Abstract
Acetylsalicylic acid (ASA) is widely used in secondary prevention of cardiovascular (CV) disease, mainly because of its antithrombotic effects. Here, we investigated whether ASA can prevent the progression of vessel wall remodelling, atherosclerosis, and CV complications in apolipoprotein E deficient (ApoE−/−) mice, a model of stable atherosclerosis, and in ApoE−/− mice with a mutation in the fibrillin-1 gene (Fbn1C1039G+/−), which is a model of elastic fibre fragmentation, accompanied by exacerbated unstable atherosclerosis. Female ApoE−/− and ApoE−/−Fbn1C1039G+/− mice were fed a Western diet (WD). At 10 weeks of WD, the mice were randomly divided into four groups, receiving either ASA 5 mg/kg/day in the drinking water (ApoE−/− (n = 14), ApoE−/−Fbn1C1039G+/− (n = 19)) or plain drinking water (ApoE−/− (n = 15), ApoE−/−Fbn1C1039G+/− (n = 21)) for 15 weeks. ApoE−/−Fbn1C1039G+/− mice showed an increased neutrophil–lymphocyte ratio (NLR) compared to ApoE−/− mice, and this effect was normalised by ASA. In the proximal ascending aorta wall, ASA-treated ApoE−/−Fbn1C1039G+/− mice showed less p-SMAD2/3 positive nuclei, a lower collagen percentage and an increased elastin/collagen ratio, consistent with the values measured in ApoE−/− mice. ASA did not affect plaque progression, incidence of myocardial infarction and survival of ApoE−/−Fbn1C1039G+/− mice, but systolic blood pressure, cardiac fibrosis and hypertrophy were reduced. In conclusion, ASA normalises the NLR, passive wall stiffness and cardiac remodelling in ApoE−/−Fbn1C1039G+/− mice to levels observed in ApoE−/− mice, indicating additional therapeutic benefits of ASA beyond its classical use.
1. Introduction
Age-related diseases such as cardiovascular disease (CVD), have a substantial impact on our quality of life. Vascular ageing is characterised by structural and functional changes in the wall of large arteries leading to arterial stiffness, thereby promoting atherosclerotic disease progression [1]. Atherosclerosis is a progressive inflammatory disease of the large and medium-sized arteries, characterised by the formation of plaques in the vessel wall. During the development of the disease, the stability of the atherosclerotic plaque plays a major role. Features of plaque instability include a large necrotic core, a high infiltration of inflammatory macrophages and a thin fibrous cap, composed of few smooth muscle cells (SMCs) and collagen fibres. When a plaque develops such an unstable phenotype, it may easily rupture, followed by thrombosis and clinical complications such as myocardial infarction (MI) and stroke [2,3,4]. Despite the significant therapeutic advances in cardiology over the past decades, atherosclerotic plaque rupture remains a leading cause of acute cardiovascular death. Delaying the process of vascular ageing and arterial stiffening might reduce the progression of atherosclerosis, thereby reducing the societal and economic burden of CVD.
Extracellular matrix remodelling plays an essential role in vascular ageing and arterial stiffness. With age, mechanical fracture of elastin and subsequent proteolysis will become more abundant, hence reducing the elasticity of the vessel wall. Furthermore, collagen synthesis will be increased, mainly because of enhanced TGF-β signalling, which will further promote arterial stiffening. In the end, this will lead to increased systolic blood pressure (BP), cardiac remodelling, and atherogenesis [5]. Another important characteristic of ageing, is chronic low-grade inflammation, termed ‘inflammaging’, which also contributes to arterial stiffness. The neutrophil–lymphocyte ratio (NLR) is a biomarker of chronic inflammation that increases with age [6,7,8]. Importantly, NLR is associated with arterial stiffness and increased morbidity and mortality in CVD patients [6,9,10,11,12,13]. Measuring NLR could therefore provide a good indication of the severity of age-related vascular stiffness and the associated progression of CVD. Overall, targeting vascular remodelling and inflammation could be a valuable approach to attenuate the progression of arterial stiffness and atherosclerosis. Current therapeutic strategies to prevent or treat vascular ageing are lifestyle interventions, such as physical activity, caloric restriction, and maintaining a healthy diet. Furthermore, pharmacological interventions with a proven effect on arterial stiffness include anti-hypertensive agents, statins, mTOR inhibitors (e.g., rapamycin), and AMPK activators (e.g., metformin), whereas PCSK9 monoclonal antibodies are promising [14,15]. Nevertheless, the need for novel therapeutic strategies remains high, due to the current ageing population and consequent higher burden of age-related diseases. In this regard, a good approach would be to evaluate known drugs, used in CVD prevention, for their potential to inhibit vascular remodelling and the development of arterial stiffness.
Acetylsalicylic acid (ASA) has been used for many years in the treatment and prevention of CVD [16,17,18]. It is an irreversible inhibitor of the enzyme cyclooxygenase and belongs to the class of the nonsteroidal anti-inflammatory drugs. Low doses of ASA (80–100 mg) are sufficient to block cyclooxygenase-1 and thus possess the ability to inhibit platelet aggregation [16]. In primary prevention, ASA is not recommended due to an increased bleeding risk (class III ESC guidelines), but in secondary prevention after MI or stroke the risk reduction is very significant even in patients with stable cardiovascular disease (class I ESC guidelines) [19,20,21,22].
The goal of the present study was to test the effect of low-dose ASA on the progression of vessel wall remodelling, atherosclerosis, and cardiovascular complications in apolipoprotein E deficient (ApoE−/−) mice, the classical model to study atherosclerosis, and ApoE−/− mice with a heterozygous mutation in the fibrillin-1 gene (Fbn1C1039G+/−). Mutations in the Fbn1 gene result in an impaired microfibrillar assembly and deposition, followed by fragmentation of elastic fibres and increased collagen deposition. This loss of structural integrity of the vessel wall leads to progressive dilatation and arterial stiffening, resembling vascular ageing [23,24,25,26,27]. In addition, the ApoE−/−Fbn1C1039G+/− mouse model shows accelerated atherosclerotic plaque progression, spontaneous plaque ruptures, MI, and sudden death [28,29]. It is therefore an excellent model to study pharmacological interventions targeting vascular ageing and CVD, because it combines two important risk factors, hypercholesterolemia (ApoE−/−) and arterial stiffness (Fbn1C1039G+/−).
