using a Bruker Avance III NMR spectrometer equipped with a Broadband decoupling inverse 1H probe, at 400 MHz and 100 MHz, respectively. Chemical shifts (δ) are given in parts per million (ppm) and refer to tetramethylsilane (TMS) as an internal standard. The coupling constants (J) are given in Hertz (Hz), and the splitting patterns are reported as: s, singlet; br s, broad singlet; d, doublet; dd, double doublet; t, triplet, and m, multiplet. Mass spectra were recorded using an ADVION Expres-sion CMSL mass spectrometer (Advion Inc., Ithaca, NY, USA). High-resolution, accurate mass measurements were performed using the ExactiveTM Plus Orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA).
4.2. General Procedure for the Synthesis of Schiff Bases (1–17)
To a solution of an appropriate aldehyde (1 EQ) in dry dichloromethane or dry methanol was added an amine (1 EQ). The resultant solution was stirred for 15 min before Na2SO4 (4 EQ) was added. The reaction mixture was then stirred at room temperature until TLC showed complete consumption of the starting material (30 min to 16 h). Next, the drying agent was removed by filtration, and the volatiles were removed under reduced pressure to afford the desired products, which were used in the next step without further purification.
N-Benzyl-1-(4-(trifluoromethyl)phenyl)methanimine (
1), quantitative yield, brown oil.
1H NMR (400 MHz, DMSO-
d6) δ 8.62 (s, 1H), 8.01 (d,
J = 8.7 Hz, 2H), 7.83 (d,
J = 8.7 Hz, 2H), 7.33 (m, 5H), 4.84 (s, 2H); Rf = 0.66 (EtOAc/Hexane = 1:1,
v/
v) as reported [
45].
N-(2,4-Dimethoxybenzyl)-1-(4-(trifluoromethyl)phenyl)methanimine (
2), quantitative yield, brown oil.
1H NMR (400 MHz, DMSO-
d6) δ 8.50 (s, 1H), 7.97 (d,
J = 8.0 Hz, 2H), 7.81 (d,
J = 8.2 Hz, 2H), 7.16 (d,
J = 8.3 Hz, 1H), 6.58 (d,
J = 2.4 Hz, 1H), 6.51 (dd,
J = 8.3, 2.4 Hz, 1H), 4.71 (s, 2H), 3.79 (s,
J = 4.7 Hz, 3H), 3.76 (s,
J = 3.6 Hz, 3H); Rf = 0.60 (EtOAc/Hexane = 1:1,
v/
v) as reported [
45].
4-(((2,4-Dimethoxybenzyl)imino)methyl)benzonitrile (
3), quantitative yield, colorless amorphous solid.
1H NMR (400 MHz, CDCl
3) δ 8.33 (s, 1H), 7.89–7.79 (m, 2H), 7.74–7.61 (m, 2H), 7.21–7.13 (m, 1H), 6.53–6.42 (m, 2H), 4.80 (s, 2H), 3.81 (app s, 3H); Rf = 0.54 (EtOAc/Hexane = 1:1,
v/
v) as reported [
45].
1-(3-Bromo-4-fluorophenyl)-N-(2,4-dimethoxybenzyl)methanimine (4), quantitative yield, yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.00 (dd, J = 6.8, 2.1 Hz, 1H), 7.68–7.58 (m, 1H), 7.21–7.14 (m, 1H), 7.15–7.05 (m, 1H), 6.50–6.46 (m, 2H), 4.74 (s, 2H), 3.80 (app s, 6H). 13C NMR (100 MHz, CDCl3) δ 161.61, 160.32, 158.86, 158.37, 133.10, 130.33, 128.99, 128.91, 119.29, 116.64, 116.41, 104.16, 98.56, 58.78, 55.40, 55.40. HRMS (ESI+) m/z calc. for C16H15BrFNO2 351.0270, found [M + H]+ 352.0338. Rf = 0.86 (EtOAc/Hexane = 1:1 v/v).
N-Benzyl-1-(4-nitrophenyl)methanimine (
5), quantitative yield, yellow amorphous solid.
1H NMR (400 MHz, CDCl
3) δ 8.46 (s, 1H), 8.26 (d,
J = 8.7 Hz, 2H), 7.94 (d,
J = 8.7 Hz, 2H), 7.43–7.15 (m, 5H), 4.88 (s, 2H); Rf = 0.66 (EtOAc/Hexane = 1:1,
v/
v) as reported [
46].
N-(2,4-Dimethoxybenzyl)-1-(4-nitrophenyl)methanimine (
6), quantitative yield, yellow amorphous solid.
1H NMR (400 MHz, DMSO-
d6) δ 8.54 (s, 1H), 8.31–8.26 (m, 2H), 8.04–7.98 (m, 2H), 7.16 (d,
J = 8.3 Hz, 1H), 6.58 (d,
J = 2.4 Hz, 1H), 6.51 (dd,
J = 8.3, 2.4 Hz, 1H), 4.73 (s, 2H), 3.79 (s, 3H), 3.76 (s, 3H); Rf = 0.46 (EtOAc/Hexane = 1:1,
v/
v) as reported [
47].
N-(2,4-Dimethoxybenzyl)-1-(4-(methylsulfonyl)phenyl)methanimine (7), quantitative yield, pale yellow amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.00 (m, 4H), 7.16 (d, J = 8.3 Hz, 1H), 6.58 (d, J = 2.4 Hz, 1H), 6.51 (dd, J = 8.3, 2.4 Hz, 1H), 4.72 (s, 2H), 3.79 (s, 3H), 3.76 (s, 3H), 3.25 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 160.40, 159.59, 158.40, 141.76, 141.24, 130.37, 128.93, 127.63, 119.01, 104.21, 98.57, 59.13, 55.41, 44.46. HRMS (ESI+) m/z calc. for C17H19NO4S 333.1035, found [M + H]+ 334.1104. Rf = 0.25 (EtOAc:Hex = 1:1, v/v).
4-(((2,4-Dimethoxybenzyl)imino)methyl)-N,N-dimethylaniline (8), quantitative yield, colorless amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 1.4 Hz, 1H), 7.63 (d, J = 8.9 Hz, 2H), 7.18 (d, J = 8.9 Hz, 2H), 6.68 (d, J = 8.9 Hz, 1H), 6.48–6.42 (m, 2H), 4.68 (s, 2H), 3.79 (s, 3H), 3.78 (s, 3H), 2.98 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 162.19, 159.96, 158.22, 152.06, 129.96, 129.70, 124.44, 120.58, 111.61, 111.01, 104.01, 98.42, 58.71, 55.37, 50.39, 40.21, 40.06. HRMS (ESI+) m/z calc. for C18H22N2O2 298.1681, found [M + H]+ 299.1751; Rf = 0.63 (EtOAc/Hexane = 1:1, v/v).
N-Benzyl-1-phenylmethanimine (
9), quantitative yield, brown oil.
1H NMR (400 MHz, DMSO-
d6) δ 8.49 (s, 1H), 7.84–7.75 (m, 2H), 7.50–7.22 (m, 8H), 4.77 (s,
J = 1.2 Hz, 2H); Rf = 0.65 (EtOAc/Hex = 1:1
v/
v) as reported [
48].
N-(2,4-Dimethoxybenzyl)-1-(furan-2-yl)methanimine (
10), quantitative yield, dark brown oil.
1H NMR (400 MHz, CDCl
3) δ 8.08 (s, 1H), 7.49 (s, 1H), 7.21–7.16 (m, 1H), 6.73 (d,
J = 3.4 Hz, 1H), 6.49–6.44 (m, 3H), 4.74 (s, 2H), 3.80 (s, 3H), 3.79 (s, 3H); Rf = 0.36 (EtOAc/Hexane = 1:1,
v/
v) as reported [
47].
N-(2,4-Dimethoxybenzyl)-1-(1H-imidazol-5-yl)methanimine (11), quantitative yield, colorless amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.72–7.69 (m, 1H), 7.41 (s, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 6.49 (dd, J = 8.3, 2.4 Hz, 1H), 4.57 (s, 2H), 3.77 (s, 3H), 3.75 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 160.21, 158.35, 130.56, 120.00, 104.92, 98.69, 58.39, 55.82, 55.63. HRMS (ESI+) m/z calc. for C13H15N3O2 245.1164, found [M + H]+ 246.1234. Rf = 0.1 (EtOAc).
1-(Benzo[
b]thiophen-2-yl)-
N-benzylmethanimine (
12), quantitative yield, yellow amorphous solid.
