
Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations



Abstract
:1. Introduction
2. Results and Discussion
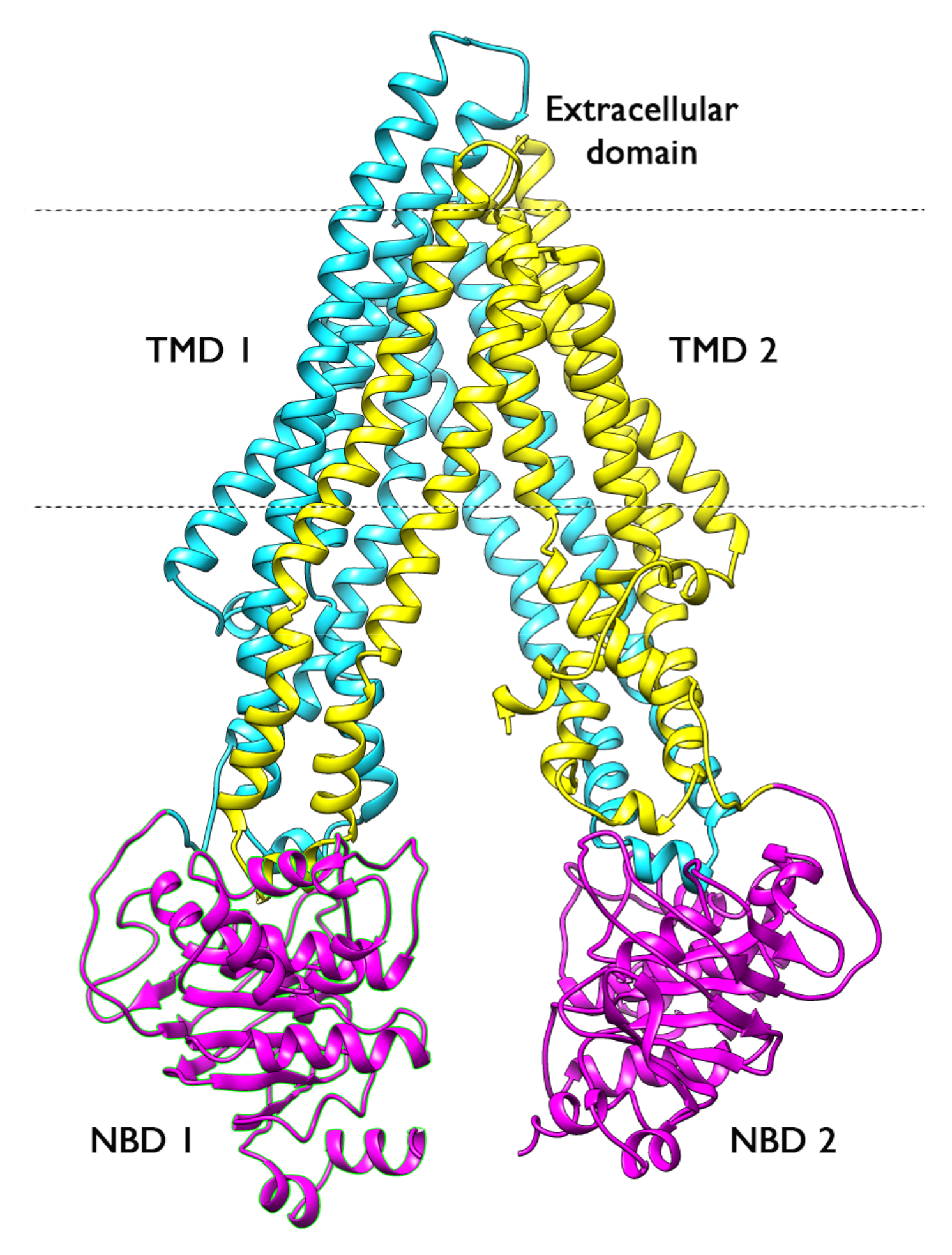
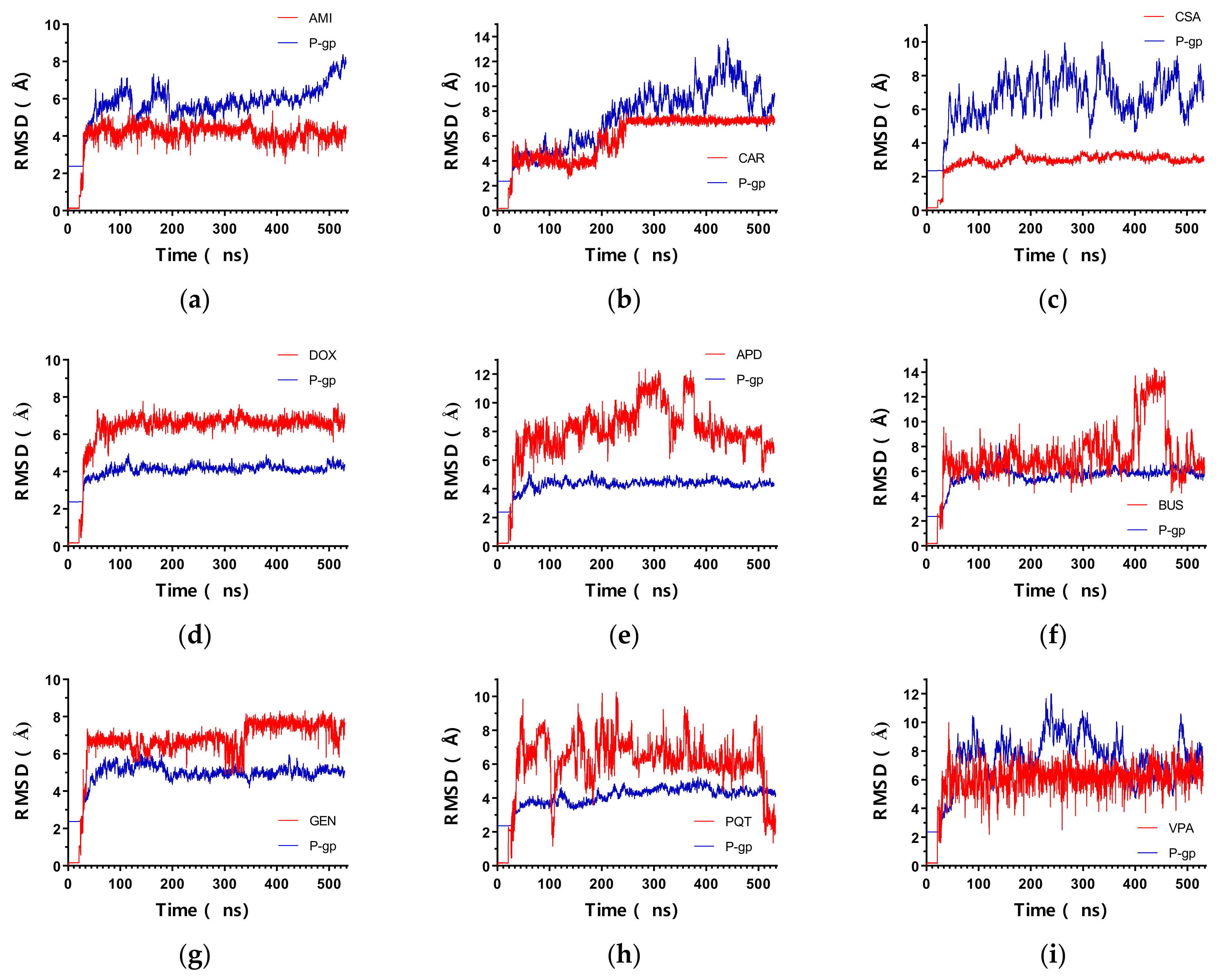
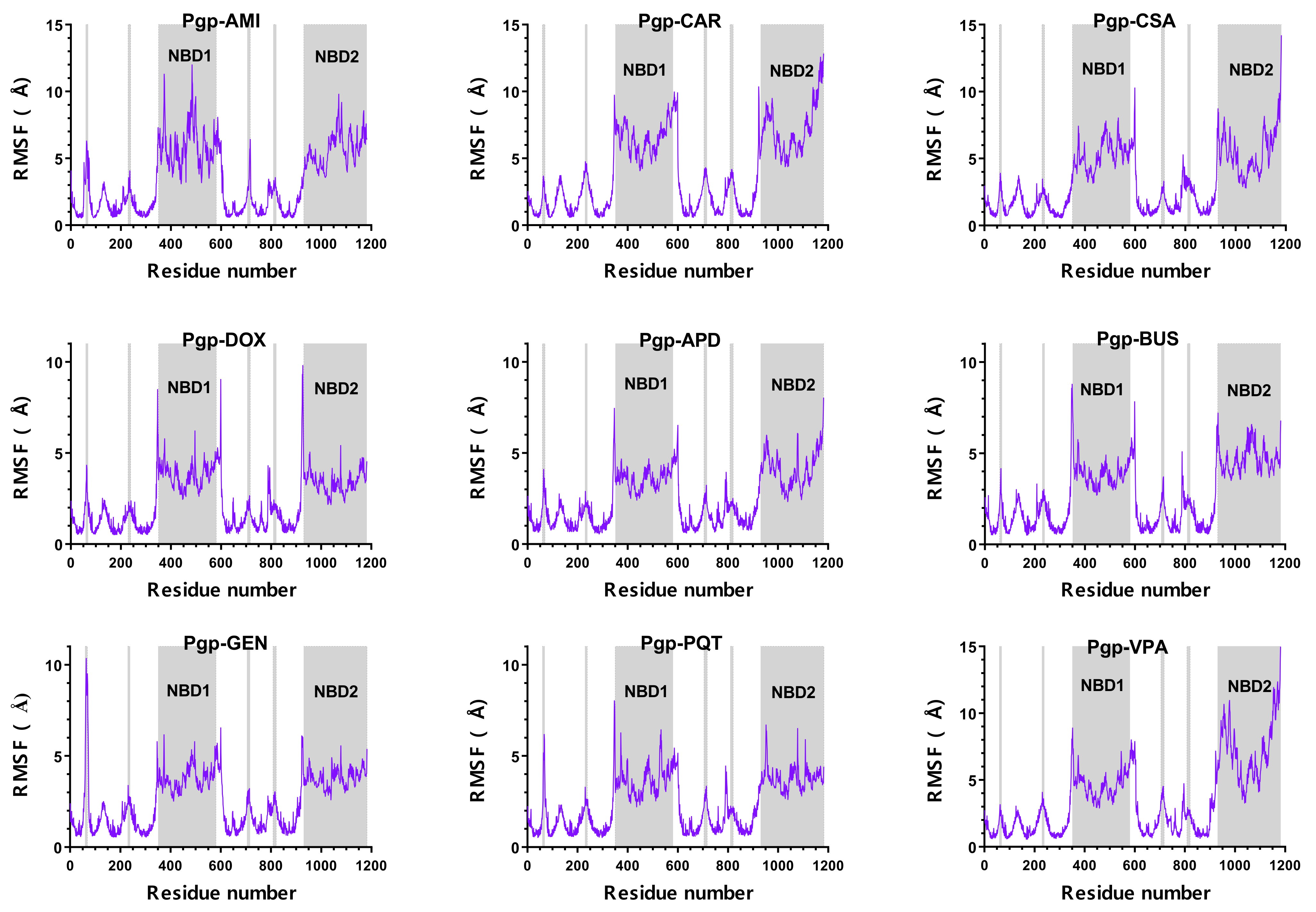
2.1. Overall Systems Dynamics
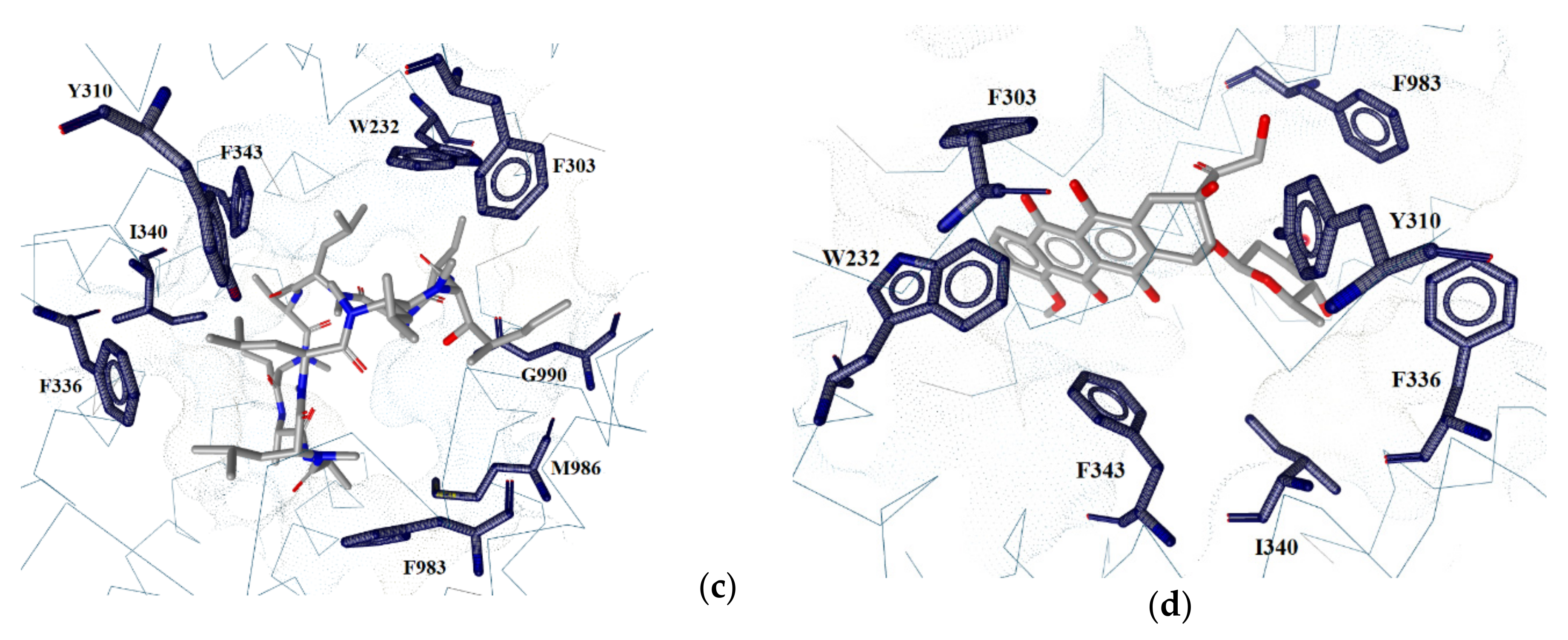
2.2. Ligand–Protein Interactions

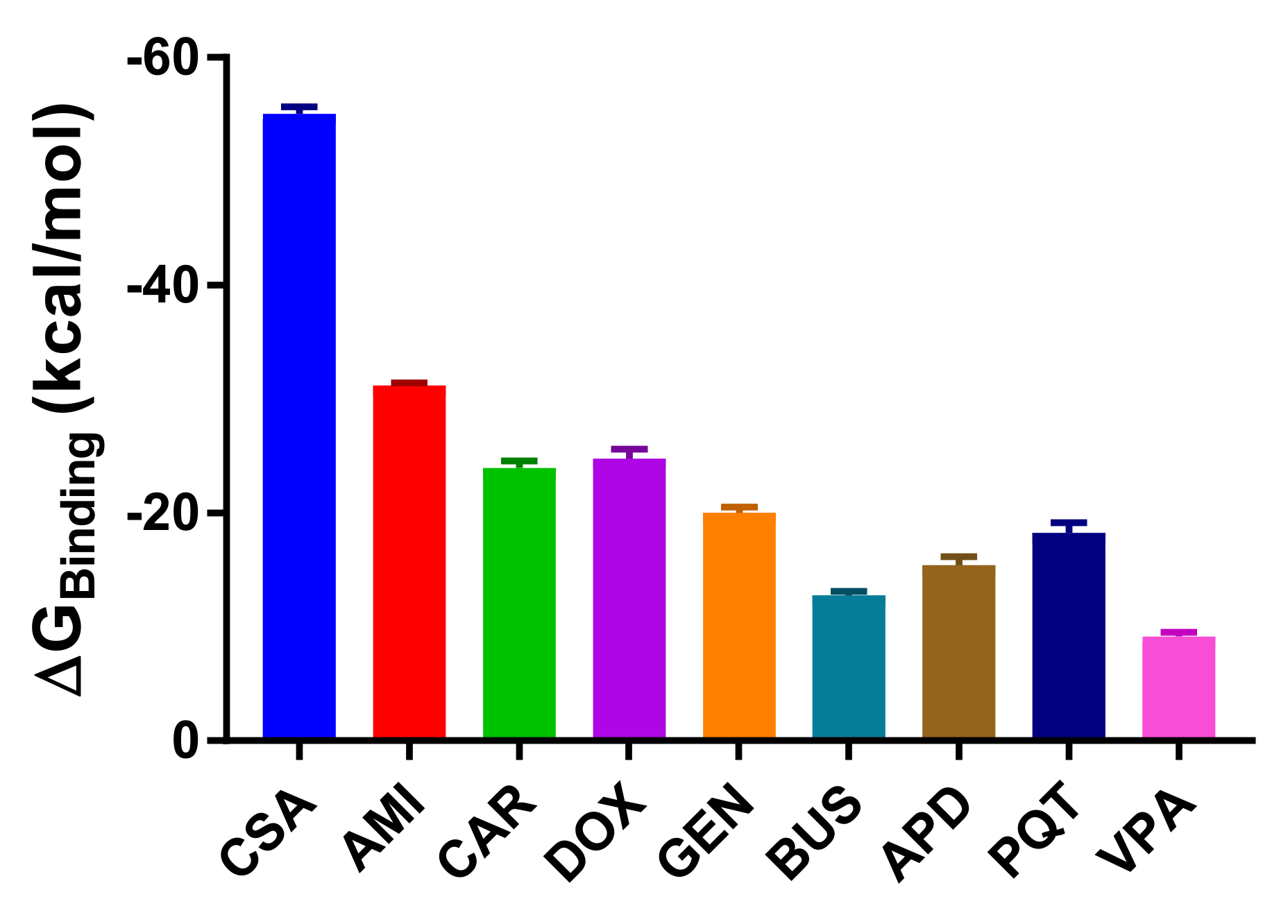
2.3. Binding Free Energy Calculations
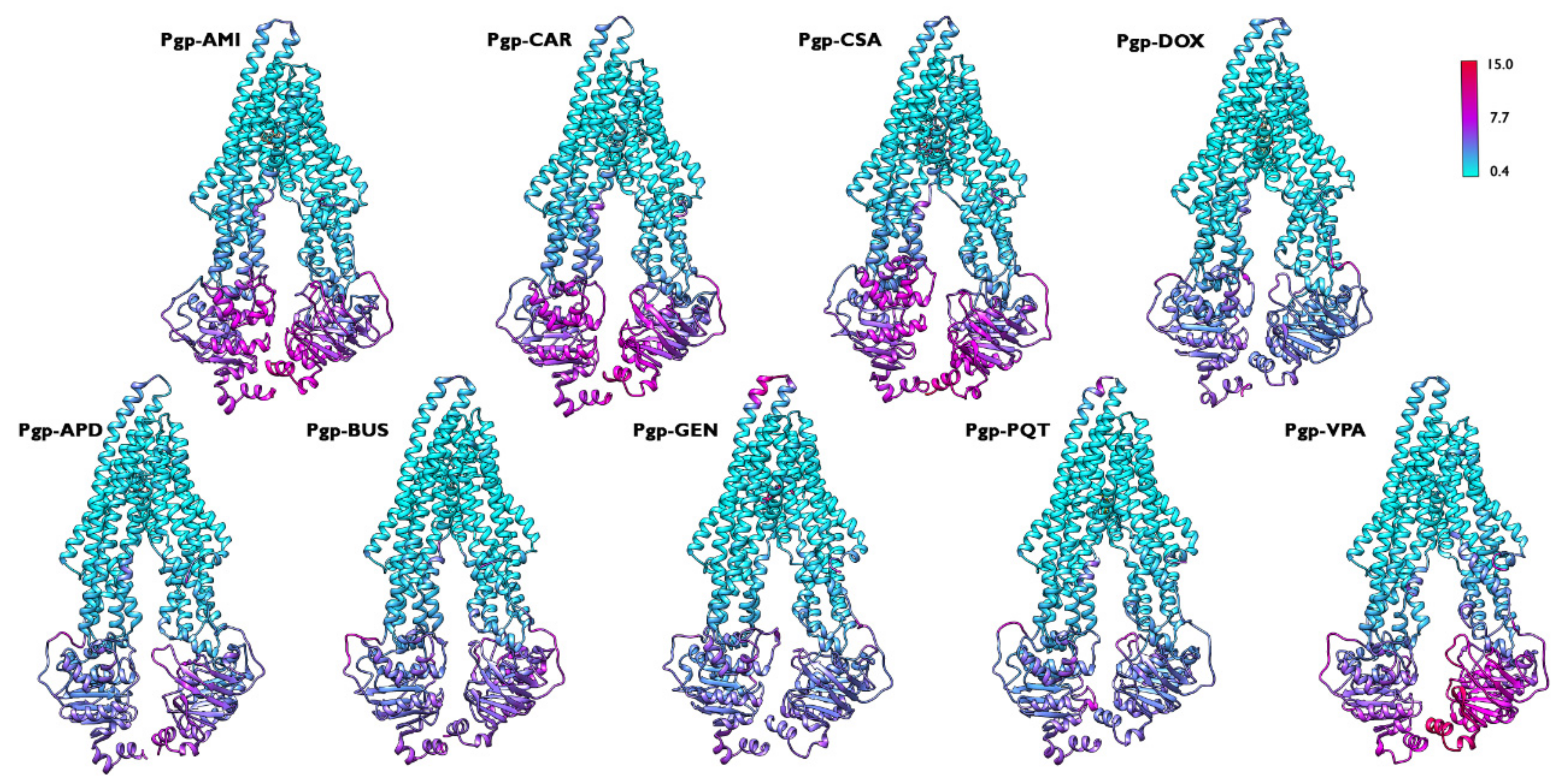
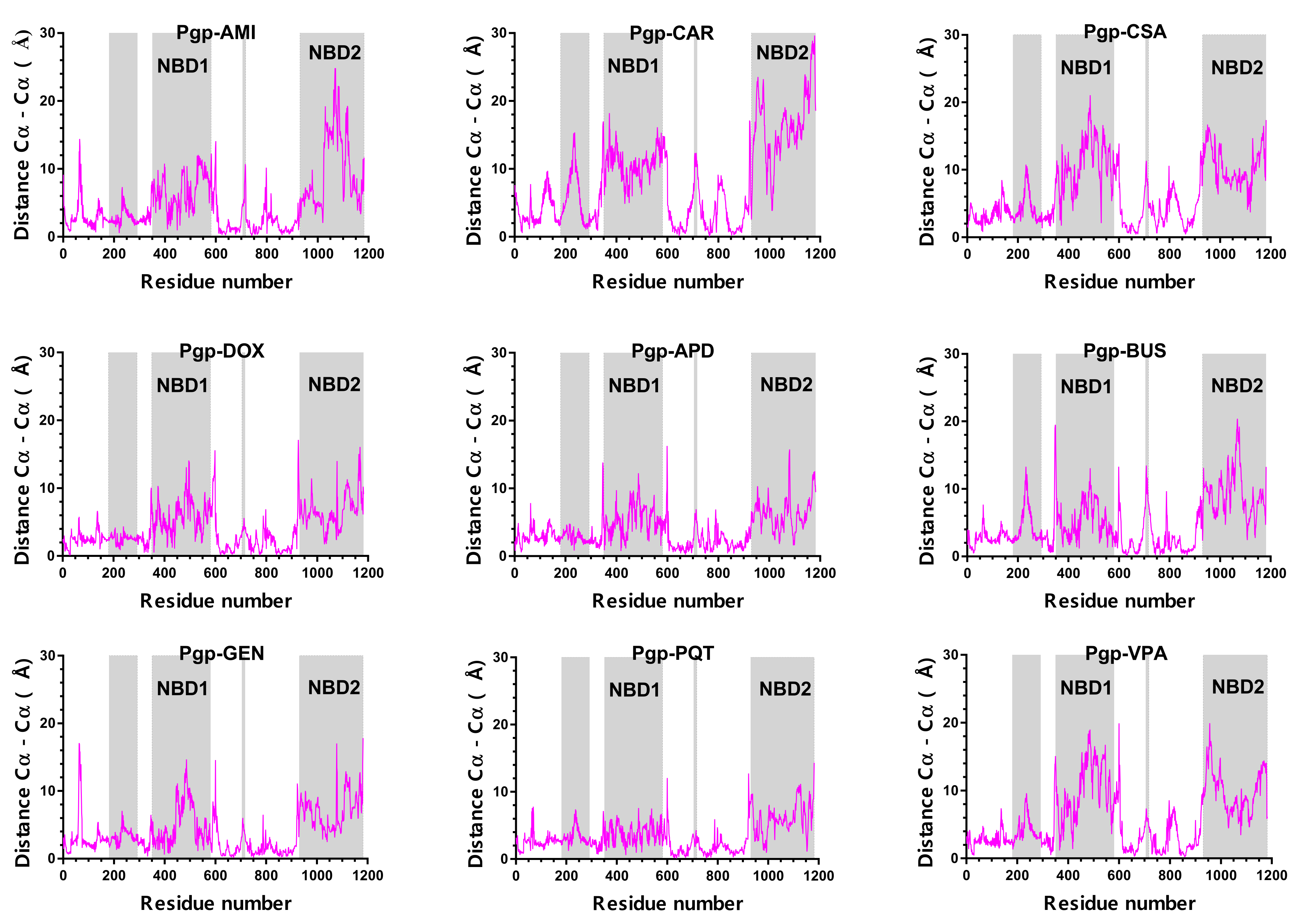
2.4. Structural Analysis
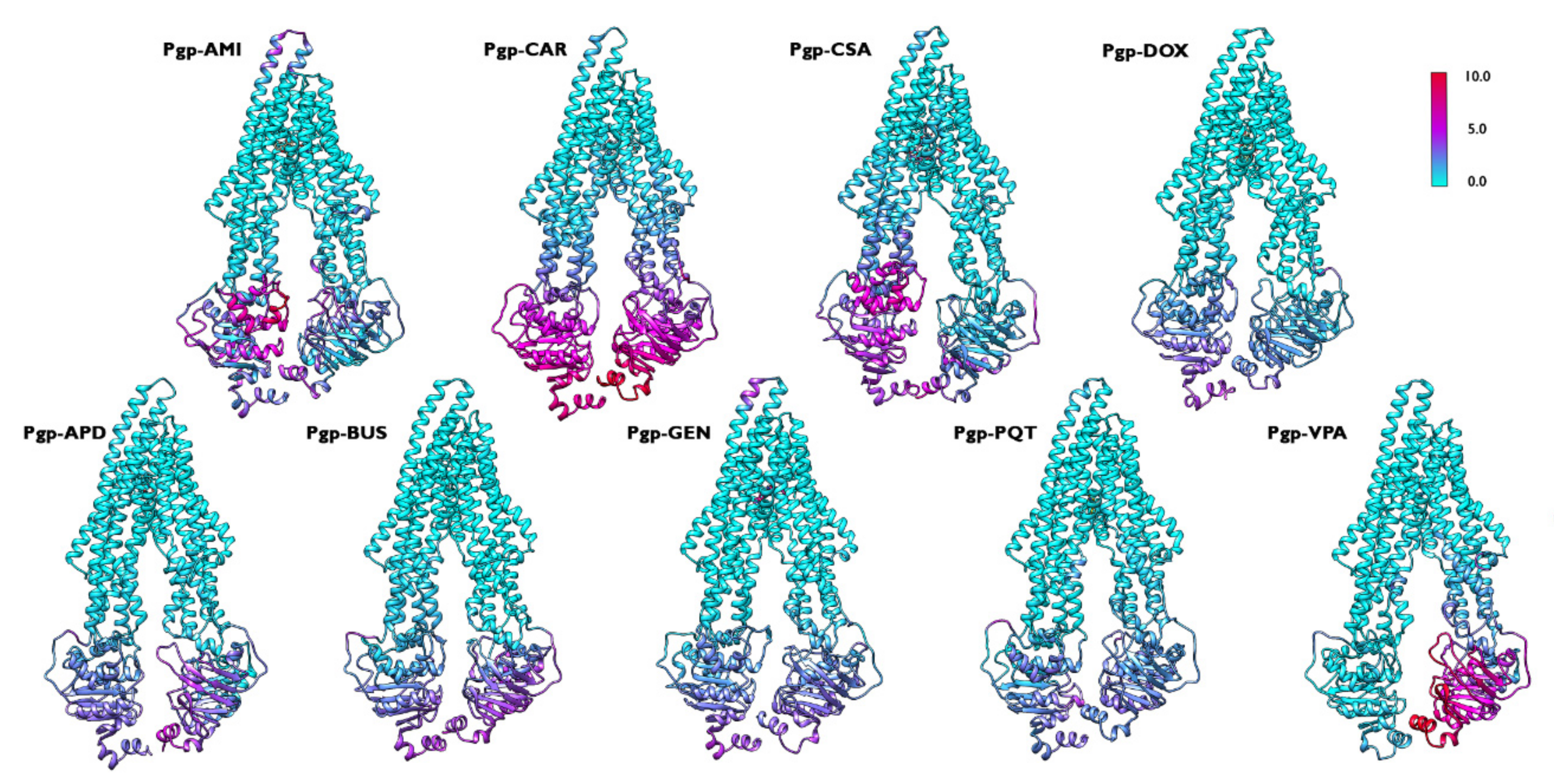
2.5. Concerted Motions in P-Glycoprotein
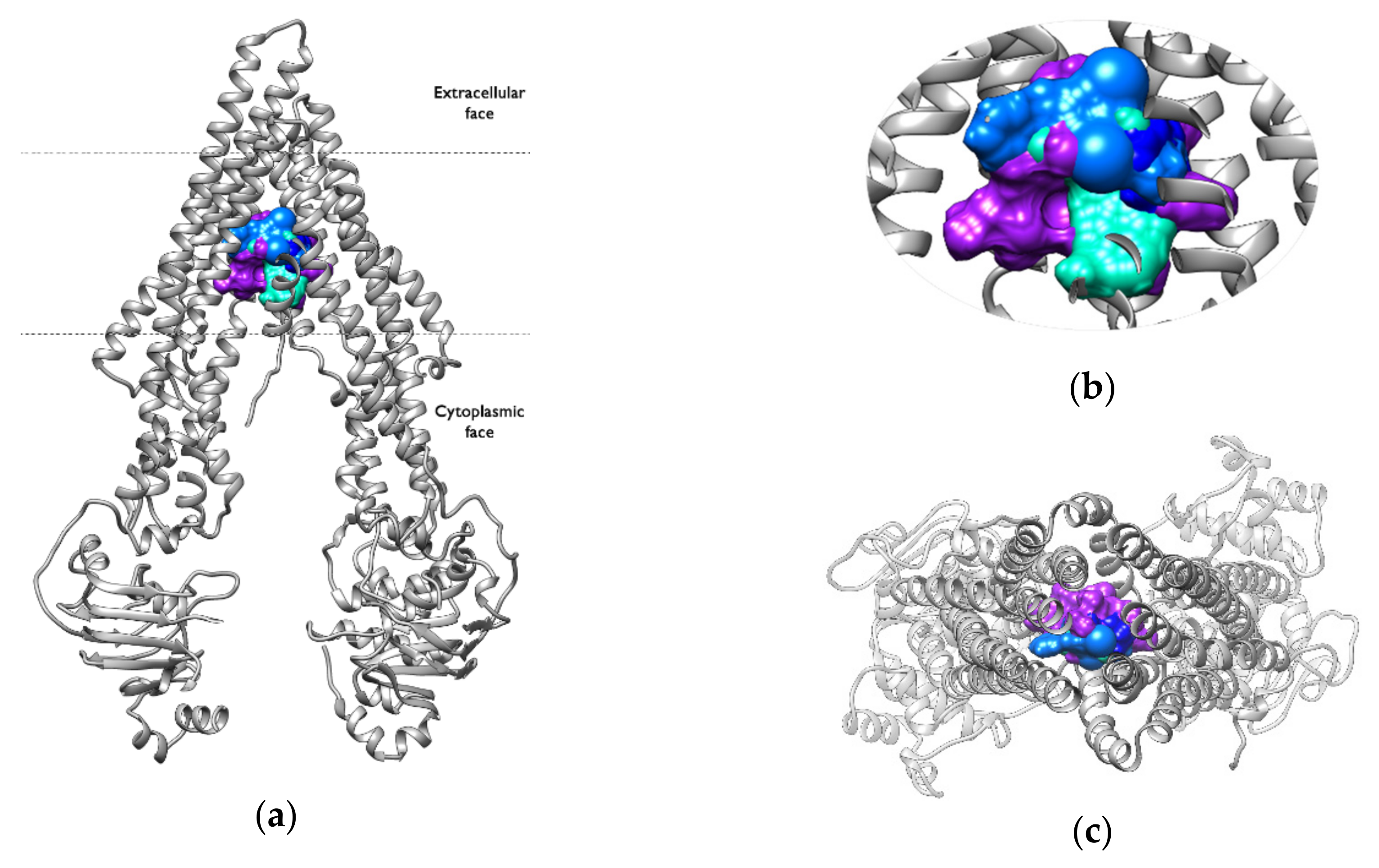
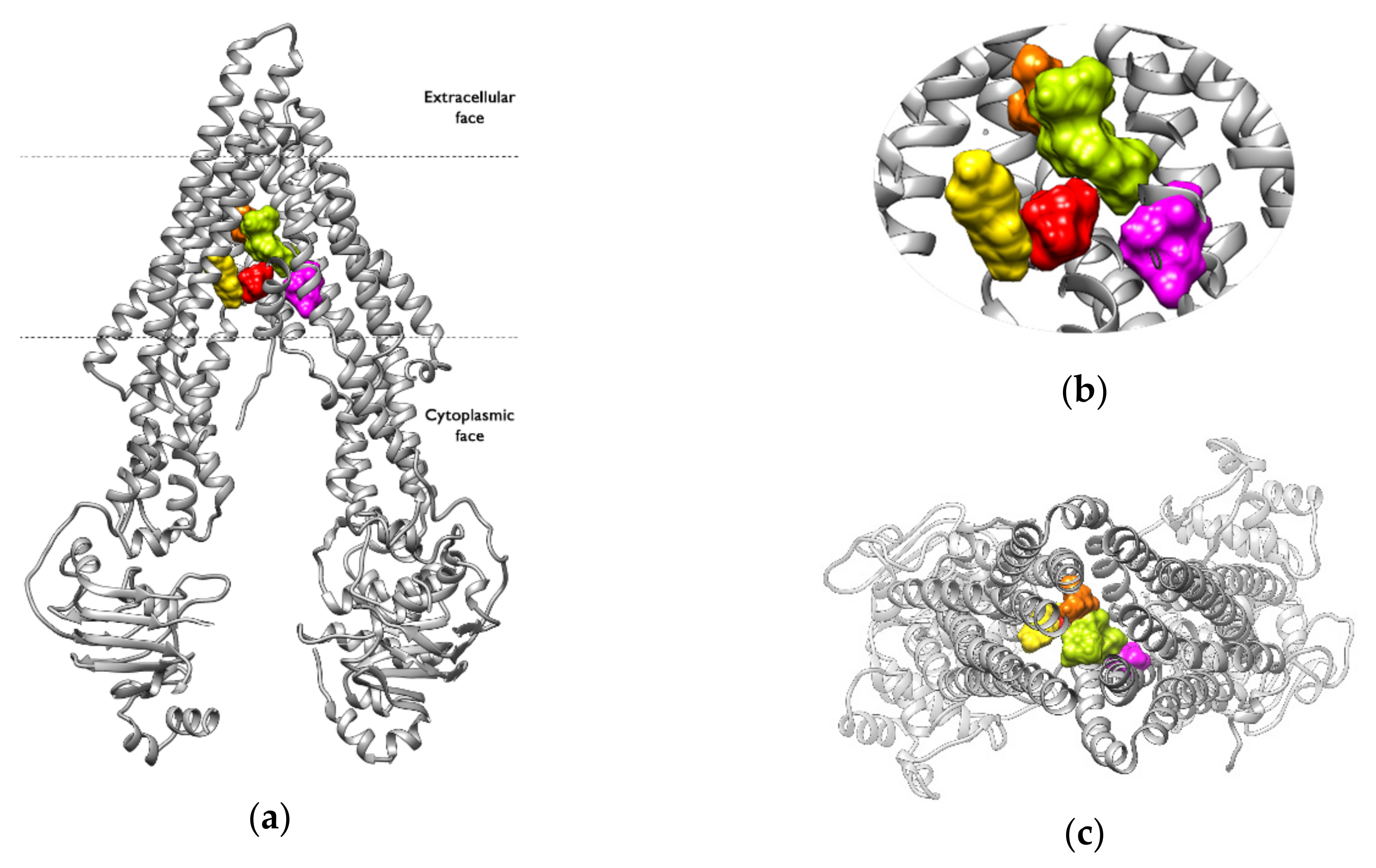
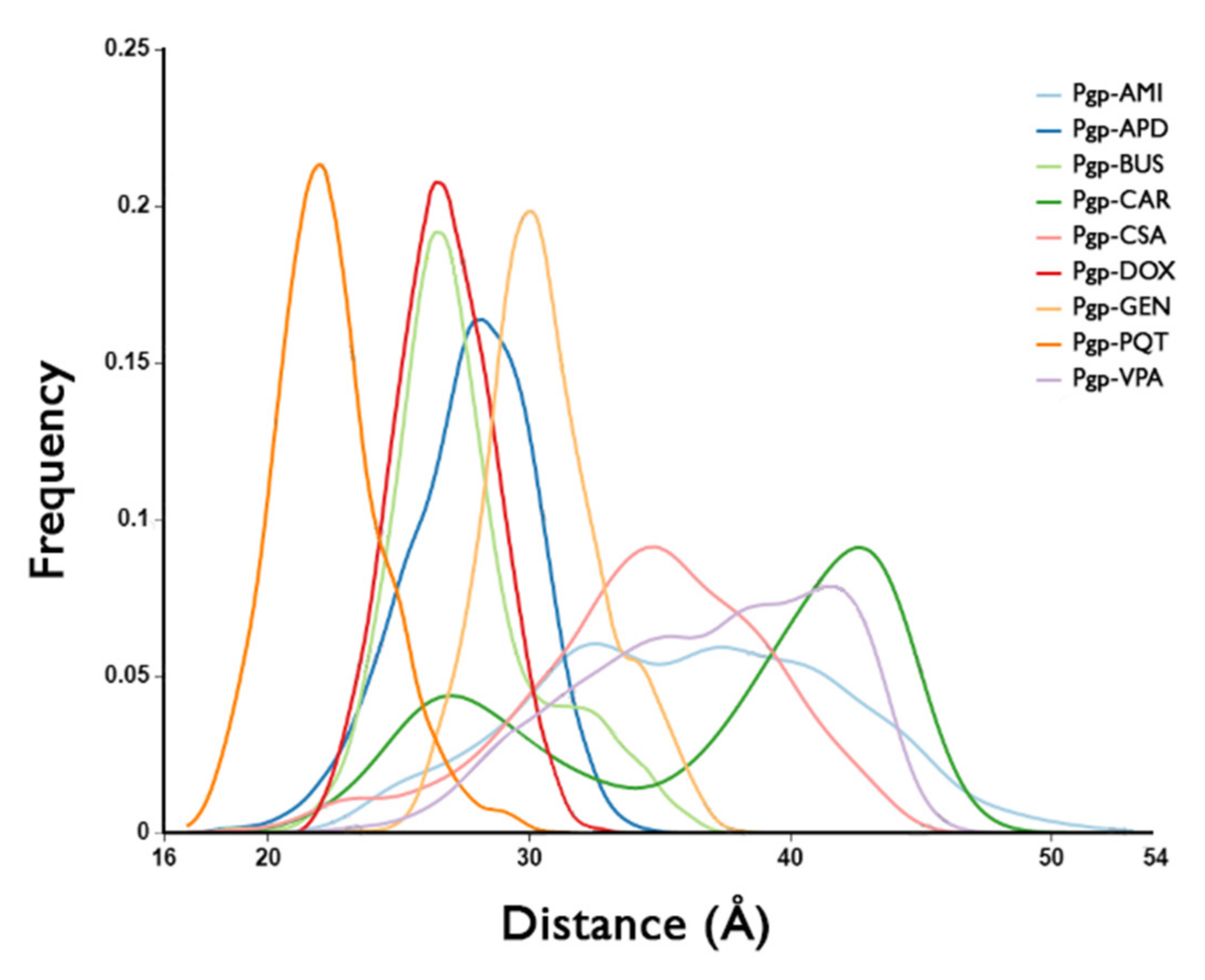
2.6. Binding Pocket

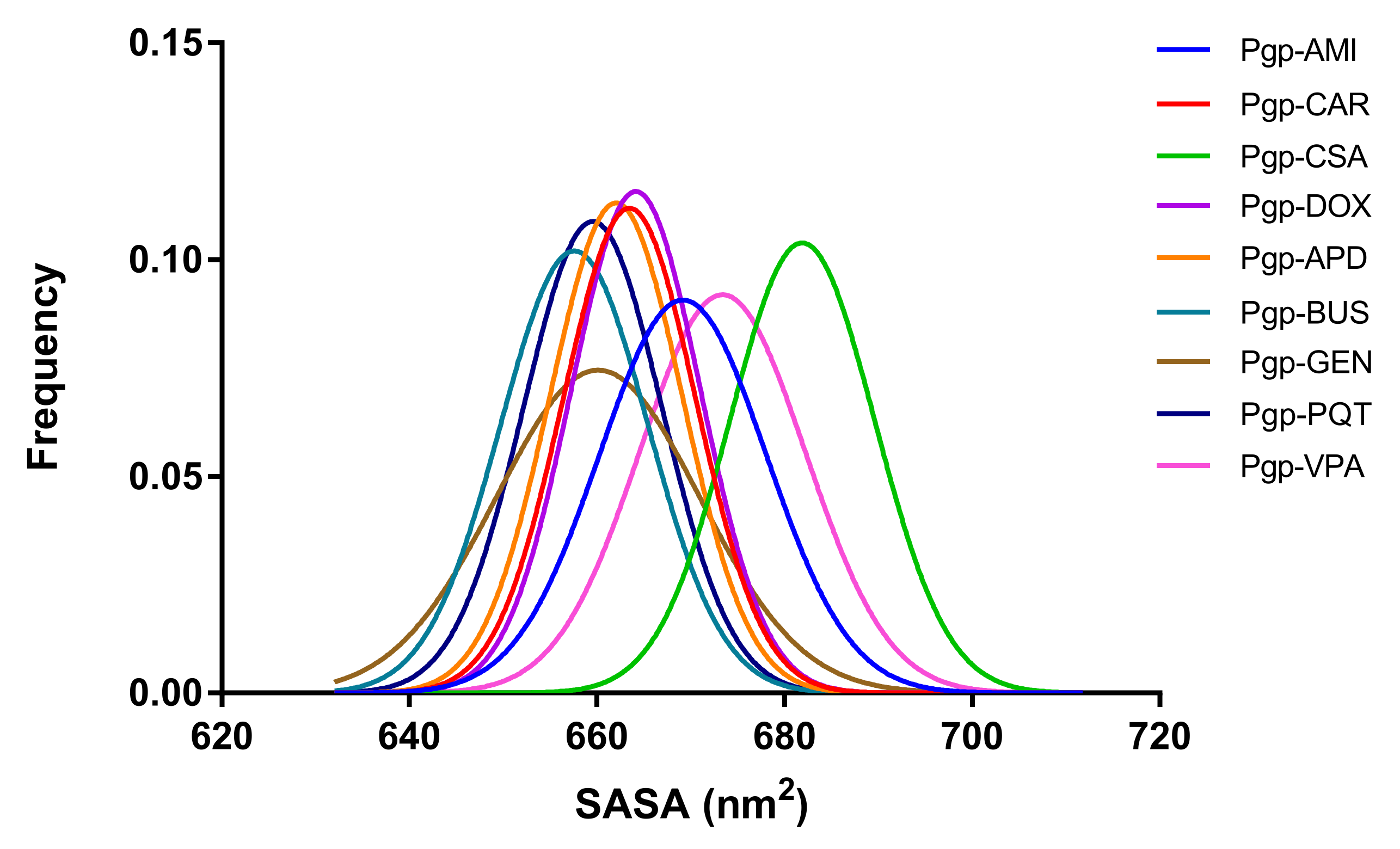
2.7. Exposure of Surfaces to the Solvent
3. Materials and Methods
3.1. Preparation of the Initial Structures
3.2. Systems Construction
3.3. Molecular Dynamics Simulations
3.3.1. Simulation Parameters
3.3.2. Energy Minimization
3.3.3. Heating
3.3.4. Equilibration
3.3.5. Production
3.4. Trajectory Analysis
3.4.1. Binding-Free Energy Calculations
3.4.2. Ligand–Protein Interactions
3.4.3. Clustering Analysis
3.4.4. Principal Component Analysis
3.4.5. Binding Pocket Volume
3.4.6. Solvent-Accessible Surface Area (SASA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lockhart, A.C.; Tirona, R.G.; Kim, R.B. Pharmacogenetics of ATP-binding cassette transporters in cancer and chemotherapy1. Mol. Cancer Ther. 2003, 2, 685–698. [Google Scholar]