2. Results
A general overview of the study outline, timing and treatment groups is visualised in Figure 1. Mortality, caused by sudden death of the mice during the timeframe of the treatment protocol, was significantly higher in ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice, but was not affected by ASA treatment (Table 1, Figure 1). Importantly, no major bleeding complications were detected in ASA-treated mice. Total plasma cholesterol levels were not different and body weight was reduced in ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− controls, without a significant treatment effect (Table 1). Heart, lung, and spleen weight were all normalised over body weight and increased in ApoE−/−Fbn1C1039G+/− mice. Furthermore, heart and spleen weight in ApoE−/−Fbn1C1039G+/− mice were reduced as a result of ASA treatment (Table 1).
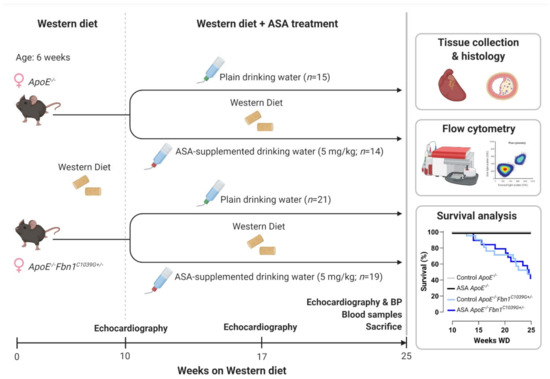
Figure 1.
Experimental outline and Kaplan–Meier survival curve. BP = blood pressure; WD = Western diet. Created with BioRender.com (accessed on 30 December 2021).

Table 1.
Main characteristics of control and ASA-treated ApoE−/− and ApoE−/−Fbn1C1039G+/− mice.
2.1. ASA Treatment Normalises Neutrophil–Lymphocyte Ratio in ApoE−/−Fbn1C1039G+/− Mice
The analysis of circulating immune cells clearly showed higher neutrophil levels in the blood of control ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice, while lymphocyte levels remained equal (Figure 2A,B). In response to ASA treatment, a trend towards lower circulating neutrophils was observed in ApoE−/−Fbn1C1039G+/− mice (Figure 2A). After calculating the neutrophil–lymphocyte ratio, a significant increase could be observed in control ApoE−/−Fbn1C1039G+/− mice. This increase was abolished in the ASA-treated group (Figure 2C). In ApoE−/−Fbn1C1039G+/− mice, the percentage of circulating Ly-6Chi monocytes was higher and ASA treatment was not able to normalise this pattern (Table 2). The levels of dendritic cells, Ly-6Clo monocytes, T cells, B cells, NK, and NKT cells were not affected by genotype or treatment (Table 2).
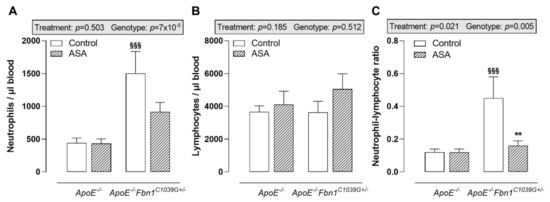
Figure 2.
Blood neutrophils and lymphocytes. (A) Circulating neutrophils were higher in control ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice and were not significantly affected by ASA treatment. (B) Absolute numbers of circulating lymphocytes remained equal between all groups of mice. (C) As a result of ASA treatment, the neutrophil–lymphocyte ratio was normalised in ApoE−/−Fbn1C1039G+/− mice. 2-way ANOVA: §§§ p < 0.001 vs. ApoE−/− control and ** p < 0.01 vs. ApoE−/−Fbn1C1039G+/− control.
Table 2.
Blood immune cell profile.
2.2. Atherosclerotic Plaque Progression and the Occurrence of Myocardial Infarctions Are Not Affected by ASA
ApoE−/−Fbn1C1039G+/− mice showed a marked increase in plaque size, stenosis, and vessel dilatation (analysed by measuring internal elastic lamina length) in the proximal ascending aorta compared to ApoE−/− mice. ASA treatment was not able to reduce these disease parameters (Table 3, Figure S1). Furthermore, the stability of the plaques remained equal between control and ASA-treated mice, since no changes in necrotic core size, percentage of smooth muscle cells, fibrous cap thickness, total collagen content, percentage of glycosaminoglycans, and the presence of leukocytes and macrophages were observed (Table 3, Figure S1). In addition, we could not observe a difference in p-SMAD2/3 positive nuclei in both control and ASA-treated ApoE−/− and ApoE−/−Fbn1C1039G+/− mice (Table 3, Figure S1).
Table 3.
Atherosclerotic plaque and MI characteristics.
In the common carotid artery, plaque formation index was higher in ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice, but it was not affected by ASA treatment (Table 3). Neither the genotype nor treatment affected the presence of intraplaque microvessels and bleeding (Table 3).
The percentage of mice showing plaques in the coronary arteries was not different between genotypes and treatment groups (Table 3). Furthermore, plaque size and stenosis were equal (Table 3). The occurrence of MI was higher in ApoE−/−Fbn1C1039G+/− mice and was not altered by ASA treatment (Table 3).