1H NMR (400 MHz, DMSO-
d6) δ 8.76 (s, 1H), 7.98–7.8 (m 2H), 7.87 (s, 1H), 7.44–7.40 (m, 2H), 7.38–7.21 (m, 5H), 4.80 (s, 2H); Rf = 0.67 (EtOAc/Hexane = 1:1,
v/
v) as reported [
49].
1-(Benzo[
d][1,3]dioxol-5-yl)-
N-benzylmethanimine (
13), quantitative yield, colorless amorphous solid.
1H NMR (400 MHz, CDCl
3) δ 8.26 (t,
J = 1.4 Hz, 1H), 7.41 (d,
J = 1.4 Hz, 1H), 7.35–7.22 (m, 5H), 7.14 (dd,
J = 8.0, 1.6 Hz, 1H), 6.82 (d,
J = 7.9 Hz, 1H), 5.98 (s, 2H), 4.77 (s, 2H); Rf = 0.60 (EtOAc/Hexane = 1:1,
v/
v) as reported [
50].
1-(Benzo[d][1,3]dioxol-5-yl)-
N-(2,4-dimethoxybenzyl)methanimine (
14), quantitative yield, colorless amorphous solid.
1H NMR (400 MHz, DMSO-
d6) δ 8.27 (s, 1H), 7.30 (d,
J = 1.5 Hz, 1H), 7.20 (dd,
J = 7.9, 1.5 Hz, 1H), 7.12 (d,
J = 8.3 Hz, 1H), 6.97 (d,
J = 8.0 Hz, 1H), 6.56 (d,
J = 2.4 Hz, 1H), 6.49 (dd,
J = 8.3, 2.4 Hz, 1H), 6.07 (s, 2H), 4.60 (s, 2H), 3.78 (s, 3H), 3.75 (s, 3H); Rf = 0.65 (EtOAc/Hexane = 1:1,
v/
v) as reported [
50].
N-Benzyl-3-methylbutan-1-imine (15), quantitative yield, light orange oil. 1H NMR (400 MHz, CDCl3) δ 7.38–7.25 (m, 5H), 6.32 (s, 1H), 3.90 (s, 2H), 2.03–1.81 (m, 3H), 0.99–0.76 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 179.39, 138.07, 128.76, 127.95, 127.81, 45.70, 44.53, 25.90, 22.58. HRMS (ESI+) m/z calc. for C12H17N 175.1361, found [M + H]+ 176.1435. Rf = 0.65 (EtOAc/Hex = 1:1, v/v).
N-(2,4-Dimethoxybenzyl)-3-methylbutan-1-imine (16), quantitative yield, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.1 Hz, 1H), 6.73 (s, 1H), 6.46–6.38 (m, 2H), 3.86 (s, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 2.00–1.88 (m, 3H), 0.88 (s, 3H), 0.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 179.20, 160.92, 158.55, 130.46, 118.49, 104.03, 98.55, 55.38, 55.29, 45.95, 40.20, 25.93, 22.64. Rf = 0.63 (EtOAc/Hex = 1:1, v/v).
N-(2,4-Dimethoxybenzyl)heptan-1-imine (17), quantitative yield, orange oil. 1H NMR (400 MHz, CDCl3) δ 9.48 (s, 1H), 7.18–7.15 (m, 1H), 6.46–6.39 (m, 2H), 3.92 (s, 2H), 3.84–3.73 (m, 6H), 1.93–1.79 (m, 4H), 1.51–1.20 (m, 6H), 0.95–0.82 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 176.84, 161.52, 158.65, 131.30, 114.65, 104.24, 98.38, 55.32, 55.27, 39.00, 22.73, 14.03. HRMS (ESI+) m/z calc. for C16H25NO2 263.1885, found [M + H]+ 264.1954. Rf = 0.85 (EtOAc/Hex = 1:1, v/v).
4.3. General Procedures for the Synthesis of Ketene Precursors
4.3.1. General Procedure for the Synthesis of Acid Chloride (18)
N-phthaloylglycine (2.00 g, 9.75 mmol, 1 EQ) was dissolved in dry dichloromethane (10 mL), and the solution was cooled to 0 °C using an ice bath before oxalyl chloride (0.95 mL, 10.73 mmol, 1.1 EQ) was added dropwise over 30 min. Upon complete addition, the reaction mixture was stirred at 0 °C for an additional 2 h, and the solvent was removed under reduced pressure without heating. The acyl chlorides thus obtained were used in the subsequent step without further purification.
2-(1,3-Dioxoisoindolin-2-yl)acetyl chloride (
18), quantitative yield, yellow amorphous solid.
1H NMR (400 MHz, CDCl
3) 7.95–7.89 (m, 1H), 7.82–7.76 (m, 1H), 4.83 (s, 1H); as reported [
51].
4.3.2. General Procedure for the Synthesis of Diazoketone (31)
N-Benzyloxycarbonylglycine (2.09 g, 10.0 mmol, 1 EQ) was dissolved in dry tetrahydrofuran (20 mL), and the resultant solution was cooled to −20 °C using a sodium chloride ice bath before triethylamine (1.39 mL, 10.0 mmol, 1 EQ) was added in one portion. Ethyl chloroformate (1.91 mL, 10.0 mmol, 1 EQ) was then added dropwise, and the reaction mixture was stirred for another 1 h. The white precipitate formed was removed by filtration. To the filtrate were slowly added dry acetonitrile (80 mL) (4:1 solution in THF) and (trimethylsilyl)diazomethane (2.0 M solution in hexane, 10 mL, 20.0 mmol, 2 EQ). The resultant reaction mixture was then stirred at 4 °C for 24–48 h. The reaction was quenched by the addition of diethyl ether and 10% (m/m) aqueous citric acid. The organic phase was then washed with saturated aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4, and the solvents were evaporated. The diazoketone was purified by silica gel column chromatography using EtOAc:Hex = 1:1, v/v as eluent.
Benzyl (3-diazo-2-oxopropyl)carbamate (
31), quantitative yield, transparent amorphous solid.
1H NMR (400 MHz, CDCl
3) δ (ppm) = 7.42–7.31 (m, 5H), 5.46 (s, 1H), 5.39 (s, 1H), 5.13 (s, 2H), 3.99 (s, 1H); as reported [
52].
4.3.3. General Procedure for the Synthesis of Mixed Anhydride
In a flame-dried flask, N-(tert-butoxycarbonyl)glycine (3.00 g, 17.13 mmol, 1 EQ) was dissolved in dry tetrahydrofuran (20 mL) and placed under an argon atmosphere. The solution was cooled to −60 °C, and triethylamine (2.62 mL, 18.84 mmol, 1.1 EQ) was added in one portion. Then ethyl chloroformate (2.13 mL, 22.27 mmol, 1.3EQ) was added dropwise over a period of 30 min. After the complete addition of the reagent, the reaction mixture was stirred at −40 °C for another 2 h. The resultant reaction mixture was then directly used in the next step without any further purification. The same reaction conditions were used for the synthesis of 2-(((benzyloxy)carbonyl)amino)acetic anhydride from ((benzyloxy)carbonyl)glycine.
4.4. General Procedure for the Synthesis of Monocyclic Beta Lactam Core I (19–30)
Schiff base (1 EQ) was dissolved in dry toluene (0.1–0.2 mmol/mL) in a flame-dried flask and placed under an argon atmosphere. Triethylamine (2.5 EQ) was then added in one portion, and the resultant solution was heated to 80 °C, before 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (1.3 EQ), dissolved in in dry toluene, was added dropwise over a period of 30 min. Upon complete addition, the reaction was stirred at 80 °C for a further 1.5–3.5 h. The reaction mixture was then cooled to room temperature, and the volatiles were removed in vacuo. The solid residue thus obtained was redissolved in ethyl acetate. The organic phase was washed with 10% aq. citric acid solution, saturated NaHCO3, and brine. The organic phase was dried (Na2SO4), filtered, then concentrated in vacuo. Some cyclized ß-lactams were purified by silica gel column chromatography using EtOAc: Hex as eluent.