- Lum, B.L.; Gosland, M.P.; Kaubisch, S.; Sikic, B.I. Molecular targets in oncology: Implications of the multidrug resistance gene. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1993, 13, 88–109. [Google Scholar]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Callaghan, R.; Ford, R.C.; Higgins, C.F. Structure of the multidrug resistance P-Glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem. 1997, 272, 10685–10694. [Google Scholar]
- Zhou, S.-F. Structure, function and regulation of P-Glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef]
- Martin, C.; Berridge, G.; Mistry, P.; Higgins, C.; Charlton, P.; Callaghan, R. Drug binding sites on P-Glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry 2000, 39, 11901–11906. [Google Scholar] [CrossRef]
- Martin, C.; Higgins, C.F.; Callaghan, R. The vinblastine binding site adopts high-and low-affinity conformations during a transport cycle of P-Glycoprotein. Biochemistry 2001, 40, 15733–15742. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Velarde, G.; Ford, R.C.; Martin, C.; Berridge, G.; Kerr, I.D.; Callaghan, R.; Schmidlin, A.; Wooding, C.; Linton, K.J.; et al. Repacking of the transmembrane domains of P-Glycoprotein during the transport ATPase cycle. EMBO J. 2001, 20, 5615–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauna, Z.E.; Ambudkar, S.V. About a switch: How P-Glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 2007, 6, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-Glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Chen, J. Molecular structure of human P-Glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Broccatelli, F.; Carosati, E.; Neri, A.; Frosini, M.; Goracci, L.; Oprea, T.I.; Cruciani, G. A novel approach for predicting P-Glycoprotein (ABCB1) inhibition using molecular interaction fields. J. Med. Chem. 2011, 54, 1740–1751. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, Y.; Zhao, Q.; Peng, H.; Hou, T. ADME evaluation in drug discovery. 10. Predictions of P-Glycoprotein inhibitors using recursive partitioning and naive Bayesian classification techniques. Mol. Pharm. 2011, 8, 889–900. [Google Scholar] [CrossRef]