2.3. Passive Wall Stiffness Is Reduced in ASA-Treated ApoE−/−Fbn1C1039G+/− Mice
Histological analysis of the proximal ascending aorta wall showed that ASA treatment was able to reduce wall thickness in ApoE−/−Fbn1C1039G+/− mice, but not in ApoE−/− mice (Figure 3A,J). The total leukocyte and macrophage content of the vessel wall was rather low and equal in all groups (Figure 3B,C,J). The percentage of α-actin positive smooth muscle cells was significantly lower in ApoE−/−Fbn1C1039G+/− mice and was not affected by ASA (Figure 3D,J). The presence of glycosaminoglycans in the vessel wall of ApoE−/−Fbn1C1039G+/− mice was increased as compared to ApoE−/− mice. In addition, ASA treatment resulted in a higher percentage of glycosaminoglycans in ApoE−/− mice, but not in ApoE−/−Fbn1C1039G+/− mice (Figure 3E,J). The percentage of p-SMAD2/3 positive nuclei was higher in ApoE−/−Fbn1C1039G+/− control mice as compared to ApoE−/− mice. In addition, ASA treatment was able to reduce p-SMAD2/3 positivity in ApoE−/−Fbn1C1039G+/− mice to the levels observed in ApoE−/− mice (Figure 3F,J). Elastin fragmentation was clearly present in ApoE−/−Fbn1C1039G+/− mice and the number of elastin breaks was significantly higher as compared to ApoE−/− mice in both control (ApoE−/−: 17 [11–23]; ApoE−/−Fbn1C1039G+/−: 114 [54–176]; p = 0.000095) and ASA-treated groups (ApoE−/−: 17 [12–22]; ApoE−/−Fbn1C1039G+/−: 118 [31–194]; p = 0.000027; Kruskal–Wallis test with Bonferroni correction for multiple comparisons). However, ASA treatment did not significantly affect the number of elastin breaks in ApoE−/− mice (p = 0.905) and in ApoE−/−Fbn1C1039G+/− mice (p = 0.977; Kruskal–Wallis test with Bonferroni correction for multiple comparisons). The percentage of elastin positive area was decreased in ApoE−/−Fbn1C1039G+/− mice as compared to ApoE−/− control mice and in accordance with the number of elastin breaks, ASA treatment did not affect elastin content of the vessel wall in both models (Figure 3G,J). A higher collagen fibre content was observed in ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice, which was normalised by treatment with ASA (Figure 3H,J). After calculating the elastin/collagen ratio, a distinct decrease was detected in ApoE−/−Fbn1C1039G+/− mice. Interestingly, ASA treatment led to higher elastin/collagen ratios in both mouse models (Figure 3I,J).
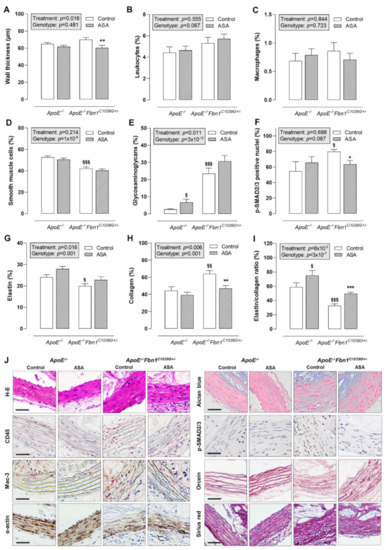
Figure 3.
Thickness and composition of the proximal ascending aorta wall. (A) H-E staining was used to evaluate the thickness of the vessel wall, which was reduced as a result of ASA treatment in ApoE−/−Fbn1C1039G+/− mice. (B,C) The percentage of leukocytes (CD45 positive cells) and macrophages (Mac-3 positive area) in the vessel wall did not change. (D) The α-actin positive area (smooth muscle cells) was significantly lower in ApoE−/−Fbn1C1039G+/− mice and was not affected by ASA treatment. (E) Alcian blue staining indicated an increase in glycosaminoglycans in ApoE−/−Fbn1C1039G+/− mice as compared to ApoE−/− mice. In addition, ASA treatment resulted in a higher percentage of glycosaminoglycans in ApoE−/− mice. (F) The percentage of p-SMAD2/3 positive nuclei were higher in ApoE−/−Fbn1C1039G+/− control mice as compared to ApoE−/− mice. ASA treatment was able to reduce p-SMAD2/3 positivity in ApoE−/−Fbn1C1039G+/− mice. (G) Orcein staining clearly showed the fragmentation of elastic fibres and a lower elastin content in ApoE−/−Fbn1C1039G+/− mice, which was not significantly affected by ASA treatment. (H) Collagen content of the vessel wall (Sirius red positive area) was higher in control ApoE−/−Fbn1C1039G+/− mice and normalised by ASA treatment. (I) The elastin/collagen ratio was reduced in ApoE−/−Fbn1C1039G+/− mice and ASA treatment led to an increase in both models. (J) Representative images of the different stainings. Scale bar = 50 µm. 2-way ANOVA: § p < 0.05, §§ p < 0.01 §§§ p < 0.001 vs. control ApoE−/− and * p < 0.05, ** p < 0.01, *** p < 0.01 vs. control ApoE−/−Fbn1C1039G+/−.
2.4. ASA Treatment Reduces Systolic Blood Pressure and Cardiac Remodelling in ApoE−/−Fbn1C1039G+/− Mice
Blood pressure (BP) measurements at 25 weeks of WD revealed a significantly lower systolic BP in ASA-treated ApoE−/−Fbn1C1039G+/− mice compared to control ApoE−/−Fbn1C1039G+/− mice (Figure 4A). In ApoE−/− mice, the same trend could be observed. Diastolic BP was not significantly changed, although a decreasing trend was present as a result of ASA treatment in both mouse models (Figure 4B).
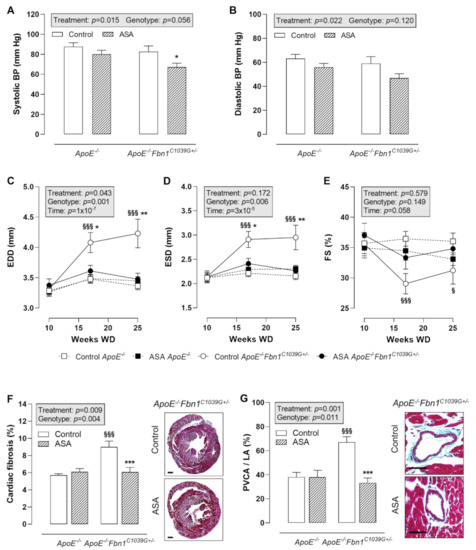
Figure 4.
