
Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges



Abstract
:1. Introduction
2. Potential Neuroprotective Mechanisms of Action of Metformin
2.1. AMPK-Mediated Autophagy Activation
2.2. Decreased Accumulation of α-Synuclein Pathological Species
2.3. Inhibition of Mitochondrial Complex I and Regulation of Mitochondrial Dynamics
2.4. Anti-Inflammatory Action
3. Metformin as a Disease Modifier for Parkinson Disease: How Effective Is It?
3.1. Evidence from Epidemiological Studies on T2DM Patients
3.2. Metformin Bioavailability in the Brain
3.3. Side Effects Associated with Prolonged Metformin Consumption
3.4. Metformin Treatments in Experimental Models versus Human Subjects: Looking for a Key of Interpretation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloemd, B.R. The emerging evidence of the Parkinson pandemic. J. Parkinson’s Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Singh, H.; Medhi, B. Old wines in new bottles: Repurposing opportunities for Parkinson’s disease. Eur. J. Pharmacol. 2018, 830, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. Drug Repurposing in Parkinson’s Disease. CNS Drugs 2018, 32, 747–761. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Outeiro, T.F.; Bubacco, L.; Bisaglia, M. Diabetes Mellitus as a Risk Factor for Parkinson’s Disease: A Molecular Point of View. Mol. Neurobiol. 2018, 55, 8754–8763. [Google Scholar] [CrossRef]
- Baker, C.; Retzik-Stahr, C.; Singh, V.; Plomondon, R.; Anderson, V.; Rasouli, N. Should metformin remain the first-line therapy for treatment of type 2 diabetes? Ther. Adv. Endocrinol. Metab. 2021, 12, 2042018820980225. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Jia, Y.; Lao, Y.; Zhu, H.; Li, N.; Leung, S.W. Is metformin still the most efficacious first-line oral hypoglycaemic drug in treating type 2 diabetes? A network meta-analysis of randomized controlled trials. Obes. Rev. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, M.C.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 614, 607–614. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, A.; Westenberger, M.; Piechotta, C.; Lehto, A.; Wirth, F.; Lau, H.; Klein, J. Cholinergic and metabolic effects of metformin in mouse brain. Brain Res. Bull. 2021, 170, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katila, N.; Bhurtel, S.; Park, P.H.; Choi, D.Y. Metformin attenuates rotenone-induced oxidative stress and mitochondrial damage via the AKT/Nrf2 pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef] [PubMed]