- Li, D.; Chen, L.; Li, Y.; Tian, S.; Sun, H.; Hou, T. ADMET evaluation in drug discovery. 13. Development of in silico prediction models for P-Glycoprotein substrates. Mol. Pharm. 2014, 11, 716–726. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Minovski, N.; Novič, M. Multiclass classifier for P-Glycoprotein substrates, inhibitors, and non-active compounds. Molecules 2019, 24, 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolghih, E.; Bryant, C.; Renslo, A.R.; Jacobson, M.P. Predicting binding to P-Glycoprotein by flexible receptor docking. PLoS Comput. Biol. 2011, 7, e1002083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology modeling of the human P-Glycoprotein (ABCB1) and insights into ligand binding through molecular docking studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; Dos Santos, D.J. Insights on P-Glycoprotein’s efflux mechanism obtained by molecular dynamics simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- O’Mara, M.L.; Mark, A.E. The effect of environment on the structure of a membrane protein: P-Glycoprotein under physiological conditions. J. Chem. Theory Comput. 2012, 8, 3964–3976. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, Z.; Zhang, J.; Kang, Y.; Liang, L.; Liu, Y.; Wang, Q. Simultaneous binding mechanism of multiple substrates for multidrug resistance transporter P-Glycoprotein. Phys. Chem. Chem. Phys. 2021, 23, 4530–4543. [Google Scholar] [CrossRef]