Blood pressure and cardiac remodelling. (A,B) Systolic blood pressure (BP) was lower after ASA treatment in ApoE−/−Fbn1C1039G+/− mice. Diastolic BP was not significantly reduced. 2-way ANOVA: * p < 0.05 vs. control ApoE−/−Fbn1C1039G+/−. (C–E) End-diastolic diameter (EDD) and end-systolic diameter (ESD) were normalised in ASA-treated ApoE−/−Fbn1C1039G+/− mice. A trend towards higher fractional shortening (FS) was observed in ApoE−/−Fbn1C1039G+/− mice treated with ASA. 3-way mixed ANOVA: § p < 0.05, §§§ p < 0.001 vs. control ApoE−/− and * p < 0.05, ** p < 0.01 vs. ASA ApoE−/−Fbn1C1039G+/− (F,G) Total cardiac fibrosis (Trichrome Masson staining; scale bar = 500 µm) was increased in control ApoE−/−Fbn1C1039G+/− mice and normalised after ASA treatment. Trichrome Masson staining of the coronary arteries (scale bar = 50 µm) revealed a significant reduction of perivascular fibrosis (perivascular collagen area divided by luminal area = PVCA/LA) after ASA treatment in ApoE−/−Fbn1C1039G+/− mice. 2-way ANOVA: §§§ p < 0.001 vs. control ApoE−/− and *** p < 0.001 vs. control ApoE−/−Fbn1C1039G+/−.
Echocardiography of the heart showed a clear increase in end-diastolic diameter (EDD) and end-systolic diameter (ESD) in control ApoE−/−Fbn1C1039G+/− mice at week 17 and 25 of WD, which was normalised to the level of ApoE−/− mice after ASA treatment (Figure 4C,D). Fractional shortening (FS) was significantly decreased at week 17 and 25 of WD in ApoE−/−Fbn1C1039G+/− vs. ApoE−/− mice. A trend towards higher values was observed after ASA treatment, equal to FS measured in ApoE−/− mice (Figure 4E). Total cardiac fibrosis (Figure 4F) and coronary perivascular fibrosis (Figure 4G) clearly normalised in ApoE−/−Fbn1C1039G+/− mice after treatment with ASA.
3. Discussion
In the present study, we investigated whether ASA therapy has an additional benefit in CVD prevention by reducing vessel wall remodelling and atheroprogression. The effects were compared between ApoE−/− and ApoE−/−Fbn1C1039G+/− mice in order to analyse two different stages of disease severity. ApoE−/− mice are a classical model to study atherosclerosis. They develop atherosclerotic plaques of different stages, but have the disadvantage that plaque rupture and complications such as MI and sudden death are rare, which are the most important aspects of human atherosclerosis [30]. Therefore, we included the ApoE−/−Fbn1C1039G+/− mouse model as well. Mutations in the Fbn1 gene lead to fragmentation of elastic fibres [31]. This results in increased arterial stiffening and progressive aortic dilatation, mimicking vascular ageing [24,25,29]. Moreover, in ApoE−/− mice, this mutation leads to the development of highly unstable plaques, resulting in spontaneous plaque rupture with end-points including MI and sudden death [28,29]. Thus, ApoE−/−Fbn1C1039G+/− mice show many features of human end-stage atherosclerosis and are therefore a good model for evaluating the therapeutic effects of drugs on age-related vessel wall remodelling, established atherosclerosis, and its complications.
Clear signs of chronic inflammation were observed in ApoE−/−Fbn1C1039G+/− mice, since the number of circulating Ly-6Chi monocytes, neutrophils and the NLR were significantly higher compared to ApoE−/− mice. The NLR is an inflammatory biomarker that independently predicts cardiovascular risk and mortality in humans [13]. It is important to note that in humans a normal NLR is between 1 and 3, while in mice these values are lower, in the range of 0.1–0.3 [32]. It was striking that ASA treatment was able to normalise this parameter. This finding is in accordance with published data, describing a lower NLR in coronary artery disease patients receiving ASA therapy [33]. Many risk factors of CVD promote neutrophilia. For instance, metabolic changes, such as hypercholesterolemia and hyperglycaemia, stress, ageing, and sleep disorders accelerate myelopoiesis. The resulting increase in circulating neutrophils plays an important role in atheroprogression by secreting reactive oxygen species, proteases, and myeloperoxidase, resulting in endothelial activation and monocyte recruitment [34,35,36]. Although we could observe a decrease in NLR in ApoE−/−Fbn1C1039G+/− mice after ASA treatment, we could not observe a similar anti-inflammatory trend in atherosclerotic plaques of these mice. In addition, no effect on plaque size and stability was observed. We initiated ASA treatment at 10 weeks of Western diet, a time point at which advanced plaques are present in ApoE−/−Fbn1C1039G+/− mice, as observed by Van Herck et al. [29]. It is possible that our treatment protocol was not sufficient to modulate plaque inflammation, due to the excessive presence of inflammatory cytokines and matrix degrading enzymes in plaques of ApoE−/−Fbn1C1039G+/− mice at the start of ASA administration [28].