2-(1-Benzyl-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-yl)isoindoline-1,3-dione (19), yield: 51%, colorless amorphous solid. The reaction was carried out according to General Procedure I using N-benzyl-1-(4-(trifluoromethyl)phenyl)methanimine (1), (1.32 g, 5 mmol, 1.0 EQ), triethylamine (1.74 mL, 12.5 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (1.45 g, 6.5 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.80–7.70 (m, 4H), 7.51 (d, J = 8.2 Hz, 2H), 7.39–7.28 (m, 7H), 5.77 (d, J = 5.4 Hz, 1H), 5.18 (d, J = 5.4 Hz, 1H), 4.82 (d, J = 15.4 Hz, 1H), 4.47 (d, J = 15.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 166.73, 163.56, 137.29, 134.53, 134.42, 131.04, 129.09, 128.64, 128.26, 127.79, 125.50, 125.47, 123.59, 60.16, 59.84, 45.83. HRMS (ESI+) m/z calc. for C25H17F3N2O3 450.1191, found [M + H]+ 451.1260. Rf = 0.42 (EtOAc/n-Hex; 2:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-yl)isoindoline-1,3-dione (20), yield: 55%, light brown amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-dimethoxybenzyl)-1-(4-(trifluoromethyl)phenyl)methanimine (2), (3.23 g, 10 mmol, 1.0 EQ), triethylamine (3.48 mL, 25 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (2.91 g, 13 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.71–7.60 (m, 4H), 7.44 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.3 Hz, 1H), 6.43 (dd, J = 8.3, 2.4 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 5.46 (d, J = 5.4 Hz, 1H), 4.90 (d, J = 14.3 Hz, 1H), 4.84 (d, J = 5.4 Hz, 1H), 4.30 (d, J = 14.3 Hz, 1H), 3.79 (s, 3H), 3.56 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.77, 163.44, 161.18, 158.59, 138.33, 134.33, 131.59, 131.07, 127.60, 125.17, 125.13, 123.49, 115.03, 104.34, 98.33, 60.65, 59.54, 55.41, 55.00, 40.82. HRMS (ESI+) m/z calc. for C27H21F3N2O5 510.1403, found [M + H]+ 511.1470. Rf = 0.31 (EtOAc/n-Hex; 1:1, v/v).
4-(1-(2,4-Dimethoxybenzyl)-3-(1,3-dioxoisoindolin-2-yl)-4-oxoazetidin-2-yl)benzonitrile (21), yield: 48%, colorless amorphous solid. The reaction was carried out according to General Procedure I using 4-(((2,4-dimethoxybenzyl)imino)methyl)benzonitrile (3) (0.32 g, 1.15 mmol, 1 EQ), triethylamine (0.40 mL, 2.87 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (0.33 g, 1.50 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.80–7.72 (m, 4H), 7.63 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.21–7.17 (m, 1H), 6.49–6.46 (m, 2H), 5.58 (d, J = 5.5 Hz, 1H), 5.01 (d, J = 5.4 Hz, 1H), 4.61 (d, J = 14.5 Hz, 1H), 4.34 (d, J = 14.6 Hz, 1H), 3.74 (s, 3H), 3.57 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 166.69, 163.32, 161.26, 158.57, 139.89, 134.49, 131.99, 131.65, 130.97, 127.88, 123.58, 118.41, 114.87, 111.87, 104.39, 98.39, 60.72, 59.57, 55.44, 55.02, 40.97. MS (ESI+, m/z), 468.4 ([M + H]+). Rf = 0.24 (EtOAc/n-Hex; 1:1, v/v).
2-(2-(4-Bromo-3-fluorophenyl)-1-(2,4-dimethoxybenzyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (22), yield: 51%, pale yellow amorphous solid. The reaction was carried out according to General Procedure I using 1-(3-bromo-4-fluorophenyl)-N-(2,4-dimethoxybenzyl)methanimine (4) (0.53 g, 1.5 mmol, 1 EQ), triethylamine (0.52 mL, 3.75 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (0.44 g, 1.95 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.73–7.64 (m, 4H), 7.37 (dd, J = 6.5, 2.2 Hz, 1H), 7.17–7.10 (m, 2H), 6.91 (t, J = 8.4 Hz, 1H), 6.43 (dd, J = 8.3, 2.4 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 5.40 (d, J = 5.3 Hz, 1H), 4.82 (d, J = 14.3 Hz, 1H), 4.74 (d, J = 5.4 Hz, 1H), 4.30 (d, J = 14.3 Hz, 1H), 3.80 (s, 3H), 3.61 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.77, 163.36, 161.19, 158.57, 134.36, 132.52, 131.60, 131.51, 131.47, 131.14, 127.91, 127.84, 123.55, 116.33, 116.10, 115.03, 104.37, 98.37, 60.22, 59.61, 55.43, 55.10, 40.71. HRMS (ESI+) m/z calc. for C26H20BrFN2O5 538.0540, found [M + H]+ 539.0606. Rf = 0.56 (EtOAc/n-Hex; 2:1, v/v).
2-(1-Benzyl-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (23), yield: 47%, pale yellow amorphous solid. The reaction was carried out according to General Procedure I using N-benzyl-1-(4-nitrophenyl)methanimine (5) (2.10 g, 10 mmol, 1 EQ), triethylamine (3.48 mL, 25 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (2.91 g, 13 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.8 Hz, 2H), 7.74–7.60 (m, 4H), 7.39 (d, J = 8.7 Hz, 2H), 7.36–7.22 (m, 5H), 5.57 (d, J = 5.5 Hz, 1H), 5.06 (d, J = 14.8 Hz, 1H), 4.93 (d, J = 5.4 Hz, 1H), 4.26 (d, J = 14.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 166.64, 163.40, 147.84, 140.76, 134.59, 134.35, 130.93, 129.16, 128.66, 128.40, 128.33, 123.72, 123.70, 60.16, 59.88, 46.08. HRMS (ESI+) m/z calc. for C24H17N3O5 427.1168, found [M + H]+ 433.1386. Rf = 0.25 (EtOAc/n-Hex; 1:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (24), yield: 51%, pale yellow amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-dimethoxybenzyl)-1-(4-nitrophenyl)methanimine (6) (3.00 g, 10 mmol, 1 EQ), triethylamine (3.48 mL, 25 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (2.91 g, 13 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.8 Hz, 2H), 7.72–7.60 (m 4H), 7.39 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.3 Hz, 1H), 6.43 (dd, J = 8.3, 2.3 Hz, 1H), 6.36 (d, J = 2.3 Hz, 1H), 5.49 (d, J = 5.5 Hz, 1H), 4.91 (s, J = 14.3 Hz, 1H), 4.88 (d, J = 5.3 Hz, 1H), 4.33 (d, J = 14.3 Hz, 1H), 3.79 (s, 3H), 3.56 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.67, 163.28, 161.29, 158.57, 147.57, 141.91, 134.50, 131.66, 130.96, 128.10, 123.64, 123.42, 114.82, 104.43, 98.41, 60.58, 59.58, 55.44, 55.06, 41.02. HRMS (ESI+) m/z calc. for C26H21N3O7 487.1380, found [M + H]+ 488.1443. Rf = 0.42 (EtOAc/n-Hex; 2:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-(4-(methylsulfonyl)phenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (25), yield: 45%, pale yellow amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-dimethoxybenzyl)-1-(4-(methylsulfonyl)phenyl)methanimine (7) (1.00 g, 3.00 mmol, 1 EQ), triethylamine (1.04 mL, 7.50 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (0.87 g, 3.90 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.86–7.69 (m, 4H), 7.68 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.8 Hz, 1H), 6.51–6.45 (m, 2H), 5.59 (d, J = 5.5 Hz, 1H), 5.03 (d, J = 5.4 Hz, 1H), 4.64 (d, J = 14.5 Hz, 1H), 4.34 (d, J = 14.6 Hz, 1H), 3.74 (s, 3H), 3.58 (s, 3H), 2.98 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.80, 163.35, 161.07, 158.68, 141.06, 140.45, 135.42, 131.54, 130.78, 128.35, 126.90, 123.79, 115.39, 105.13, 98.72, 60.68, 59.76, 55.69, 55.64, 43.71, 41.04. HRMS (ESI+) m/z calc. for C27H24N2O7S 520.1304, found [M + H]+ 521.1378. Rf = 0.36 (EtOAc/n-Hex; 2:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-(furan-2-yl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (26), yield: 51%, colorless amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-dimethoxybenzyl)-1-(furan-2-yl)methanimine (10) (1.23 g, 5 mmol, 1 EQ), triethylamine (1.74 mL, 12.5 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (1.45 g, 6.5 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.81–7.63 (m, 4H), 7.21–7.18 (m, 1H), 7.15 (d, J = 8.1 Hz, 1H), 6.48–6.40 (m, 2H), 6.29 (d, J = 3.3 Hz, 1H), 6.18 (dd, J = 3.3, 1.8 Hz, 1H), 5.42 (d, J = 5.0 Hz, 1H), 4.84 (d, J = 5.0 Hz, 1H), 4.80 (d, J = 14.5 Hz, 1H), 4.22 (d, J = 14.5 Hz, 1H), 3.80 (s, 3H), 3.71 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.73, 163.17, 161.01, 158.61, 148.22, 142.85, 134.20, 131.45, 131.38, 123.51, 115.39, 110.56, 109.87, 104.20, 98.41, 59.03, 55.41, 55.29, 55.12, 40.18. HRMS (ESI+) m/z calc. for C24H20N2O6 432.1321, found [M + H]+ 433.1386. Rf = 0.23 (EtOAc/n-Hex; 2:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-(1H-imidazol-5-yl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (27), yield: 33%, light brown amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-dimethoxybenzyl)-1-(1H-imidazol-5-yl)methanimine (11) (1.19 g, 7.7 mmol, 1 EQ), triethylamine (1.35 mL, 15.4 mmol, 2 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (2.23 g, 10 mmol, 1.3EQ). Product was purified by silica gel column chromatography using DKM: MeOH = 15:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H), 7.90–7.79 (m, 4H), 7.57–7.47 (m, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.38–6.23 (m, 2H), 4.92 (d, J = 16.7 Hz, 1H), 4.78 (d, J = 7.5 Hz, 2H), 4.74 (d, J = 7.3 Hz, 1H), 4.62 (d, J = 16.6 Hz, 1H), 3.81 (s, 3H), 3.75 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 185.51, 184.92, 167.93, 161.04, 158.25, 140.99, 139.31, 136.01, 134.21, 132.14, 123.59, 104.24, 98.57, 63.70, 55.41, 55.38, 55.21, 39.94. HRMS (ESI+) m/z calc. for C23H20N4O5 432.1434, found [M + H]+ 433.1500. Rf = 0.42 (DKM/MeOH; 9:1, v/v).