- Saeki, T.; Ueda, K.; Tanigawara, Y.; Hori, R.; Komano, T. Human P-Glycoprotein transports cyclosporin A and FK506. J. Biol. Chem. 1993, 268, 6077–6080. [Google Scholar] [CrossRef]
- Wigler, P.W. PSC833, cyclosporinA, and dexniguldipine effects on cellular calcein retention and inhibition of the multidrug resistance pump in human leukemic lymphoblasts. Biochem. Biophys. Res. Commun. 1999, 257, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein inhibitory potential using a rhodamine 123 accumulation assay. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Murase, O.; Schowen, R.L.; Aubé, J.; Borchardt, R.T. A functional assay for quantitation of the apparent affinities of ligands of P-Glycoprotein in Caco-2 cells. Pharm. Res. 2001, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Takara, K.; Tanigawara, Y.; Komada, F.; Nishiguchi, K.; Sakaeda, T.; Okumura, K. Cellular pharmacokinetic aspects of reversal effect of itraconazole on P-Glycoprotein-mediated resistance of anticancer drugs. Biol. Pharm. Bull. 1999, 22, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Lacher, S.E.; Gremaud, J.N.; Skagen, K.; Steed, E.; Dalton, R.; Sugden, K.D.; Cardozo-Pelaez, F.; Sherwin, C.M.; Woodahl, E.L. Absence of P-Glycoprotein transport in the pharmacokinetics and toxicity of the herbicide paraquat. J. Pharmacol. Exp. Ther. 2014, 348, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltes, S.; Fedrowitz, M.; Tortos, C.L.; Potschka, H.; Löscher, W. Valproic acid is not a substrate for P-Glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J. Pharmacol. Exp. Ther. 2007, 320, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Eyal, S.; Lamb, J.G.; Smith-Yockman, M.; Yagen, B.; Fibach, E.; Altschuler, Y.; White, H.S.; Bialer, M. The antiepileptic and anticancer agent, valproic acid, induces P-Glycoprotein in human tumour cell lines and in rat liver. Br. J. Pharmacol. 