The vessel wall of ApoE−/−Fbn1C1039G+/− mice showed clear signs of remodelling. We observed a significantly lower elastin/collagen ratio compared to ApoE−/− mice, which is an important indicator of passive wall stiffness. This was mainly due to a marked increase in total collagen content. In addition, the presence of glycosaminoglycans was dramatically increased. TGF-β is considered an important mediator in this remodelling process. Microfibrils interact with TGF-β-binding proteins. Thus, a mutation in the Fbn1 gene results in increased TGF-β levels and activity, due to disturbed sequestration of this cytokine. Ultimately, this will lead to a higher collagen and glycosaminoglycan production in the vessel wall [37,38,39]. We observed an increase in p-SMAD2/3 positive nuclei in the vessel wall of ApoE−/−Fbn1C1039G+/− mice, clearly demonstrating enhanced TGF-β signalling. Interestingly, ASA treatment resulted in a lower percentage of p-SMAD2/3 positive nuclei in the proximal ascending aorta wall of ApoE−/−Fbn1C1039G+/− mice. In contrast, we also observed a slight increase in glycosaminoglycans. Whether this will be harmful or affect vascular function after chronic treatment, remains to be investigated. Apart from this finding, we observed a clear reduction in passive wall stiffness, as seen by a lower collagen content and a subsequent increase in the elastin/collagen ratio. It has been described that ASA treatment is able to reduce aortic stiffness in humans [40,41]. Thus, based upon our findings, it is plausible that the decrease in SMAD signalling plays a major role in this beneficial property. This is also in accordance with a study by Franken et al., that investigated the effect of anti-inflammatory therapies on aortic dilatation and remodelling in Fbn1C1039G+/− mice and found that reducing inflammation was not sufficient to improve disease progression. Only the use of losartan, which is an Angiotensin II type 1 receptor blocker, was able to induce a therapeutic benefit by reducing both inflammation and SMAD signalling [42]. It has been demonstrated that ASA is able to reduce the transcription of the Angiotensin II type 1 receptor in cardiac fibroblasts. This resulted in a reduction in collagen synthesis, which was comparable to the effect of losartan [43]. Another publication described that ASA was able to reduce SMAD signalling in the kidney of diabetic rats, which further supports our data [44].
Subsequent to the effects on vessel wall remodelling, we investigated related blood pressure and cardiac parameters. Arterial stiffness is known to be both a cause and consequence of hypertension [45]. Although we could not observe a higher systolic BP in ApoE−/−Fbn1C1039G+/− mice compared to ApoE−/− mice, we did see a decrease after treatment with ASA, which may be the result of the lower passive wall stiffness. Due to the positive feedback loop between hypertension and arterial stiffness, the reduction in systolic BP may also have contributed to the observed decrease in passive stiffness [45]. In addition, a marked reduction in left ventricular dilatation, cardiac fibrosis and heart weight was observed in ASA-treated ApoE−/−Fbn1C1039G+/− mice. This finding is not unexpected due to the lower systolic BP and reduced stiffness of the proximal ascending aorta wall, thereby minimizing hemodynamic overload. It is also plausible that, in accordance to the vessel wall, a reduction in SMAD signalling contributed to the diminished cardiac fibrosis [43].
Besides very clear signs of left ventricular dilatation and an increase in heart weight in ApoE−/−Fbn1C1039G+/− mice, also spleen and lung weight were significantly higher as compared to ApoE−/− mice. It is well known that left ventricular dysfunction results in pulmonary congestion and increased lung weight [46]. In addition, it has been previously described that mice with ischemic heart failure consistently showed splenomegaly and splenic remodelling. This resulted in a mobilization of splenic proinflammatory monocytes to the heart, thereby enhancing disease progression [47,48]. Interestingly, ASA treatment was able to reduce both cardiac remodelling and spleen weight in ApoE−/−Fbn1C1039G+/− mice, thus it might diminish the detrimental effects related to the cardiosplenic axis.
Our finding that ASA failed to reduce MI and improve survival contrasts with the results in humans, where ASA has been used for many years in the prevention of cardiovascular disease [16,17,18]. This discrepancy may be attributed to the fibrinolytic system of mice. In these animals, plasma levels of plasminogen activator inhibitor and thrombin activatable fibrinolysis inhibitor are lower than in humans, shifting the fibrinolytic balance towards enhanced lysis of thrombi [49]. Thus, if plaque rupture occurs in ApoE−/−Fbn1C1039G+/− mice, this will not always give rise to occlusive thrombi that provoke MI or stroke. The observed complications are more likely the result of stenotic occlusion of arteries. Another possibility to explain the difference could be the duration of the study and/or the duration of the ASA treatment.
In conclusion, the present study showed that ASA treatment significantly improves passive wall stiffness, systolic BP and cardiac remodelling in a mouse model of elastic fibre fragmentation and advanced atherosclerosis. Furthermore, a marked reduction of NLR was observed, which is an important biomarker of chronic inflammation and is associated with adverse outcome in CVD patients. These data point towards additional benefits of ASA treatment in the prevention of age-related arterial stiffness. Furthermore, ASA might also have the potential to reduce vessel wall remodelling observed in Marfan syndrome. In order to determine the clinical significance of our findings, larger trials and more in-depth research is required.
4. Materials and Methods
4.1. Mice
Female ApoE−/− and ApoE−/−Fbn1C1039G+/− mice were fed a Western diet (WD, 4021.90, AB Diets, Woerden, The Netherlands) starting at the age of six weeks. The animals were group-housed (5–7 per cage) in standard mouse cages in the animal facility of the University of Antwerp, in a 12-h light/dark cycle. Cages were stored at constant room temperature (20–24 °C) and humidity (45%). Food and water were supplied ad libitum. Cases of sudden death were documented. At the end of the experiment (25 weeks of WD), blood samples were obtained from the retro-orbital plexus of anesthetised mice (ketamine 100 mg/kg, xylazine 10 mg/kg, i.p.). Subsequently, the mice were sacrificed with sodium pentobarbital (250 mg/kg, i.p.). The spleen, lung, and heart weights were determined. Analysis of total plasma cholesterol was performed via a commercially available kit (Randox, Crumlin, UK). The animal procedures were performed conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and all experiments were approved by the ethics committee of the University of Antwerp.
4.2. Acetylsalicylic Acid Treatment
At 10 weeks of WD, ApoE−/− and ApoE−/−Fbn1C1039G+/− mice were randomly divided into four groups, receiving either ASA (Aspegic®, 9 mg lysine acetylsalicylate is equivalent to 5 mg ASA, 14 ApoE−/− mice and 19 ApoE−/−Fbn1C1039G+/− mice) at a dose of 5 mg/kg/day added to the drinking water or plain drinking water (control, 15 ApoE−/− mice and 21 ApoE−/−Fbn1C1039G+/− mice) for 15 weeks. For stability reasons, the ASA drinking water was changed every day.