2-(1-Benzyl-2-isobutyl-4-oxoazetidin-3-yl)isoindoline-1,3-dione (28), yield: 31%, colorless amorphous solid. The reaction was carried out according to General Procedure I using N-Benzyl-3-methylbutan-1-imine (15) (2 g, 11.6 mmol, 1 EQ), triethylamine (3.21 mL, 23 mmol, 2 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (3.1 g, 15 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.95–7.66 (m, 4H), 7.34–7.14 (m, 5H), 6.41 (d, J = 13.8 Hz, 1H), 5.26 (dd, J = 13.8, 7.3 Hz, 1H), 4.84–4.44 (m, 4H), 2.38–2.22 (m, 1H), 0.98 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.98, 164.64, 136.60, 134.07, 132.27, 128.52, 127.37, 125.63, 123.56, 60.38, 48.50, 39.81, 29.57, 22.85, 22.68. HRMS (ESI+) m/z calc. for C22H22N2O3 362.1630, found [M + H]+ 363.1694. Rf = 0.54 (EtOAc/n-Hex; 1:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-isobutyl-4-oxoazetidin-3-yl)isoindoline-1,3-dione (29), yield: 25%, colorless amorphous solid. The reaction was carried out according to General Procedure I using N-(2,4-Dimethoxybenzyl)-3-methylbutan-1-imine (16) (2.73 g, 11.6 mmol, 1 EQ), triethylamine (3.21 mL, 23 mmol, 2 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (3.36 g, 15 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.99–7.82 (m, 4H), 7.01–6.93 (m, 1H), 6.84–6.74 (m, 1H), 6.63–6.51 (m, 1H), 6.50–6.44 (m, 1H), 5.14–5.00 (m, 1H), 4.83–4.55 (m, 4H), 3.90–3.66 (m, 6H), 2.37–2.13 (m, 1H), 0.95 (d, J = 6.7 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 168.00, 165.35, 160.05, 157.83, 135.21, 132.07, 127.88, 123.80, 120.61, 116.68, 105.05, 98.63, 65.35, 55.86, 55.62, 43.67, 41.94, 29.24, 23.36. HRMS (ESI+) m/z calc. for C24H26N2O5 422.1842, found [M + H]+ 423.1910. Rf = 0.45 (EtOAc/n-Hex; 1:1, v/v).
2-(1-(2,4-Dimethoxybenzyl)-2-hexyl-4-oxoazetidin-3-yl)isoindoline-1,3-dione (30), yield: 11%, brown oil. The reaction was carried out according to General Procedure I using N-(2,4-Dimethoxybenzyl)heptan-1-imine (17) (2.31 g, 8.76 mmol, 1EQ), triethylamine (3.05 mL, 21.9 mmol, 2.5 EQ) and 2-(1,3-dioxoisoindolin-2-yl)acetyl chloride (18) (2.53 g, 11.3 mmol, 1.3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.93–7.66 (m, 4H), 7.07–6.94 (m, 1H), 6.59–6.39 (m, 2H), 5.32–5.15 (m, 1H), 4.80–4.69 (m, 1H), 4.67–4.54 (m, 1H), 3.92–3.87 (m, 2H), 3.84–3.75 (m, 6H), 2.04–1.93 (m, 1H), 1.39–1.04 (m, 8H), 0.92–0.79 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 168.03, 164.42, 160.42, 157.82, 134.02, 132.33, 123.53, 119.31, 113.91, 104.26, 98.29, 60.42, 55.39, 55.31, 42.61, 39.88, 31.14, 30.29, 29.37, 28.29, 22.46, 14.02. HRMS (ESI+) m/z calc. for C26H30N2O5 450.2155, found [M + H]+ 451.2229. Rf = 0.42 (EtOAc/n-Hex; 1:1, v/v).
4.5. General Procedure for the Synthesis of Monocyclic Beta Lactam Core II (32–34)
A solution of an appropriate Schiff base (2 EQ) and diazoketone (1 EQ) in 1,2 dimethoxyethane (3 mL) was stirred for 20–30 min at 180 °C in a microwave reactor. The volatiles were then removed in vacuo, and the crude product thus obtained was purified by silica gel column chromatography using EtOAc: Hexane (1:1) as an eluent.
Benzyl ((1-benzyl-2-oxo-4-phenylazetidin-3-yl)methyl)carbamate (32), yield: 31%, brown oil. The reaction was carried out according to General Procedure II using N-benzyl-1-phenylmethanimine (9) (250 mg, 1.28 mmol, 2 EQ) and benzyl (3-diazo-2-oxopropyl)carbamate (31) (179 mg, 0.64 mmol, 1 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 1.6 Hz, 1H), 7.40–7.30 (m, 10H), 7.25–7.21 (m, 3H), 7.12 (dd, J = 6.9, 2.5 Hz, 2H), 5.18–5.13 (m, 1H), 5.11 (d, J = 12.3 Hz, 1H), 4.98 (d, J = 12.3 Hz, 1H), 4.83 (d, J = 15.0 Hz, 1H), 4.28 (d, J = 2.0 Hz, 1H), 3.76 (d, J = 14.7 Hz, 1H), 3.64–3.59 (m, 1H), 3.20–3.15 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 166.69, 156.61, 137.00, 135.31, 134.51, 128.99, 128.79, 128.54, 128.39, 128.33, 128.17, 128.01, 127.73, 126.51, 69.10, 60.36, 57.72, 48.85, 44.57. HRMS (ESI+) m/z calc. for C25H24N2O3 400.1787, found [M + H]+ 401.1856. Rf = 0.27. (EtOAc/n-Hex; 1:1, v/v).
Benzyl ((2-(benzo[b]thiophen-2-yl)-1-benzyl-4-oxoazetidin-3-yl)methyl)carbamate (33), yield: 35%, yellow oil. The reaction was carried out according to General Procedure II using 1-(benzo[b]thiophen-2-yl)-N-benzylmethanimine (12) (250 mg, 1.00 mmol, 2 EQ) and benzyl (3-diazo-2-oxopropyl)carbamate (31) (117 mg, 0.50 mmol, 1 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:2 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.38–7.30 (m, 13H), 5.13 (s, 2H), 4.98 (d, J = 12.2 Hz, 1H), 4.87 (d, J = 15.1 Hz, 1H), 4.67 (d, J = 1.0 Hz, 1H), 4.33 (s, 1H), 3.86 (d, J = 15.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 167.35, 156.67, 141.88, 139.57, 139.44, 136.25, 135.16, 128.88,128.80, 128.78, 128.75, 128.56, 128.38, 128.21, 128.14, 128.02, 127.87, 124.82, 124.65, 123.68, 123.26, 122.60, 66.99, 60.92, 54.14, 52.36, 44.81. HRMS (ESI+) m/z calc. for C27H24N2O3S 456.1508, found [M + H]+ 457.1579. Rf = 0.24 (EtOAc/n-Hex; 1:2, v/v).