2006, 149, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Liu, H.Y.; Liu, X.; Zhang, D.M.; Liu, Y.C.; Liu, X.D.; Wang, G.J.; Xie, L. Increased P-Glycoprotein function and level after long-term exposure of four antiepileptic drugs to rat brain microvascular endothelial cells in vitro. Neurosci. Lett. 2008, 434, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-Glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.S.; Oldham, M.L.; Zhang, Q.; Chen, J. Crystal structure of the multidrug transporter P-Glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekins, S.; Kim, R.B.; Leake, B.F.; Dantzig, A.H.; Schuetz, E.G.; Lan, L.B.; Yasuda, K.; Shepard, R.L.; Winter, M.A.; Schuetz, J.D.; et al. Three-dimensional quantitative structure-activity relationships of inhibitors of P-Glycoprotein. Mol. Pharmacol. 2002, 61, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; Pajeva, I.K. Structure-activity relationships of multidrug resistance reversers. Curr. Med. Chem. 2001, 8, 685–713. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Nicklisch, S.C.T.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-Glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, S.I.; Cornwell, M.M.; Kuwano, M.I.C.H.I.H.I.K.O.; Pastan, I.; Gottesman, M.M. Most drugs that reverse multidrug resistance also inhibit photoaffinity labeling of P-Glycoprotein by a vinblastine analog. Mol. Pharmacol. 1988, 33, 144–147. [Google Scholar]