- Tayara, K.; Espinosa-Oliva, A.M.; García-Domínguez, I.; Ismaiel, A.A.; Boza-Serrano, A.; Deierborg, T.; Machado, A.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Divergent effects of metformin on an inflammatory model of Parkinson’s disease. Front. Cell. Neurosci. 2018, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ou, Y.; Li, Y.; Hu, S.; Shao, L.W.; Liu, Y. Metformin extends C. Elegans lifespan through lysosomal pathway. eLife 2017, 6, 1–17. [Google Scholar] [CrossRef]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Foley, A.; Partridge, L. Activation of AMPK by the Putative Dietary Restriction Mimetic Metformin Is Insufficient to Extend Lifespan in Drosophila. PLoS ONE 2012, 7, e47699. [Google Scholar] [CrossRef]
- Na, H.J.; Park, J.S.; Pyo, J.H.; Jeon, H.J.; Kim, Y.S.; Arking, R.; Yoo, M.A. Metformin inhibits age-related centrosome amplification in Drosophila midgut stem cells through AKT/TOR pathway. Mech. Ageing Dev. 2015, 149, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Pollak, M.; Zhang, Y.; Yu, Y.; Becker, K.G.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 100–106. [Google Scholar] [CrossRef]
- Saewanee, N.; Praputpittaya, T.; Malaiwong, N.; Chalorak, P.; Meemon, K. Neuroprotective effect of metformin on dopaminergic neurodegeneration and α-synuclein aggregation in C. elegans model of Parkinson’s disease. Neurosci. Res. 2021, 162, 13–21. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ozbey, G.; Nemutlu-Samur, D.; Parlak, H.; Yildirim, S.; Aslan, M.; Tanriover, G.; Agar, A. Metformin protects rotenone-induced dopaminergic neurodegeneration by reducing lipid peroxidation. Pharmacol. Rep. 2020, 72, 1397–1406. [Google Scholar] [CrossRef]
- Ryu, Y.K.; Go, J.; Park, H.Y.; Choi, Y.K.; Seo, Y.J.; Choi, J.H.; Rhee, M.; Lee, T.G.; Lee, C.H.; Kim, K.S. Metformin regulates astrocyte reactivity in Parkinson’s disease and normal aging. Neuropharmacology 2020, 175, 108173. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy & metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [PubMed]
- Muraleedharan, R.; Dasgupta, B. AMPK in the brain: Its roles in glucose and neural metabolism. FEBS J. 2021. [Google Scholar] [CrossRef]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK signaling as a neuroprotective strategy in Parkinson’s disease. J. Parkinson’s Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, X.W.; Chen, D.Z.; Hao, F.; Tao, S.X.; Yu, H.Y.; Cheng, R.; Liu, H. Neuro-Protective role of metformin in patients with acute stroke and type 2 diabetes mellitus via ampk/mammalian target of rapamycin (mTOR) signaling pathway and oxidative stress. Med. Sci. Monit. 2019, 25, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. MTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model. Mech. 2019, 12, dmm038596. [Google Scholar] [CrossRef] [Green Version]
- Vega-Rubin-de-Celis, S.; Peña-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Stancu, A.L. AMPK activation can delay aging. Discoveries 2015, 3, e53. [Google Scholar] [CrossRef] [Green Version]
