
Modulation of miR-29a and ADAM12 Reduces Post-Ischemic Skeletal Muscle Injury and Improves Perfusion Recovery and Skeletal Muscle Function in a Mouse Model of Type 2 Diabetes and Peripheral Artery Disease


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
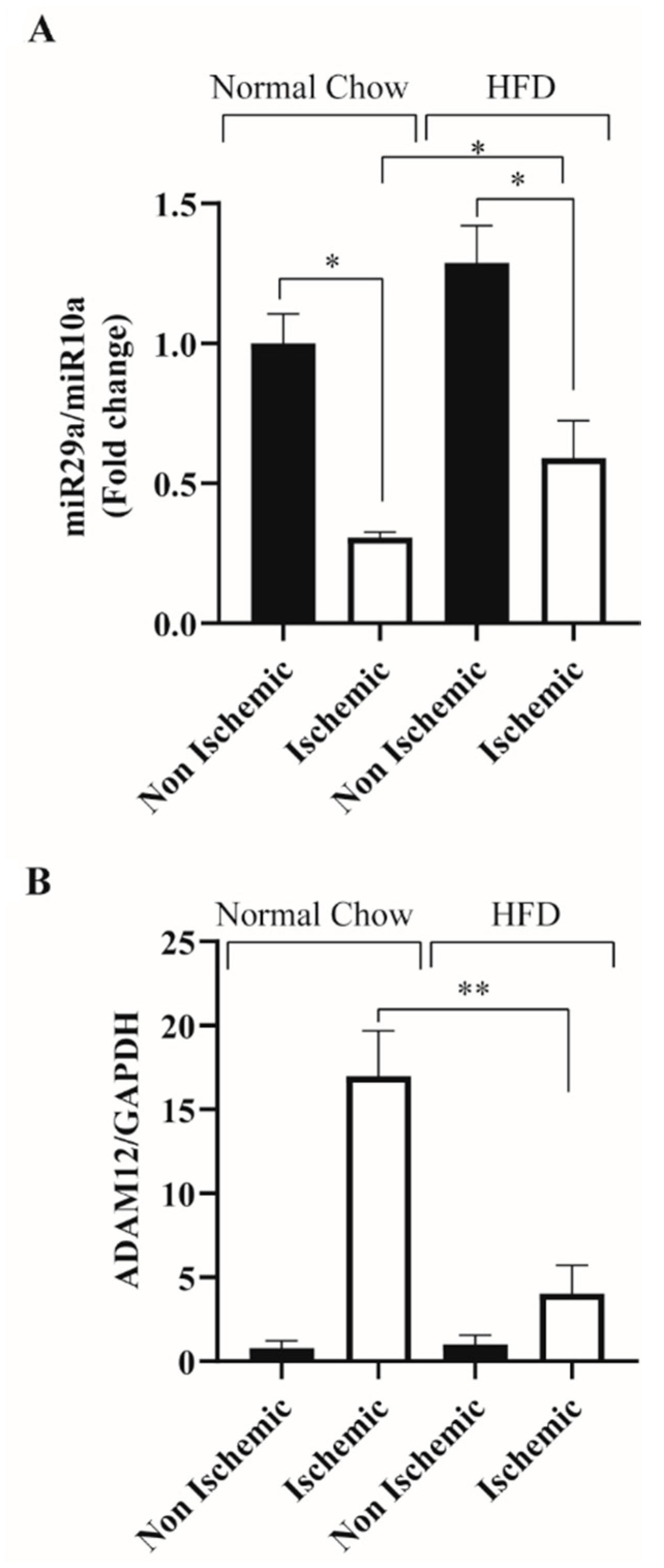
2.1. HFD Feeding Impairs Ischemia Induced ADAM12 Upregulation and miR-29a Downregulation in C57BL/6 Mice
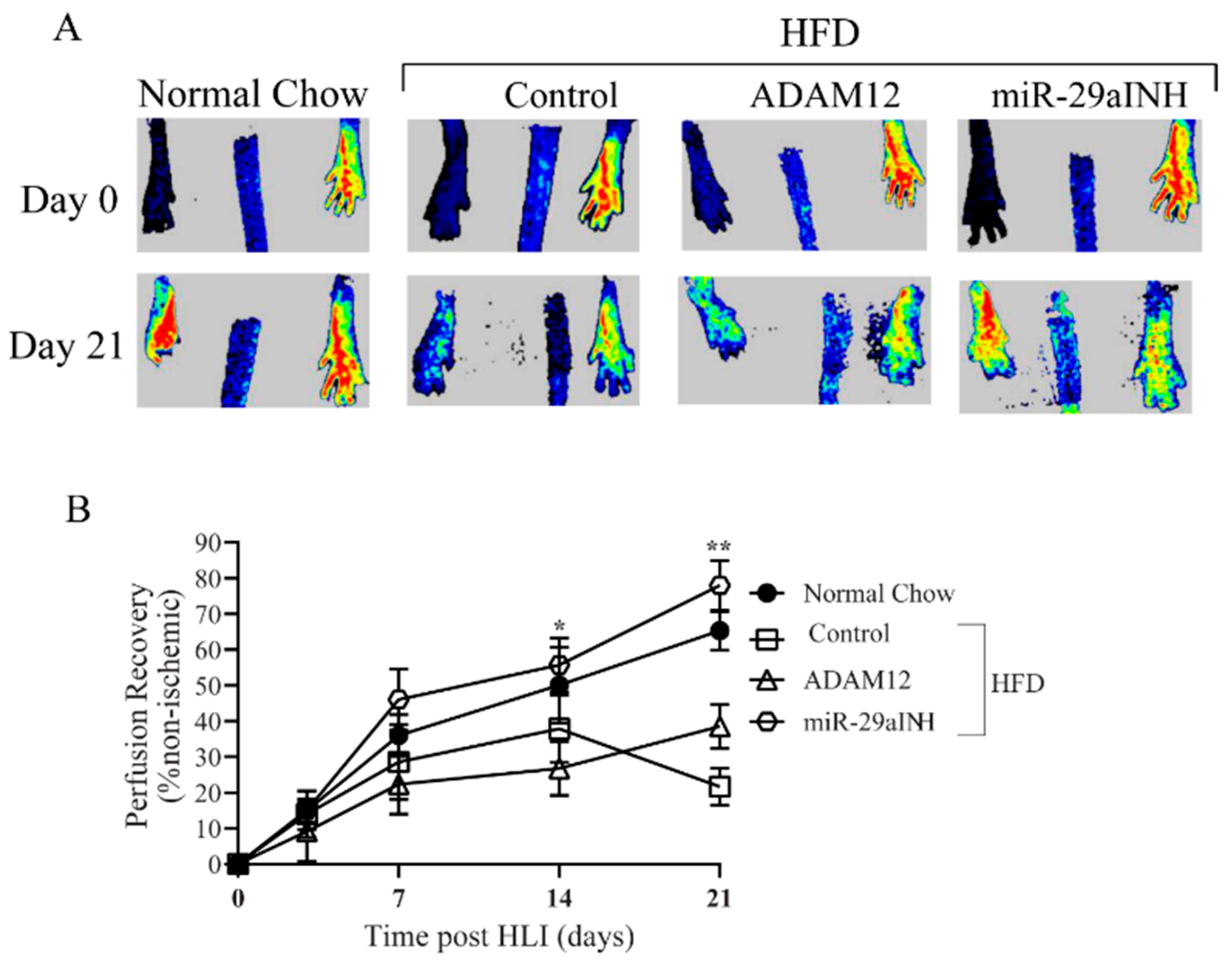