4.3. Echocardiography
Transthoracic echocardiograms were performed at the start of the ASA treatment (10 weeks of WD), at 17 weeks of WD and at the end of the experiment. The procedure was performed on anesthetised mice (sevoflurane; 8% for induction and 4.5% for maintenance, SevoFlo®, Penlon vaporiser) using a Toshiba diagnostic ultrasound system (SSA-700A), equipped with a 15-MHz transducer. EDD and ESD were measured and FS was calculated as follows: (EDD − ESD)/EDD × 100.
4.4. Blood Pressure Measurements
Peripheral BP was measured at 25 weeks of WD in anesthetised mice (ketamine 100 mg/kg, xylazine 10 mg/kg, i.p.) via a tail cuff (CodaTM High throughput, Kent Scientific, Torrington, CT, USA). Mice were placed on a warming platform (37 °C) and 10 min of acclimatization was allowed before measurements were initiated. The mean of at least 10 measurements per mouse was used.
4.5. Flow Cytometry
EDTA-treated blood (500 µL) was lysed using red blood cell lysing buffer Hybri-Max (Sigma-Aldrich, St. Louis, MO, USA). Subsequently, the remaining leukocytes were labelled with the following antibodies (BioLegend, San Diego, CA, USA): anti-CD11c (N418), anti-I-Ab (KH74), anti-CD11b (M1/70), anti-CD3ε (145-2C11), anti-CD19 (6D5), anti-NK1.1 (PK136), anti-Ly-6C (HK1.4), and anti-Gr-1 (RB6-8C5). Labelling occurred in the dark at 4 °C in FACS buffer (PBS + 0.1% BSA (Sigma Aldrich, St. Louis, MO, USA) + 0.05% NaN3 (Merck, Kenilworth, NJ, USA)) containing CD16/32 Fc-receptor blocker (BioLegend, San Diego, CA, USA). Next, cells were analysed on a BD Accuri C6 cytometer equipped with a blue and red laser (Becton Dickinson, Erembodegem, Belgium). Dead cells were excluded based on forward scatter, side scatter and positive staining for propidium iodide (Invitrogen, Waltham, MA, USA). Data analysis was performed with FCS Express 4 (De Novo Software, Glendale, CA, USA).
4.6. Histology
After sudden death or sacrifice of the mice, the proximal ascending aorta and the heart were collected. Tissues were fixed in 4% formaldehyde for 24 h, dehydrated overnight in 60% isopropanol and embedded in paraffin. Serial cross sections (5 µm) of the proximal ascending aorta and heart were prepared for histological analyses. Atherosclerotic plaque size, stenosis (plaque size/total vessel area), necrotic core (defined as acellular areas with a threshold of 3000 µm2), internal elastic lamina length and wall thickness of the proximal ascending aorta were analysed using haematoxylin-eosin (H-E) stained sections. Immunohistochemical staining of the proximal ascending aorta with primary antibodies against CD45 (ab10558, Abcam, Cambridge, UK) and Mac-3 (553322, Pharmingen, San Diego, CA, USA) was used to determine the percentage of leukocytes and macrophages, respectively. Collagen content was measured in Sirius red stained sections. An α-SMC actin (F3777, Sigma, St. Louis, MO, USA) immunohistochemical staining was used to define the total vascular smooth muscle cell content in the plaque and vessel wall. Fibrous cap thickness was determined as the median value of 10 measurements of the luminal α-SMC actin positive layer of the atherosclerotic plaque. Vessel wall elastin content was measured using orcein stained sections and alcian blue staining was performed to visualise glycosaminoglycans. p-SMAD2/3 (3101, Cell Signalling, Danvers, MA, USA) immunohistochemical staining was used to determine the percentage of p-SMAD2/3 positive nuclei over the total number of nuclei in the plaque and proximal ascending aorta wall. The occurrence of MI, coronary plaques, total cardiac fibrosis, and perivascular fibrosis, measured as the mean perivascular collagen area divided by the luminal area (PVCA/LA) of 10 coronary arteries per mouse, was analysed on Masson’s trichrome stained sections of the heart. If plaques were present in the coronary arteries, plaque size and percentage stenosis were measured using Masson’s trichrome stained sections. Longitudinal sections of the carotid artery were used to analyse plaque formation index, the occurrence of intraplaque microvessels and bleeding. The plaque formation index was determined on H-E stained sections and calculated by using the following formula: (∑ total plaque length/∑ total vessel length) × 100. Immunohistochemical staining for anti-VWF (von Willebrand factor; PC054; Binding Site, San Diego, CA, USA) and Ter-119 (anti–Ter-119, 550565; BD Biosciences, Franklin Lakes, NJ, USA) were performed to detect intraplaque endothelial cells (microvessels) and erythrocytes (bleeding), respectively. All images were acquired with Universal Grab 6.1 software using an Olympus BX40 microscope and were quantified with ImageJ software (NIH, Bethesda, MA, USA).
4.7. Statistical Analyses
Normally distributed data are expressed as mean ± SEM and non-normally distributed variables are represented as median [min-max]. Statistical analyses were performed using SPSS software (version 28, SPSS Inc., Chicago, IL, USA). Statistical tests are specified in the figure and table legends. If a two-way ANOVA or a three-way mixed ANOVA was used, a significant main effect was further analysed by performing a simple main effect analysis, including a Bonferroni correction for multiple comparisons. Mice that died during the experiment were excluded from body and organ weight analyses. Histological data of the proximal ascending aorta, coronary arteries and the heart only include mice that survived until 20 weeks of WD in order to avoid disease stage related variations. Differences were considered significant at p < 0.05.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23010404/s1.