- Litman, T.; Zeuthen, T.; Skovsgaard, T.; Stein, W.D. Competitive, non-competitive and cooperative interactions between substrates of P-Glycoprotein as measured by its ATPase activity. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 1997, 1361, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, A.; Chung, G.; Lynn, A.; Yalvigi, A.; Brown, C.; Ellens, H.; O’Connor, M.; Lee, C.; Bentz, J. Derivation of a System-Independent Ki for P-Glycoprotein Mediated Digoxin Transport from System-Dependent IC50 Data. Drug Metab. Dispos. 2018, 46, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Melchior, D.L.; Sharom, F.J.; Evers, R.; Wright, G.E.; Chu, J.W.; Wright, S.E.; Chu, X.; Yabut, J. Determining P-Glycoprotein–drug interactions: Evaluation of reconstituted P-Glycoprotein in a liposomal system and LLC-MDR1 polarized cell monolayers. J. Pharmacol. Toxicol. Methods 2012, 65, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharom, F.J.; Liu, R.; Romsicki, Y. Spectroscopic and biophysical approaches for studying the structure and function of the P-Glycoprotein multidrug transporter. Biochem. Cell Biol. 1998, 76, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Domicevica, L.; Biggin, P.C. Homology modelling of human P-Glycoprotein. Biochem. Soc. Trans. 2015, 43, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Case, D.A.; Walker, R.C.; Cheatham, T.E., III; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T.; Wang, J.; Duke, R.E.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Accelrys, P.E. Discovery Studio Modeling Environment; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2017. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. [Google Scholar]
- Zhang, L.; Hermans, J. Hydrophilicity of cavities in proteins. Proteins Struct. Funct. Bioinform. 1996, 24, 433–438. [Google Scholar] [CrossRef]