Benzyl ((2-(benzo[d][1,3]dioxol-5-yl)-1-benzyl-4-oxoazetidin-3-yl)methyl)carbamate (34), yield: 29%, colorless amorphous solid. The reaction was carried out according to General Procedure II using 1-(benzo[d][1,3]dioxol-5-yl)-N-benzylmethanimine (13) (250 mg, 1.05 mmol, 2 EQ) and benzyl (3-diazo-2-oxopropyl)carbamate (31) (123 mg, 0.53 mmol, 1 EQ) Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.51 (t, J = 6.0 Hz, 1H), 7.38–7.25 (m, 9H), 7.16 (d, J = 6.7 Hz, 2H), 6.85 (d, J = 7.9 Hz, 1H), 6.79 (s, 1H), 6.67 (d, J = 7.9 Hz, 1H), 6.01 (d, J = 1.3 Hz, 2H), 5.02 (d, J = 12.6 Hz, 1H), 4.96 (d, J = 12.6 Hz, 1H), 4.65 (d, J = 15.6 Hz, 1H), 4.31 (d, J = 1.8 Hz, 1H), 3.81 (d, J = 15.6 Hz, 1H), 3.70–3.52 (m, 1H), 3.40–3.34 (m, 1H), 3.09 (ddd, J = 7.3, 5.1, 1.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.59, 156.79, 148.26, 147.60, 137.54, 136.50, 132.07, 129.07, 128.79, 128.23, 128.07, 127.81, 120.55, 108.85, 106.72, 101.59, 65.81, 60.66, 58.45, 51.93, 44.12. HRMS (ESI+) m/z calc. for C26H24N2O5 444.1685, found [M + H]+ 445.1752. Rf = 0.31 (EtOAc/n-Hex; 1:1, v/v).
4.6. General Procedure for the Synthesis of Monocyclic Beta Lactam Core III (35–37)
To a solution of an appropriate Schiff base (1 EQ) in dry dichloromethane (2 mL/mmol) was added dry tetrahydrofuran (5 mL/mmol), and the reaction mixture was cooled to −60 °C before mixed acid anhydride (1.3 EQ) was added slowly. Next, triethylamine (1.5 EQ) was added dropwise over a period of 30 min. Upon complete addition, the reaction mixture was allowed to warm to room temperature with stirring overnight. The volatiles were removed in vacuo, and the solid residue thus obtained dissolved in ethyl acetate. The organic phase was washed with 0.1 M HCl (aq), saturated NaHCO3 (aq) and brine, dried (Na2SO4), filtered, then concentrated in vacuo. The crude product thus obtained was then purified by silica gel column chromatography using EtOAc: Hex as eluent or recrystallized from methyl tert-butyl ether.
tert-butyl (2-(4-cyanophenyl)-1-(2,4-dimethoxybenzyl)-4-oxoazetidin-3-yl)carbamate (35), yield: 11%, colorless amorphous solid. The reaction was carried out according to General Procedure III using 4-(((2,4-dimethoxybenzyl)imino)methyl)benzonitrile (3), (14.16 g, 50.5 mmol, 1 EQ), N-(tert-butoxycarbonyl)glycine (11.5 g, 65.6 mmol, 1.3 EQ) and triethylamine (10.5 mL, 75.7 mmol, 1.5 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:2 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.0 Hz, 1H), 6.46–6.39 (m, 2H), 4.93 (dd, J = 8.4, 5.0 Hz, 1H), 4.70 (d, J = 5.0 Hz, 1H), 4.43 (d, J = 14.5 Hz, 1H), 4.09–3.97 (m, 1H), 3.72 (s, 3H), 3.56 (s, 3H), 1.14 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 166.08, 160.96, 158.59, 155.06, 141.81, 132.00, 131.42, 129.23, 119.33, 115.50, 110.46, 105.06, 98.63, 78.80, 62.76, 61.72, 60.22, 55.68, 55.60, 28.24. HRMS (ESI+) m/z calc. for C24H27N3O5 437.1951, found [M + H]+ 438.2015. Rf = 0.54 (EtOAc/n-Hex; 1:1, v/v).
Benzyl (1-(2,4-dimethoxybenzyl)-2-(4-(dimethylamino)phenyl)-4-oxoazetidin-3-yl)carbamate (36), yield: 31%, colorless amorphous solid. The reaction was carried out according to General Procedure III using 4-(((2,4-dimethoxybenzyl)imino)methyl)-N,N-dimethylaniline (8), (1.27 g, 4.25 mmol, 1 EQ), ((benzyloxy)carbonyl)glycine (1. 15 g, 5.52 mmol, 1.3 EQ,) and triethylamine (0.88 mL, 6.37 mmol, 1.5 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:3 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.39–7.28 (m, 4H), 7.24–6.93 (m, 5H), 6.76–6.60 (m, 2H), 6.48–6.36 (m, 2H), 5.71 (d, J = 47.4 Hz, 1H), 5.14 (dd, J = 21.9, 9.5 Hz, 1H), 4.94 (d, J = 12.0 Hz, 1H), 4.84 (d, J = 14.5 Hz, 1H), 4.18 (d, J = 20.5 Hz, 2H), 3.79 (app s, 6H), 2.98 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 167.19, 160.87, 158.60, 153.11, 151.08, 131.78, 128.71, 128.52, 128.42, 128.22, 128.02, 127.72, 116.06, 112.03, 104.09, 98.45, 75.00, 67.19, 67.13, 55.41, 55.25, 48.33, 40.41, 38.28. HRMS (ESI+) m/z calc. for C28H31N3O5 489.2264, found [M + H]+ 490.2325. Rf = 0.45 (EtOAc/n-Hex; 1:1, v/v).
Benzyl (2-(benzo[d][1,3]dioxol-5-yl)-1-(2,4-dimethoxybenzyl)-4-oxoazetidin-3-yl)carbamate (37), yield: 33%, colorless amorphous solid. The reaction was carried out according to General Procedure III using 1-(benzo[d][1,3]dioxol-5-yl)-N-(2,4-dimethoxybenzyl)methanimine (14) (1.85 g, 6.18 mmol, 1 EQ), ((benzyloxy)carbonyl)glycine (1.68 g, 8.04 mmol, 1.3 EQ,) and triethylamine (1.29 mL, 9.27 mmol, 1.5 EQ). Product was precipitated from methyl tert-butyl ether. 1H NMR (400 MHz, CDCl3) δ 7.32–7.25 (m, 4H), 7.15 (d, J = 7.2 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 6.62–6.58 (m, 2H), 6.39 (d, J = 7.2 Hz, 2H), 5.96 (s, 2H), 5.18 (dd, J = 9.4, 5.0 Hz, 1H), 4.98–4.94 (m, 2H), 4.70 (d, J = 14.4 Hz, 1H), 4.62 (d, J = 4.9 Hz, 1H), 3.99 (d, J = 14.4 Hz, 1H), 3.79 (s, 3H), 3.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.42, 161.02, 158.56, 155.32, 148.15, 147.66, 135.99, 131.21, 128.46, 128.13, 127.86, 120.64, 115.31, 108.62, 107.02, 104.08, 101.27, 98.40, 77.35, 77.03, 76.72, 66.99, 61.82, 61.13, 55.40, 55.17, 45.81, 39.71, 8.63. HRMS (ESI+) m/z calc. for C27H26N2O7 490.1740, found [M + H]+ 491.1803. Rf = 0.28 (EtOAc/n-Hex; 1:1, v/v).