Author Contributions
Conceptualization, L.R., W.M. and G.R.Y.D.M.; Formal analysis, L.R. and M.R.; Funding acquisition, W.M. and G.R.Y.D.M.; Investigation, L.R., M.R., D.M.S. and B.E.V.; Project administration, L.R.; Supervision, D.M.S., W.M. and G.R.Y.D.M.; Visualization, L.R.; Writing—original draft, L.R.; Writing—review & editing, L.R., M.R., D.M.S., B.E.V., W.M. and G.R.Y.D.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the University of Antwerp (BOF-GOA project ID 33931 and BOF-SEP project ID 40183). Besa Emini Veseli is a PhD fellow of the Horizon 2020 program of the European Union—Marie Skłodowska-Curie Actions, Innovative Training Networks (ITN), Call: H2020-MSCA-ITN-2015, NUMBER-675527-MOGLYNET.
Institutional Review Board Statement
The study was conducted conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes, and approved by the Ethics Committee of the University of Antwerp (protocol code 2014-15 and date of approval: March 2014).
Informed Consent Statement
Not appliable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors like to thank Rita Van den Bossche, Hermine Fret, Anne-Elise Van Hoydonck, Mandy Vermont, Ammar Kurdi, Bieke Van der Veken, Hadis Shakeri, Ilse Van Brussel, and Katrien Lemmens for technical support. The authors are grateful to Bronwen Martin for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palombo, C.; Kozakova, M. Arterial stiffness, atherosclerosis and cardiovascular risk: Pathophysiologic mechanisms and emerging clinical indications. Vascul. Pharmacol. 2016, 77, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S.; Bentzon, J.F.; Daemen, M.; Falk, E.; Garcia-Garcia, H.M.; Herrmann, J.; Hoefer, I.; Jauhiainen, S.; Jukema, J.W.; Krams, R.; et al. Stabilization of atherosclerotic plaques: An update. Eur. Heart J. 2013, 34, 3251–3258. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Ageing. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Mozos, I.; Malainer, C.; Horbanczuk, J.; Gug, C.; Stoian, D.; Luca, C.T.; Atanasov, A.G. Inflammatory Markers for Arterial Stiffness in Cardiovascular Diseases. Front. Immunol. 2017, 8, 1058. [Google Scholar] [CrossRef]
- Li, J.; Chen, Q.; Luo, X.; Hong, J.; Pan, K.; Lin, X.; Liu, X.; Zhou, L.; Wang, H.; Xu, Y.; et al. Neutrophil-to-Lymphocyte Ratio Positively Correlates to Age in Healthy Population. J. Clin. Lab. Anal. 2015, 29, 437–443. [Google Scholar] [CrossRef]
- Valiathan, R.; Ashman, M.; Asthana, D. Effects of Ageing on the Immune System: Infants to Elderly. Scand. J. Immunol. 2016, 83, 255–266. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Huang, L.; Lu, J. Association between neutrophil–lymphocyte ratio and arterial stiffness in patients with acute coronary syndrome. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef]
- Chen, C.; Cong, B.L.; Wang, M.; Abdullah, M.; Wang, X.L.; Zhang, Y.H.; Xu, S.J.; Cui, L. Neutrophil to lymphocyte ratio as a predictor of myocardial damage and cardiac dysfunction in acute coronary syndrome patients. Integr. Med. Res. 2018, 7, 192–199. [Google Scholar] [CrossRef]
- Ozyilmaz, S.; Akgul, O.; Uyarel, H.; Pusuroglu, H.; Gul, M.; Satilmisoglu, M.H.; Bolat, I.; Ozyilmaz, I.; Ucar, H.; Yildirim, A.; et al. The importance of the neutrophil-to-lymphocyte ratio in patients with hypertrophic cardiomyopathy. Rev. Port. Cardiol. 2017, 36, 239–246. [Google Scholar] [CrossRef]
- Kaya, H.; Ertas, F.; Soydinc, M.S. Association between neutrophil to lymphocyte ratio and severity of coronary artery disease. Clin. Appl. Thromb. Hemost. 2014, 20, 221. [Google Scholar] [CrossRef]
- Adamstein, N.H.; MacFadyen, J.G.; Rose, L.M.; Glynn, R.J.; Dey, A.K.; Libby, P.; Tabas, I.A.; Mehta, N.N.; Ridker, P.M. The neutrophil–lymphocyte ratio and incident atherosclerotic events: Analyses from five contemporary randomized trials. Eur. Heart J. 2021, 42, 896–903. [Google Scholar] [CrossRef]
- Nowak, K.L.; Rossman, M.J.; Chonchol, M.; Seals, D.R. Strategies for Achieving Healthy Vascular Ageing. Hypertension 2018, 71, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Toth, P.P.; Fogacci, F.; Virdis, A.; Borghi, C. Improvement in arterial stiffness after short-term treatment with PCSK9 inhibitors. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Picker, S.M. Antiplatelet therapy in the prevention of coronary syndromes: Mode of action, benefits, drawbacks. Cardiovasc. Hematol. Agents Med. Chem. 2013, 11, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.S.; Brown, D.L.; Becker, R.C. Low-dose aspirin in patients with stable cardiovascular disease: A meta-analysis. Am. J. Med. 2008, 121, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hennekens, C.H. Aspirin in the treatment and prevention of cardiovascular disease: Current perspectives and future directions. Curr. Atheroscler. Rep. 2007, 9, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Brotons, C.; Coppolecchia, R.; Cricelli, C.; Darius, H.; Gorelick, P.B.; Howard, G.; Pearson, T.A.; Rothwell, P.M.; Ruilope, L.M.; et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): A randomised, double-blind, placebo-controlled trial. Lancet 2018, 392, 1036–1046. [Google Scholar] [CrossRef]
- McNeil, J.J.; Woods, R.L.; Nelson, M.R.; Reid, C.M.; Kirpach, B.; Wolfe, R.; Storey, E.; Shah, R.C.; Lockery, J.E.; Tonkin, A.M.; et al. Effect of Aspirin on Disability-free Survival in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1499–1508. [Google Scholar] [CrossRef]