- Heller, H.; Schaefer, M.; Schulten, K. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal phase. J. Phys. Chem. 1993, 97, 8343–8360. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Crowley, M.F.; Williamson, M.J.; Walker, R.C. CHAMBER: Comprehensive support for CHARMM force fields within the AMBER software. Int. J. Quantum Chem 2009, 109, 3767–3772. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.R.; Sørensen, J.; Hensley, N.; Wong, C.; Zhu, C.; Perison, T.; Amaro, R.E. POVME 3.0: Software for mapping binding pocket flexibility. J. Chem. Theory Comput. 2017, 13, 4584–4592. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate−DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Morris, J.H.; Huang, C.C.; Babbitt, P.C.; Ferrin, T.E. StructureViz: Linking cytoscape and UCSF chimera. Bioinformatics 2007, 23, 2345–2347. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [Green Version]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Sekhar, A.; Vallurupalli, P.; Kay, L.E. Defining a length scale for millisecond-timescale protein conformational exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 11391–11396. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Simulation Contacts | Type of Residue | |||
|---|---|---|---|---|---|
| Non-Bonded | Hydrogen Bond | Aromatic | Aliphatic | Polar | |
| AMI 1 | 16 | 1 | 8 | 8 | 5 |
| CAR 2 | 11 | 2 | 6 | 5 | 2 |
| CSA 3 | 25 | 2 | 8 | 17 | 9 |
| DOX 4 | 12 | 1 | 6 | 6 | 2 |
| APD 5 | 5 | 1 | 1 | 4 | 3 |
| BUS 6 | 7 | 0 | 4 | 2 | 1 |
| GEN 7 | 11 | 1 | 5 | 6 | 2 |
| PQT 8 | 3 | 0 | 0 | 3 | 2 |
| VPA 9 | 3 | 0 | 1 | 1 | 0 |
| Eigenvector | Cumulated Eigenvalues Expressed in Percent (%) 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| AMI 2 | CAR 3 | CSA 4 | DOX 5 | APD 6 | BUS 7 | GEN 8 | PQT 9 | VPA 10 | |