4.7. General Procedure for the Synthesis of Phthalimide Deprotected ß Lactam (38–41)
4.7.1. Hydrazine Hydrate
3-Amino-4-substituted monocyclic ß-lactams with aromatic substituents:
2-(1-Benzyl-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-yl)isoindoline-1,3-dione (19) (225 mg, 0.44 mmol, 1 EQ) was dissolved in dried methanol and placed under argon atmosphere. Hydrazine hydrate (0.046 mL, 0.76 mmol, 1.7 EQ) was added dropwise. The mixture was stirred for 2 h at room temperature. Then, the solvent was evaporated. Anhydrous methanol and 3 drops of concentrated aqueous HCl were added to the solid. After the solid was completely dissolved again, the solvent was evaporated. The solid was again dissolved in anhydrous methanol and stirred for 16 h at room temperature. The precipitate formed was filtered off and the solvent evaporated. The solid was dissolved in dichloromethane and washed with saturated aqueous NaHCO3. The aqueous phase was extracted three times with dichloromethane, the combined organic phases were dried over Na2SO4 and the solvents evaporated. The deprotected amine was used directly in the next step without purification.
2-(2-((1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamoyl)benzoyl)hydrazin-1-ide (38), colorless amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 9.04 (d, J = 7.9 Hz, 1H), 8.15 (d, J = 8.8 Hz, 2H), 7.45 (d, J = 8.7 Hz, 2H), 7.39–7.27 (m, 2H), 7.20 (td, J = 7.5, 1.4 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.49–6.39 (m, 4H), 5.39 (dd, J = 7.8, 5.1 Hz, 1H), 4.91 (d, J = 5.0 Hz, 1H), 4.50 (d, J = 14.5 Hz, 1H), 4.30 (s, 1H), 4.17 (d, J = 14.4 Hz, 1H), 3.72 (s, 3H), 3.59 (s, 3H). MS (ESI+, m/z) 518.1 ([M − H]−). Rf = 0.05 (EtOAc).
1-Benzyl-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-aminium chloride (39), yield: 91%, colorless amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 3H), 7.74 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 7.38–7.17 (m, 4H), 5.06 (d, J = 5.4 Hz, 1H), 4.92 (d, J = 5.4 Hz, 1H), 4.69 (d, J = 15.4 Hz, 1H), 4.17 (d, J = 15.4 Hz, 1H).13C NMR (100 MHz, DMSO-d6) δ 163.03, 136.83, 135.32, 130.06, 129.63, 129.11, 128.75, 128.19, 125.84, 123.23, 58.90, 57.79, 45.10.HRMS (ESI+) m/z calc. for C17H15F3N2O 320.1136, found [M + H]+ 321.1208. Rf = 0.61 (EtOAc).
1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-aminium chloride (40), yield: 94%, light brown oil. The reaction was carried out according to the General Procedure using 2-(1-(2,4-dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (24) (1.01 g, 2.1 mmol, 1 EQ) and hydrazine hydrate (0.213 mL, 3.5 mmol, 1.7 EQ). Product was used directly in the next step without purification. 1H NMR (400 MHz, DMSO-d6) δ 8.23–8.13 (m, 2H), 7.48–7.38 (m, 2H), 7.03 (dd, J = 11.5, 6.3 Hz, 1H), 6.48–6.37 (m, 2H), 4.68 (d, J = 5.1 Hz, 1H), 4.45 (d, J = 14.7 Hz, 1H), 4.43 (d, J = 5.3 Hz, 1H), 4.00 (d, J = 14.5 Hz, 1H), 3.71 (s, 3H), 3.56 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.45, 160.89, 158.58, 147.23, 145.34, 131.29, 129.27, 123.55, 115.89, 105.07, 98.64, 71.70, 66.04, 62.52, 55.66, 55.61. HRMS (ESI+) m/z calc. for C18H19N3O5 357.1325, found [M + H]+ 358.1390. Rf = 0.51 (DCM/ iPrOH; 11:1, v/v).
3-Amino-4-substituted monocyclic ß-lactams with aliphatic substituents:
2-(1-(2,4-Dimethoxybenzyl)-2-isobutyl-4-oxoazetidin-3-yl)isoindoline-1,3-dione (29) (200 mg, 0.47 mmol, 1 EQ) in was dissolved in dried methanol and placed under argon atmosphere. Hydrazine hydrate (0.088 mL, 1.42 mmol, 3 EQ) was added dropwise. The mixture was stirred for 2 h at room temperature. Then the solvent was evaporated, and the residue was redissolved in ethyl acetate. The organic phase was washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4 and the solvent was evaporated. Mixture of isomers cis and trans 1-(2,4-dimethoxybenzyl)-2-isobutyl-4-oxoazetidin-3-aminium chloride (41), quantitative yield, colorless amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 14.6 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.48–6.37 (m, 4H), 6.32 (d, J = 13.9 Hz, 1H), 5.09 (dd, J = 13.9, 7.3 Hz, 1H), 4.93 (dd, J = 14.6, 7.2 Hz, 1H), 4.78 (s, 2H), 4.55 (s, 2H), 3.83 (d, J = 6.3 Hz, 6H), 3.79 (d, J = 5.4 Hz, 6H), 3.64 (s, 2H), 3.43 (s, 2H), 2.34 (td, J = 13.7, 7.0 Hz, 1H), 2.26 (td, J = 13.5, 6.8 Hz, 1H), 1.02–0.95 (m, 6H), 0.95–0.89 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 172.10, 171.86, 160.14, 159.82, 157.74, 157.36, 128.01, 126.41, 124.38, 123.70, 123.44, 119.97, 117.29, 116.10, 104.07, 104.01, 98.37, 98.22, 55.39, 55.34, 55.29, 55.26, 44.05, 43.74, 42.80, 42.13, 29.50, 29.45, 23.11, 22.92. MS (ESI+) m/z calc. for C16H24N2O3 292.1787, found [M + H]+ 293.1853. Rf = 0.07 (EtOAc).
4.7.2. Methylhydrazine
2-(1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl) isoindoline-1,3-dione (24) (250 mg, 0.51 mmol, 1 EQ) was dissolved in dry methanol and methylhydrazine (0.081 mL, 1.54 mmol, 3 EQ) was added. The reaction was stirred at room temperature. After 4 h, additional methylhydrazine (0.11 mL, 2.12 mmol, 4 EQ) was added and stirred overnight. As the reaction was not yet complete, further methylhydrazine (0.11 mL, 2.12 mmol, 4 EQ) was added, and the reaction was left at room temperature for an additional 72 h. The reaction was allowed to proceed to completion. The organic phase was washed with saturated NaHCO3 solution and brine and dried over Na2SO4. The solvent was evaporated, and the product was purified by silica gel column chromatography using DCM:iPrOH = 11:1 as eluent.
4.7.3. Ethanolamine
2-(1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl) isoindoline-1,3-dione (24) (250 mg, 0.51 mmol, 1 EQ) was dissolved in ethyl acetate. Ethanolamine (0.46 mL, 7.7 mmol, 15 EQ) was added, and the reaction mixture was refluxed (80 °C) for 2 h. Then the reaction mixture was cooled to room temperature, and a saturated solution of NaHCO3 and additional ethyl acetate were added. The organic phase was washed with brine and dried over Na2SO4. The solvent was evaporated, and the product was purified by column chromatography using DCM:iPrOH = 11:1 as eluent.
4.7.4. Ethylenediamine
2-(1-(2,4-Dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl) isoindoline-1,3-dione (24) (250 mg, 0.51 mmol, 1 EQ) was dissolved in ethyl acetate. Ethylendiamine solution (1 M in ethyl acetate; 0.67 mL in 10 mL of ethyl acetate, 10 mmol, 19.5 EQ) was added, and the reaction mixture was stirred overnight at room temperature. After 16 h saturated solution of NaHCO3 and additional ethyl acetate were added. The organic phase was washed with brine and dried over Na2SO4. The solvent was evaporated and the product purified by silica gel column chromatography using DCM:iPrOH = 11:1 as eluent.