- Group, A.S.C.; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Ferruzzi, J.; Collins, M.J.; Yeh, A.T.; Humphrey, J.D. Mechanical assessment of elastin integrity in fibrillin-1-deficient carotid arteries: Implications for Marfan syndrome. Cardiovasc. Res. 2011, 92, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Medley, T.L.; Cole, T.J.; Gatzka, C.D.; Wang, W.Y.; Dart, A.M.; Kingwell, B.A. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation 2002, 105, 810–815. [Google Scholar] [CrossRef]
- Mariko, B.; Pezet, M.; Escoubet, B.; Bouillot, S.; Andrieu, J.P.; Starcher, B.; Quaglino, D.; Jacob, M.P.; Huber, P.; Ramirez, F.; et al. Fibrillin-1 genetic deficiency leads to pathological ageing of arteries in mice. J. Pathol. 2011, 224, 33–44. [Google Scholar] [CrossRef]
- Sherratt, M.J. Tissue elasticity and the ageing elastic fibre. Age 2009, 31, 305–325. [Google Scholar] [CrossRef]
- Tsamis, A.; Krawiec, J.T.; Vorp, D.A. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: A review. J. R. Soc. Interface 2013, 10, 20121004. [Google Scholar] [CrossRef]
- Van der Donckt, C.; Van Herck, J.L.; Schrijvers, D.M.; Vanhoutte, G.; Verhoye, M.; Blockx, I.; Van Der Linden, A.; Bauters, D.; Lijnen, H.R.; Sluimer, J.C.; et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur. Heart J. 2015, 36, 1049–1058. [Google Scholar] [CrossRef]
- Van Herck, J.L.; De Meyer, G.R.Y.; Martinet, W.; Van Hove, C.E.; Foubert, K.; Theunis, M.H.; Apers, S.; Bult, H.; Vrints, C.J.; Herman, A.G. Impaired Fibrillin-1 Function Promotes Features of Plaque Instability in Apolipoprotein E-Deficient Mice. Circulation 2009, 120, 2478–2487. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.L. Evaluation of the neutrophil:lymphocyte ratio as an indicator of chronic distress in the laboratory mouse. Lab. Anim. 2017, 46, 303–307. [Google Scholar] [CrossRef]
- Mayyas, F.A.; Al-Jarrah, M.I.; Ibrahim, K.S.; Alzoubi, K.H. Level and significance of plasma myeloperoxidase and the neutrophil to lymphocyte ratio in patients with coronary artery disease. Exp. Ther. Med. 2014, 8, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, H.; Silvestre Roig, C.; Daemen, M.; Lutgens, E.; Soehnlein, O. Neutrophils in atherosclerosis. A brief overview. Hamostaseologie 2015, 35, 121–127. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Santos, H.O.; Izidoro, L.F.M. Neutrophil–lymphocyte Ratio in Cardiovascular Disease Risk Assessment. Int. J. Cardiovasc. Sci. 2018, 31, 532–537. [Google Scholar] [CrossRef]
- Kaartinen, V.; Warburton, D. Fibrillin controls TGF-beta activation. Nat. Genet. 2003, 33, 331–332. [Google Scholar] [CrossRef]
- Buday, A.; Orsy, P.; Godo, M.; Mozes, M.; Kokeny, G.; Lacza, Z.; Koller, A.; Ungvari, Z.; Gross, M.L.; Benyo, Z.; et al. Elevated systemic TGF-beta impairs aortic vasomotor function through activation of NADPH oxidase-driven superoxide production and leads to hypertension, myocardial remodeling, and increased plaque formation in apoE(-/-) mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H386–H395. [Google Scholar] [CrossRef] [PubMed]
- Nataatmadja, M.; West, J.; West, M. Overexpression of transforming growth factor-beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aortic aneurysm. Circulation 2006, 114, I371–I377. [Google Scholar] [CrossRef] [PubMed]
- Pietri, P.; Vlachopoulos, C.; Terentes-Printzios, D.; Xaplanteris, P.; Aznaouridis, K.; Petrocheilou, K.; Stefanadis, C. Beneficial effects of low-dose aspirin on aortic stiffness in hypertensive patients. Vasc. Med. 2014, 19, 452–457. [Google Scholar] [CrossRef]
- Lee, S.H.; Pekas, E.J.; Lee, S.; Headid, R.J., 3rd; Park, S.Y. The Impact of Aspirin Intake on Lactate Dehydrogenase, Arterial Stiffness, and Oxidative Stress During High-Intensity Exercise: A Pilot Study. J. Hum. Kinet. 2020, 72, 101–113. [Google Scholar] [CrossRef]
- Franken, R.; Hibender, S.; den Hartog, A.W.; Radonic, T.; de Vries, C.J.; Zwinderman, A.H.; Groenink, M.; Mulder, B.J.; de Waard, V. No beneficial effect of general and specific anti-inflammatory therapies on aortic dilatation in Marfan mice. PLoS ONE 2014, 9, e107221. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, J.; Khaidakov, M.; Mitra, S.; Ding, Z.; Raina, S.; Goyal, T.; Mehta, J.L. Aspirin suppresses cardiac fibroblast proliferation and collagen formation through downregulation of angiotensin type 1 receptor transcription. Toxicol. Appl. Pharmacol. 2012, 259, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Gaikwad, A.B.; Tikoo, K. Combination of aspirin with telmisartan suppresses the augmented TGFbeta/smad signalling during the development of streptozotocin-induced type I diabetic nephropathy. Chem. Biol. Interact. 2010, 185, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Harrison, D.G.; Figueroa, C.A.; Lacolley, P.; Laurent, S. Central Artery Stiffness in Hypertension and Ageing: A Problem With Cause and Consequence. Circ. Res. 2016, 118, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, H.; Xu, D.; Xu, X.; Wang, H.; Hu, X.; Lu, Z.; Kwak, D.; Xu, Y.; Gunther, R.; et al. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: Heart failure causes severe lung disease. Hypertension 2012, 59, 1170–1178. [Google Scholar] [CrossRef]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef]
- Prabhu, S.D. The Cardiosplenic Axis Is Essential for the Pathogenesis of Ischemic Heart Failure. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 202–214. [Google Scholar]
- Jackson, C.L.; Bennett, M.R.; Biessen, E.A.L.; Johnson, J.L.; Krams, R. Assessment of unstable atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 714–720. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).