| 1 | 34.02 | 58.04 | 33.35 | 28.39 | 36.07 | 37.58 | 32.61 | 30.24 | 40.62 |
| 2 | 54.10 | 71.55 | 54.74 | 44.49 | 50.55 | 55.95 | 50.48 | 50.83 | 60.08 |
| 3 | 72.36 | 84.42 | 73.97 | 65.63 | 68.96 | 73.99 | 66.34 | 69.08 | 79.22 |
| 4 | 77.40 | 87.92 | 79.90 | 71.06 | 73.86 | 77.89 | 71.59 | 73.77 | 84.27 |
| 5 | 81.31 | 90.28 | 83.37 | 74.68 | 77.73 | 80.48 | 75.29 | 76.59 | 87.40 |
| 6 | 84.36 | 91.67 | 85.85 | 78.10 | 80.73 | 82.94 | 77.94 | 78.86 | 88.90 |
| 7 | 86.74 | 92.75 | 87.37 | 80.78 | 82.66 | 85.18 | 80.06 | 80.59 | 90.12 |
| 8 | 88.31 | 93.48 | 88.43 | 82.37 | 84.11 | 86.38 | 81.76 | 82.18 | 91.14 |
| 9 | 89.34 | 94.09 | 89.39 | 83.78 | 85.41 | 87.52 | 83.39 | 83.35 | 92.01 |
| 10 | 90.14 | 94.66 | 90.14 | 85.05 | 86.46 | 88.43 | 84.60 | 84.44 | 92.69 |
| 11 | 90.70 | 95.13 | 90.86 | 86.00 | 87.32 | 89.11 | 85.57 | 85.42 | 93.25 |
| 12 | 91.20 | 95.43 | 91.49 | 86.81 | 88.06 | 89.76 | 86.40 | 86.29 | 93.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 362. https://doi.org/10.3390/ijms23010362
Mora Lagares L, Pérez-Castillo Y, Minovski N, Novič M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2022; 23(1):362. https://doi.org/10.3390/ijms23010362
Chicago/Turabian StyleMora Lagares, Liadys, Yunierkis Pérez-Castillo, Nikola Minovski, and Marjana Novič. 2022. "Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations" International Journal of Molecular Sciences 23, no. 1: 362. https://doi.org/10.3390/ijms23010362
APA StyleMora Lagares, L., Pérez-Castillo, Y., Minovski, N., & Novič, M. (2022). Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. International Journal of Molecular Sciences, 23(1), 362. https://doi.org/10.3390/ijms23010362