4.8. General Procedure for the Synthesis of tert-Butyloxycarbonyl Protected ß Lactam (43, 51–53)
In a flame-dried flask, 3-amino ß-lactam (1 EQ) was dissolved in dry dichloromethane. Triethylamine (1.1 EQ), di-tert-butyl dicarbonate (1.5 EQ) and 4-(dimethylamino)pyridine (catalytic amount) were added, and the solution was stirred overnight at room temperature. The solvent was evaporated, and the crude product was purified by silica gel column chromatography using EtOAc: Hex as eluent.
tert-butyl (1-benzyl-2-isobutyl-4-oxoazetidin-3-yl)carbamate (43), yield: 51%, colorless amorphous solid. The reaction was carried out according to General Procedure using 3-amino-1-(benzyl)-4-isobutylazetidin-2-one (160 mg, 0.69 mmol, 1 EQ), triethylamine (0.11 mL, 0.76 mmol, 1.1 EQ), di-tert-butyl dicarbonate (226 mg, 1.03 mmol, 1.5 EQ) and 4-(dimethylamino)pyridine (catalytic amount). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:2 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.37–7.08 (m, 5H), 5.60–5.41 (m, 1H), 5.21–4.93 (m, 1H), 4.88–4.65 (m, 2H), 4.17–3.91 (m, 2H), 2.39–2.19 (m, 1H), 1.52–1.38 (m, 9H), 0.98–0.90 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 167.47, 155.82, 136.71, 128.52, 127.10, 123.16, 79.76, 47.86, 42.96, 29.54, 28.35, 27.91, 23.01, 22.74. HRMS (ESI+) m/z calc. for C19H28N2O3 332.2100, found [M + H]+ 333.2166. Rf = 0.63 (EtOAc/n-Hex; 1:1, v/v). tert-butyl (1-benzyl-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-yl)carbamate (51), yield: 47%, colorless amorphous solid. The reaction was carried out according to General Procedure using 1-benzyl-2-oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-aminium chloride (39) (143 mg, 0.4 mmol, 1 EQ), triethylamine (0.061 mL, 0.44 mmol, 1 EQ), di-tert-butyl dicarbonate (130 mg, 0.6 mmol, 1.5 EQ) and 4-(dimethylamino)pyridine (catalytic amount). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:2 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 7.64 (d, J = 8.1 Hz, 2H), 7.55 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.34–7.25 (m, 3H), 7.22 (d, J = 6.6 Hz, 2H), 5.05 (dd, J = 8.3, 5.0 Hz, 1H), 4.85 (d, J = 4.9 Hz, 1H), 4.66 (d, J = 15.4 Hz, 1H), 4.13 (d, J = 15.4 Hz, 1H), 1.39 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 166.14, 154.27, 138.46, 134.44, 128.99, 128.53, 128.20, 127.83, 125.56, 125.54, 80.39, 62.46, 60.92, 45.15, 27.86. HRMS (ESI+) m/z calc. for C22H23F3N2O3 420.1661, found [M + Na]+ 443.1550. Rf = 0.58 (EtOAc/n-Hex; 1:1, v/v).
tert-butyl (1-(2,4-dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamate (52), yield: 60%, colorless amorphous solid. The reaction was carried out according to General Procedure using 1-(2,4-dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-aminium chloride (40) (143 mg, 0.4 mmol, 1 EQ), triethylamine (0.061 mL, 0.44 mmol, 1 EQ), di-tert-butyl dicarbonate (130 mg, 0.6 mmol, 1.5 EQ) and 4-(dimethylamino)pyridine (catalytic amount). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:2 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.3 Hz, 1H), 6.39 (dd, J = 8.3, 2.3 Hz, 1H), 6.33 (d, J = 2.3 Hz, 1H), 5.15 (dd, J = 8.1, 5.0 Hz, 1H), 4.86 (d, J = 8.1 Hz, 1H), 4.77 (d, J = 4.9 Hz, 1H), 4.69 (d, J = 14.3 Hz, 1H), 4.13 (d, J = 14.3 Hz, 1H), 3.78 (s, 3H), 3.57 (s, 3H), 1.19 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 165.89, 161.27, 158.50, 154.33, 147.63, 143.08, 131.45, 128.10, 123.40, 114.79, 104.28, 98.36, 80.46, 62.34, 61.42, 55.42, 55.03, 40.41, 27.90. HRMS (ESI+) m/z calc. for C23H27N3O7 457.1849, found [M + H]+ 458.1917. Rf = 0.57 (EtOAc/n-Hex; 1:1, v/v).
tert-butyl (1-(2,4-dimethoxybenzyl)-2-isobutyl-4-oxoazetidin-3-yl)carbamate (53), yield: 54%, colorless amorphous solid. The reaction was carried out according to General Procedure using 3-amino-1-(2,4-dimethoxybenzyl)-4-isobutylazetidin-2-one (41) (138 mg, 0.47 mmol, 1 EQ), triethylamine (0.072 mL, 0.52 mmol, 1.1 EQ), di-tert-butyl dicarbonate (155 mg, 0.71 mmol, 1.5 EQ) and 4-(dimethylamino)pyridine (catalytic amount). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 6.88 (d, J = 8.3 Hz, 1H), 6.47–6.22 (m, 3H), 5.52 (br s, 1H), 5.20–4.90 (m, 1H), 4.80–4.50 (m, 2H), 4.17–3.92 (m, 2H), 3.85–3.75 (m, 6H), 2.38–2.17 (m, 1H), 1.49–1.44 (m, 9H), 0.96 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.35, 159.93, 157.42, 154.13, 128.19, 123.12, 104.07, 104.01, 98.26, 83.65, 55.26, 43.29, 43.01, 42.77, 42.40, 29.47, 27.94, 22.79. HRMS (ESI+) m/z calc. for C21H32N2O5 392.2311, found [M + H]+ 393.2384. Rf = 0.63 (EtOAc/n-Hex; 1:1, v/v).
4.9. General Procedure for the Synthesis of N1-Benzyl Deprotected ß Lactam (42–44)
Birch Reduction
In a flame-dried flask, Na dispersion in mineral oil (25 wt%, TCI, 6 EQ) and 15-crown-5 (6 EQ) were dissolved in dry tetrahydrofuran. The solution was warmed to room temperature under argon and stirred vigorously for 5 min. Then, the reaction mixture was cooled to 0 °C before a solution of ß-lactam (1 EQ), and isopropanol (6 EQ) in tetrahydrofuran was slowly added. After 15 min, the reaction was stopped by the addition of a saturated aqueous solution of NaHCO3 and diethyl ether. The aqueous phase was extracted with diethyl ether (2 × 30 mL). The combined organic phases were dried (Na2SO4), filtered, then concentrated in vacuo. The crude product thus obtained was purified by silica gel column chromatography using EtOAc:Hex as eluent.
N-(2,4-dimethoxybenzyl)-3-(p-tolyl)propanamide (42), colorless amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 8.0 Hz, 1H), 7.05 (s, 4H), 6.46–6.37 (m, 2H), 5.76 (br s, 1H), 4.32 (d, J = 5.7 Hz, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 2.94–2.85 (m, 2H), 2.47–2.38 (m, 2H), 2.30 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 171.69, 160.48, 158.52, 137.86, 135.54, 130.53, 129.12, 128.21, 118.88, 103.88, 98.55, 55.41, 55.28, 38.86, 38.72, 31.29, 21.00. HRMS (ESI+) m/z calc. for C19H23NO3 313.1678, found [M + H]+ 314.1745. Rf = 0.51 (EtOAc/n-Hex; 1:1, v/v).
tert-butyl (2-isobutyl-4-oxoazetidin-3-yl)carbamate (44), yield: 89%, transparent oil. The reaction was carried out according to General Procedure using Na dispersion in mineral oil (25 wt%, TCI, 131 mg, 13.5 mmol, 6 EQ),15-crown-5 were (0.283 mL, 13.5 mmol, 6 EQ), tert-butyl (1-benzyl-2-isobutyl-4-oxoazetidin-3-yl) carbamate (43) (75 mg, 2.25 mmol, 1 EQ) and isopropanol (0,109 mL, 13.5 mmol, 6 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent.. 1H NMR (400 MHz, CDCl3) δ 7.64 (br s, 1H), 6.69 (ddd, J = 14.3, 10.5, 1.2 Hz, 1H), 5.19 (dd, J = 14.3, 7.0 Hz, 1H), 5.15–5.07 (m, 1H), 3.82 (d, J = 5.9 Hz, 2H), 2.33 (dqd, J = 13.5, 6.8, 1.3 Hz, 1H), 1.47 (s, 9H), 1.01 (s, 3H), 1.00 (s, 3H). HRMS (ESI+) m/z calc. for C12H22N2O3 242.1630, found [M + H]+ 243.1685. Rf = 0.36 (EtOAc/n-Hex; 1:1, v/v).