- Sinnett, S.E.; Brenman, J.E. The Role of AMPK in Drosophila melanogaster. Exp. Suppl. 2016, 107, 389–401. [Google Scholar] [PubMed] [Green Version]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal to Form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente Miranda, H.; Szegő, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masato, A.; Sandre, M.; Antonini, A.; Bubacco, L. Patients Stratification Strategies to Optimize the Effectiveness of Scavenging Biogenic Aldehydes: Towards a Neuroprotective Approach for Parkinson’s Disease. Curr. Neuropharmacol. 2021, 19, 1618–1639. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Guo, Y.J.; Xiong, H.; Chen, K.; Zou, J.J.; Lei, P. Brain regions susceptible to alpha-synuclein spreading. Mol. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. To stage, or not to stage. Curr. Opin. Neurobiol. 2020, 61, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinson’s Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 2014, 1–18. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Ismaiel, A.A.K.; Espinosa-Oliva, A.M.; Santiago, M.; García-Quintanilla, A.; Oliva-Martín, M.J.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases mptp-induced damage to the nigrostriatal dopaminergic system. Toxicol. Appl. Pharmacol. 2016, 298, 19–30. [Google Scholar] [CrossRef]
- Wang, D.X.; Chen, A.D.; Wang, Q.J.; Xin, Y.Y.; Yin, J.; Jing, Y.H. Protective effect of metformin against rotenone-induced parkinsonism in mice. Toxicol. Mech. Methods 2020, 30, 350–357. [Google Scholar] [CrossRef]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free Radic. Biol. Med. 2015, 88, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Habib, S.L.; Yadav, A.; Kidane, D.; Weiss, R.H.; Liang, S. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle 2016, 15, 3048–3059. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rõlova, T.; Lehtonen, Š.; Goldsteins, G.; Kettunen, P.; Koistinaho, J. Metabolic and immune dysfunction of glia in neurodegenerative disorders: Focus on iPSC models. Stem Cells 2021, 39, 256–265. [Google Scholar] [CrossRef]
- Olsen, A.L.; Feany, M.B. Parkinson’s disease risk genes act in glia to control neuronal α-synuclein toxicity. Neurobiol. Dis. 2021, 159, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin alleviates high glucose-induced ER stress and inflammation by inhibiting the interaction between caveolin1 and AMPKα in rat astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.L.; Lee, M.-S.; Hsu, C.-C.; Chuang, S.-Y.; Lee, J.-T.; Tsai, H.-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Brakedal, B.; Flønes, I.; Reiter, S.F.; Torkildsen, Ø.; Dölle, C.; Assmus, J.; Haugarvoll, K.; Tzoulis, C. Glitazone use associated with reduced risk of Parkinson’s disease. Mov. Disord. 2017, 32, 1594–1599. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Han, K.D.; Kwon, H.; Park, S.E.; Park, Y.G.; Kim, Y.H.; Yoo, S.J.; Rhee, E.J.; Lee, W.Y. Association between Glycemic Status and the Risk of Parkinson Disease: A Nationwide Population-Based Study. Diabetes Care 2020, 43, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 1155. [Google Scholar] [CrossRef]
- Lamoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Lv, W.S.; Wen, J.P.; Li, L.; Sun, R.X.; Wang, J.; Xian, Y.X.; Cao, C.X.; Wang, Y.L.; Gao, Y.Y. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012, 1444, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic cation transporters in human physiology, pharmacology, and toxicology. Int. J. Mol. Sci. 2020, 21, 7890. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Campanelli, F.; Chapy, H.; Gomez-Zepeda, D.; Glacial, F.; Smirnova, M.; Taghi, M.; Pallud, J.; Perrière, N.; Declèves, X.; et al. An interspecies molecular and functional study of organic cation transporters at the blood-brain barrier: From rodents to humans. Pharmaceutics 2020, 12, 308. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Tai, Y.; Huang, M.T.; Tsai, Y.F.; Hsu, H.J.; Tzen, K.Y.; Liou, H.H. Cellular localization of the organic cation transporters, OCT1 and OCT2, in brain microvessel endothelial cells and its implication for MPTP transport across the blood-brain barrier and MPTP-induced dopaminergic toxicity in rodents. J. Neurochem. 2010, 114, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, N.; Kato, Y. Physiological Roles of Carnitine/Organic Cation Transporter OCTN1/SLC22A4 in Neural Cells. Biol. Pharm. Bull. 2017, 40, 1146–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyanaraman, B. Teaching the basics of repurposing mitochondria-targeted drugs: From Parkinson’s disease to cancer and back to Parkinson’s disease. Redox Biol. 2020, 36, 101665. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Sikora, A.; Zielonka, J.; Dwinell, M. Mitochondria-targeted metformins: Anti-tumour and redox signalling mechanisms. Interface Focus 2017, 7, 20160109. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Zimprich, A.; Berrio, D.A.C.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.K.; Kübler, M.; Lewin, R.; et al. Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain 2017, 140, 2444–2459. [Google Scholar] [CrossRef] [PubMed]
- Schlichtmann, B.W.; Kalyanaraman, B.; Schlichtmann, R.L.; Panthani, M.G.; Anantharam, V.; Kanthasamy, A.G.; Mallapragada, S.K.; Narasimhan, B. Functionalized polyanhydride nanoparticles for improved treatment of mitochondrial dysfunction. J. Biomed. Mater. Res. Part B Appl. Biomater. 2022, 110, 450–459. [Google Scholar] [CrossRef]