4.10. General Procedure for the Synthesis of N1-Dimethoxybenzyl Deprotected ß Lactam (46–50)
Cerium Ammonium Nitrate
ß-Lactam (1 EQ) was dissolved in acetonitrile (25 mL/mmol) and distilled water (20 mL/mmol) and placed under argon. The solution was cooled to −10 °C with a sodium chloride ice bath. Cerium ammonium nitrate (3 EQ) was dissolved in distilled water and added dropwise to the vigorously stirring reaction mixture. The reaction was stirred at −10 °C for 1–2 h and then transferred to a separation funnel containing diethyl ether and saturated aqueous NaHCO3. The aqueous phase was washed with diethyl ether. The combined organic phases were dried over Na2SO4, and the solvents were evaporated. The solid was purified by silica gel column chromatography, using the gradient EtOAc: Hex as eluent.
tert-butyl (1-(2,4-dimethoxybenzoyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamate (44), light orange amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 8.7 Hz, 2H), 7.55–7.51 (m, 1H), 7.52 (d, J = 8.4, 2H), 6.61–6.51 (m, 2H), 5.59 (d, J = 6.4 Hz, 1H), 5.36–5.27 (m, 1H), 4.70 (d, J = 8.4 Hz, 1H), 3.91 (s, 3H), 3.88 (s, 3H), 1.28 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 164.61, 163.87, 160.10, 154.18, 147.80, 141.44, 132.04, 127.90, 123.80, 115.51, 105.18, 98.65, 81.20, 60.79, 59.56, 55.93, 55.58, 27.95. HRMS (ESI+) m/z calc. for C23H25N3O8 471.1642, found [M + Na]+ 494.1532. Rf = 0.43 (EtOAc/n-Hex; 2:1, v/v).
2-(2-Oxo-4-(4-(trifluoromethyl)phenyl)azetidin-3-yl)isoindoline-1,3-dione (46), yield: 65%, colorless amorphous solid. The reaction was carried out according to General Procedure using 4-(1-(2,4-dimethoxybenzyl)-3-(1,3-dioxoisoindolin-2-yl)-4-oxoazetidin-2-yl)benzonitrile (21) (156 mg, 0.31 mmol, 1 EQ) and cerium ammonium nitrate (502 mg, 0.92 mmol, 3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 1:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.72–7.60 (m, 4H), 7.54–7.41 (m, 4H), 7.28 (br s, 1H), 5.71 (dd, J = 5.4, 1.8 Hz, 1H), 5.26 (d, J = 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ = 166.66, 164.59, 138.82, 134.48, 131.00, 130.45, 130.13, 127.11, 125.43, 123.62, 60.37, 57.23. HRMS (ESI+) m/z calc. for C18H11F3N2O3 360.0722, found [M + H]+ 361.0791. Rf = 0.43 (EtOAc/n-Hex; 1:1, v/v).
2-(2-(3-Bromo-4-fluorophenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (47), yield: 40%, light yellow amorphous solid. The reaction was carried out according to General Procedure using 2-(2-(4-bromo-3-fluorophenyl)-1-(2,4-dimethoxybenzyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (22) (151 g, 0,29 mmol, 1 EQ) and cerium ammonium nitrate (473 mg, 0.86 mmol, 3EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.76–7.65 (m, 4H), 7.53 (dd, J = 6.4, 2.0 Hz, 1H), 7.29–7.23 (m, 1H), 7.03 (br s, 1H), 6.99 (t, J = 8.4 Hz, 1H), 5.63 (dd, J = 5.3, 1.9 Hz, 1H), 5.14 (d, J = 5.3 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 166.65, 164.37, 159.93, 157.46, 134.52, 132.15, 132.12, 131.98, 131.04, 127.46, 127.38, 123.69, 116.66, 116.44, 109.29, 109.07, 60.45, 56.58. HRMS (ESI+) m/z calc. for C17H10BrFN2O3 387.9859, found [M + H]+ 388.9936. Rf = 0.43 (EtOAc/n-Hex; 2:1, v/v).
tert-butyl (2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamate (48), yield: 51%, light red amorphous solid. The reaction was carried out according to General Procedure using tert-butyl (1-(2,4-dimethoxybenzyl)-2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamate (52) (2.5 g, 5.4 mmol, 1 EQ) and cerium ammonium nitrate (9 g, 16.4 mmol, 3 EQ). Product was purified by silica gel column chromatography using the gradient EtOAc: Hex = 1:1 to 4:1 as eluent. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (br s, 1H), 8.22 (d, J = 8.7 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 8.4 Hz, 1H), 5. 41–5.36 (m, 1H), 5.05–4.93 (m, 1H), 1.18 (s, J = 12.9 Hz, 9H). 13C NMR (100 MHz, DMSO-d6) δ 167.10, 155.14, 147.19, 145.99, 128.87, 123.40, 78.84, 63.68, 56.77, 28.27. HRMS (ESI+) m/z calc. for C14H17N3O5 307.1168, found [M + H]+ 308.1240. Rf = 0.23 (EtOAc/n-Hex; 2:1, v/v).
2-(2-(4-(Methylsulfonyl)phenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (49), yield: 43%, yellow amorphous solid. The reaction was carried out according to General Procedure using 2-(1-(2,4-Dimethoxybenzyl)-2-(4-(methylsulfonyl)phenyl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (25) (72 mg, 0.14 mmol, 1 EQ) and cerium ammonium nitrate (228 mg, 0.42 mmol, 3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.4 Hz, 2H), 7.74–7.64 (m, 4H), 7.54 (d, J = 8.2 Hz, 2H), 7.14 (br s, 1H), 5.72 (dd, J = 5.5, 1.9 Hz, 1H), 5.27 (d, J = 5.5 Hz, 1H), 2.90 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.58, 164.18, 141.21, 140.20, 134.62, 130.95, 127.79, 127.56, 123.74, 60.48, 57.18, 44.33. HRMS (ESI+) m/z calc. for C18H14N2O5S 370.0623, found [M + Na]+ 393.0530. Rf = 0.33 (EtOA).
2-(2-(Furan-2-yl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (50), yield: 15%, light brown oil. The reaction was carried out according to General Procedure 2-(1-(2,4-dimethoxybenzyl)-2-(furan-2-yl)-4-oxoazetidin-3-yl)isoindoline-1,3-dione (26) (206 mg, 0.48 mmol, 1 EQ) and cerium ammonium nitrate (0.84 g, 1.43 mmol, 3 EQ). Product was purified by silica gel column chromatography using EtOAc: n-Hex = 2:1 (v/v) as eluent. 1H NMR (400 MHz, DMSO-d6) δ 8.04–7.80 (m, 4H), 7.02 (dd, J = 10.6, 3.7 Hz, 1H), 6.91 (d, J = 7.3 Hz, 1H), 6.08 (dd, J = 10.6, 1.4 Hz, 1H), 5.87 (d, J = 6.0 Hz, 1H), 5.64 (dd, J = 6.0, 3.7 Hz, 1H), 4.55 (d, J = 6.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 191.40, 167.88, 165.11, 146.54, 135.84, 131.53, 128.34, 124.56, 67.63, 56.54, 53.78. HRMS (ESI+) m/z calc. for C15H10N2O4 282.0641, found [M + H]+ 283.0721. Rf = 0.33 (EtOAc/n-Hex; 2:1, v/v).
4.11. General Procedure for the Synthesis of tert-Butyloxycarbonyl Deprotected ß Lactam (54)
With Use of Trifluoroacetic Acid
tert-butyl (2-(4-nitrophenyl)-4-oxoazetidin-3-yl)carbamate (48) (150 mg, 0.5 mmol, 1 EQ) was dissolved in dry dichloromethane (2 mL), anisole (0.49 mL, 4.5 mmol, 9 EQ) was added, and the solution was cooled to −5 °C on a sodium chloride ice bath. Trifluoroacetic acid (1.53 mL, 20 mmol, 40 EQ) was added, and the solution was slowly warmed to room temperature. After stirring for 1.5 h, the solvent and the excess trifluoroacetic acid were evaporated. The residue was precipitated from methyl tert-butyl ether. The solid was used in the next step without further purification.
2-(4-Nitrophenyl)-4-oxoazetidin-3-aminium trifluoroacetate (54), yield: 83%, brown oil. 1H NMR (400 MHz, DMSO-d6) δ 9.24 (s, 1H), 8.46 (br s, 3H), 8.30 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 5.19 (d, J = 5.4 Hz, 1H), 4.86 (dd, J = 5.4, 1.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 163.43, 148.06, 142.37, 129.63, 123.92, 59.90, 49.18. HRMS (ESI+) m/z calc. for C9H9N3O3 207.0644, found [M + H]+ 208.0716. Rf = 0.08 (EtOAc/n-Hex; 9:1, v/v).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}