- Martin, D.; Thaker, J.; Shreve, M.; Lamerato, L.; Budzynska, K. Assessment of vitamin B 12 deficiency and B 12 screening trends for patients on metformin: A retrospective cohort case review. BMJ Nutr. Prev. Health 2021, 4, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-term metformin therapy and vitamin B12 deficiency: An association to bear in mind. World J. Diabetes 2021, 12, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Q.; Gao, Y.; Nie, K.; Liang, Y.; Zhang, Y.; Wang, L. Serum Folate, Vitamin B12 Levels, and Systemic Immune-Inflammation Index Correlate with Motor Performance in Parkinson’s Disease: A Cross-Sectional Study. Front. Neurol. 2021, 12, 665075. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Choi, I.Y.; Jang, W.; Jeong, S.M.; Park, S.; Han, K.; Lee, Y.; Lee, D.H.; Shin, D.W. Gastrectomy, vitamin B12 supplementation and the risk of Parkinson’s disease: A nationwide cohort study. Parkinsonism Relat. Disord. 2021, 83, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shoulson, I. Deprenyl and tocopherol antioxidative therapy of parkinsonism (DATATOP). Parkinson Study Group. Acta Neurol. Scand. Suppl. 1989, 126, 171–175. [Google Scholar] [CrossRef]
- Luthra, N.S.; Marcus, A.H.; Hills, N.K.; Christine, C.W. Vitamin B12 measurements across neurodegenerative disorders. J. Clin. Mov. Disord. 2020, 7, 3. [Google Scholar] [CrossRef]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinson’s Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Wang, Y.; Wei, W.; Zhao, W.; Lu, F.; Liu, F. Vitamin B12 inhibits α-synuclein fibrillogenesis and protects against amyloid-induced cytotoxicity. Food Funct. 2019, 10, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Katila, N.; Bhurtel, S.; Park, P.H.; Hong, J.T.; Choi, D.Y. Activation of AMPK/aPKCζ/CREB pathway by metformin is associated with upregulation of GDNF and dopamine. Biochem. Pharmacol. 2020, 180, 114193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chaudhari, K.; Shetty, R.; Winters, A.; Gao, X.; Hu, Z.; Ge, W.P.; Sumien, N.; Forster, M.; Liu, R.; et al. Metformin alters locomotor and cognitive function and brain metabolism in normoglycemic mice. Aging Dis. 2019, 10, 949–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
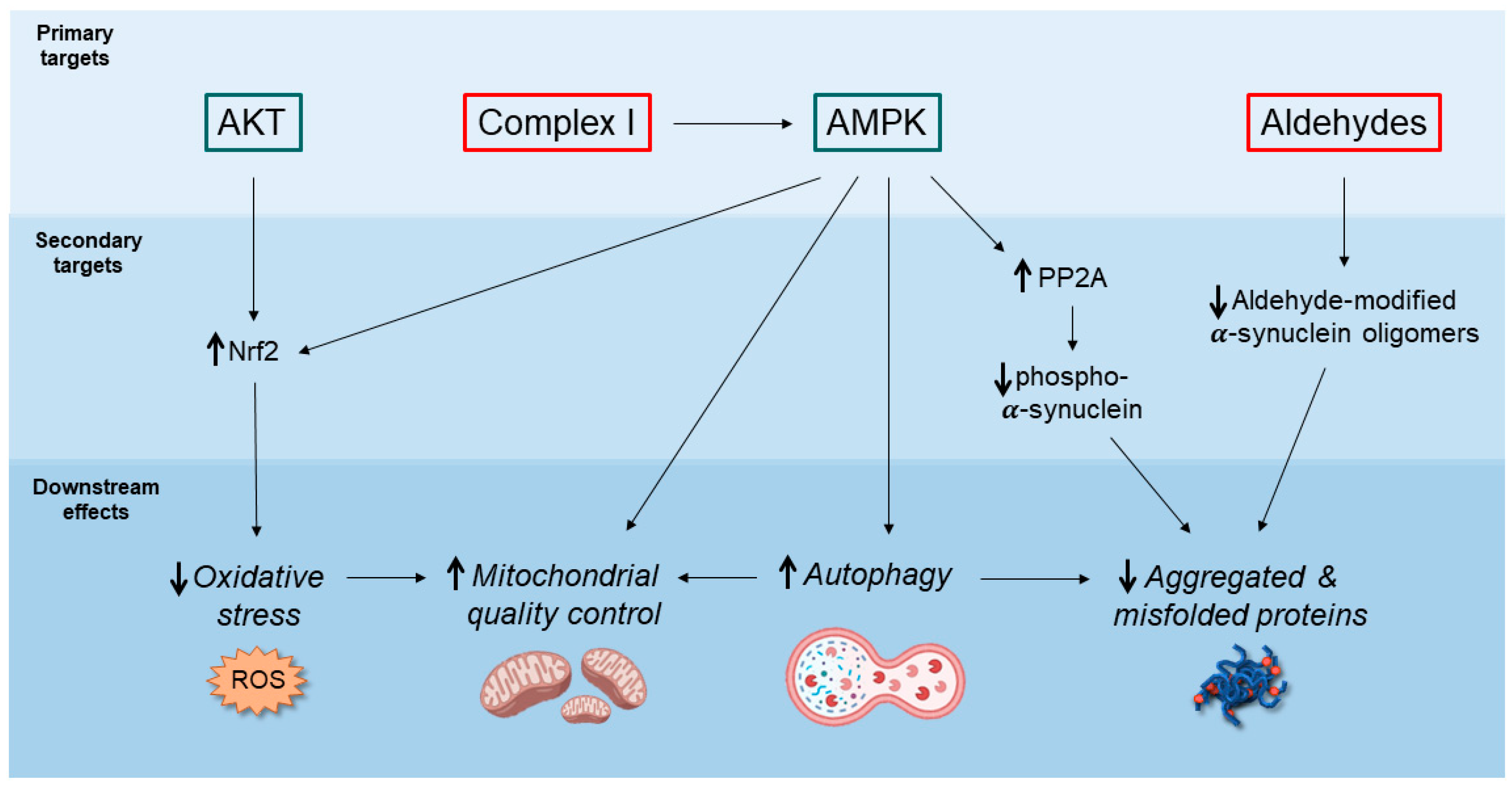

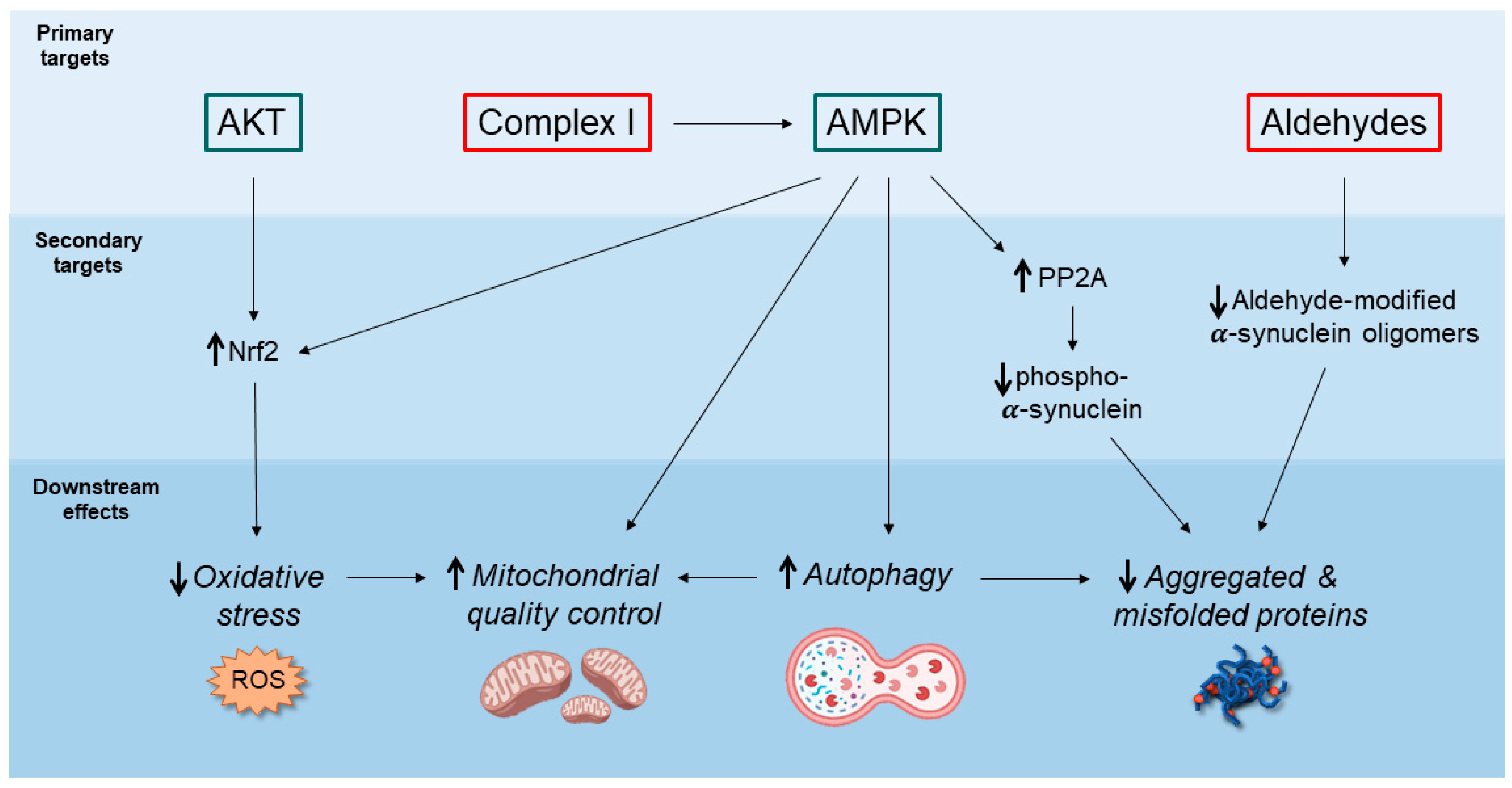{kind=link}
| Cellular Model | Concentration | Duration of Treatment | Read-Out | Ref. |
| Human neuronal stem cell | 1 mM | 48 h |
| [10] |
| Rat hepatocytes | 0.05–0.1 mM | 24–60 h |
| [11] |
| Rat hepatocytes | From 0.02 to 2 mM | 1–7–39 h |
| [12] |
| Mitochondria isolated from frontal mouse brain | 1 mM | 15 min |
| [13] |
| Mouse primary hepatocytes | 0.5–1 mM | 22 h |
| [14] |
| Mouse primary hepatocytes | 0.075 mM | 22 h |
| [14] |
| In vitroPD cellular models | ||||
| SH-SY5Y overexpressing α-syn | 0.5–1.0–2.5 mM | 16–24 h |
| [15] |
| SH-SY5Y neuroblastoma cells treated with rotenone | From 0.01 to 10 mM | 2–3–6 h |
| [16] |
| BV2 cells treated with LPS or IL-4 | 1 mM | 3–12–24 h |
| [17] |
| Animal Model | Dosage | Duration | Administration Route | Read-Out | Ref. |
|---|---|---|---|---|---|
| C. elegans | 50 mM | Oral |
| [18] | |
| C. elegans bcat-1 knock-down | 0.05 mM | 4 days | Oral |
| [19] |
| D. melanogaster | From 1 to 100 mM | 7 days | Oral |
| [20] |
| D. melanogaster | 5 mM | 7 days | Oral |
| [21] |
| M. musculus | 0.1–1% w/w | Oral |
| [22] | |
| M. musculus | 200 mg/Kg | Intraperitoneal injection |
| [13] | |
| M. musculus | 1–10 mM | Hypothalamus infusion |
| [13] | |
| M. musculus | From 6.25 to 50 mg/kg | Once a day for 12 weeks | Oral |
| [14] |
| R. norvegicus | 50–150 mg/ml | Once a day for 5 days | Oral intubation |
| [11] |
| In vivoPD animal models | |||||
| C. elegans treated with 50 mM 6-OHDA | 5–10 mM | 72 h | Oral |
| [23] |
| M. musculus injected with MPTP (30 mg/kg/day) for 7 days | 200 mg/kg/day | 7 days (following MPTP) | Intraperitoneal injection |
| [24] |
| M. musculus injected with rotenone (2.5 mg/kg/day) for 10 days | 300 mg/kg/day | 10 days (co-admin.) | Intraperitoneal injection |
| [25] |
| M. musculus treated with 15 μg 6-OHDA | 100–200 mg/kg | Once a day for 4 weeks | Oral |
| [26] |
| R. norvegicus injected with 2 μg of LPS | 150 mg/Kg | Twice a day for 7 days | Oral |
| [17] |
| Location | Study Period | Sample Size | Medication Users | Mean Age (Years) | HR [95% C.I.] | Follow-Up (Years) | Reference |
|---|---|---|---|---|---|---|---|
| Taiwan | 1996–2007 | 11,730 | 1879 | 64.3 ± 9.6 | 0.95 [0.53–1.71] | 11 or until PD onset | [65] |
| Norway | 2004–2014 | 102,745 | 94,349 | 63.4 ± 11.1 | 1.39 [1.06–1.82] | 6.95 | [66] |
| South Korea | 2009–2010 | 1,308,089 | 644,921 | 60.8 ± 10.0 | 1.22 [1.10–1.36] | 6.3 | [67] |
| USA | 2004–2010 | 5530 | 2774 | 63.2 ± 10.9 | 0.19 [0.12–0.31] | 5.2 | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostini, F.; Masato, A.; Bubacco, L.; Bisaglia, M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. Int. J. Mol. Sci. 2022, 23, 398. https://doi.org/10.3390/ijms23010398
Agostini F, Masato A, Bubacco L, Bisaglia M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. International Journal of Molecular Sciences. 2022; 23(1):398. https://doi.org/10.3390/ijms23010398
Chicago/Turabian StyleAgostini, Francesco, Anna Masato, Luigi Bubacco, and Marco Bisaglia. 2022. "Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges" International Journal of Molecular Sciences 23, no. 1: 398. https://doi.org/10.3390/ijms23010398
APA StyleAgostini, F., Masato, A., Bubacco, L., & Bisaglia, M. (2022). Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. International Journal of Molecular Sciences, 23(1), 398. https://doi.org/10.3390/ijms23010398