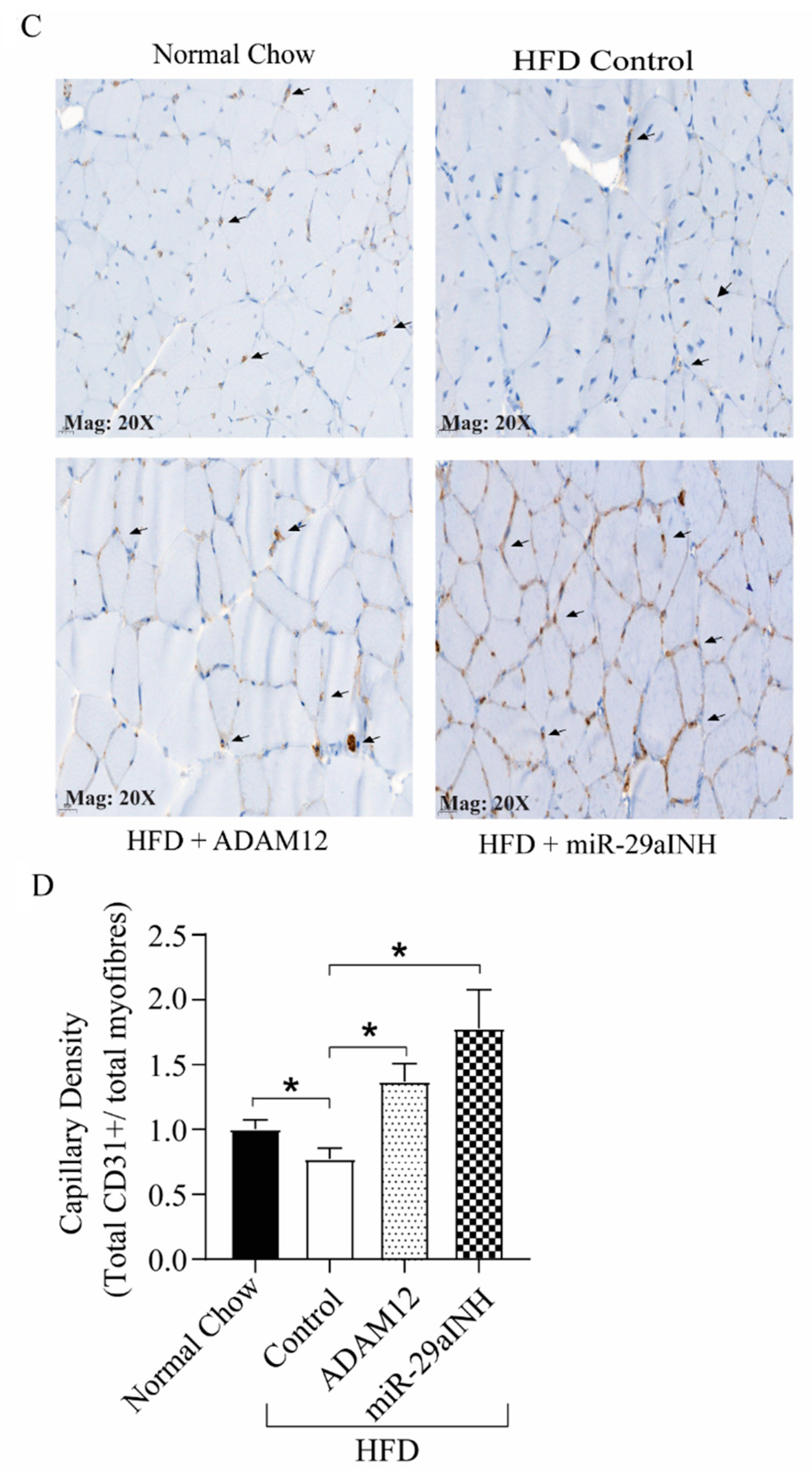
2.2. ADAM12 Gene Transfer and miR-29a Inhibition in Ischemic GA Muscle of HFD Mice Increased ADAM12 Expression, Improved Perfusion Recovery and Capillary Density
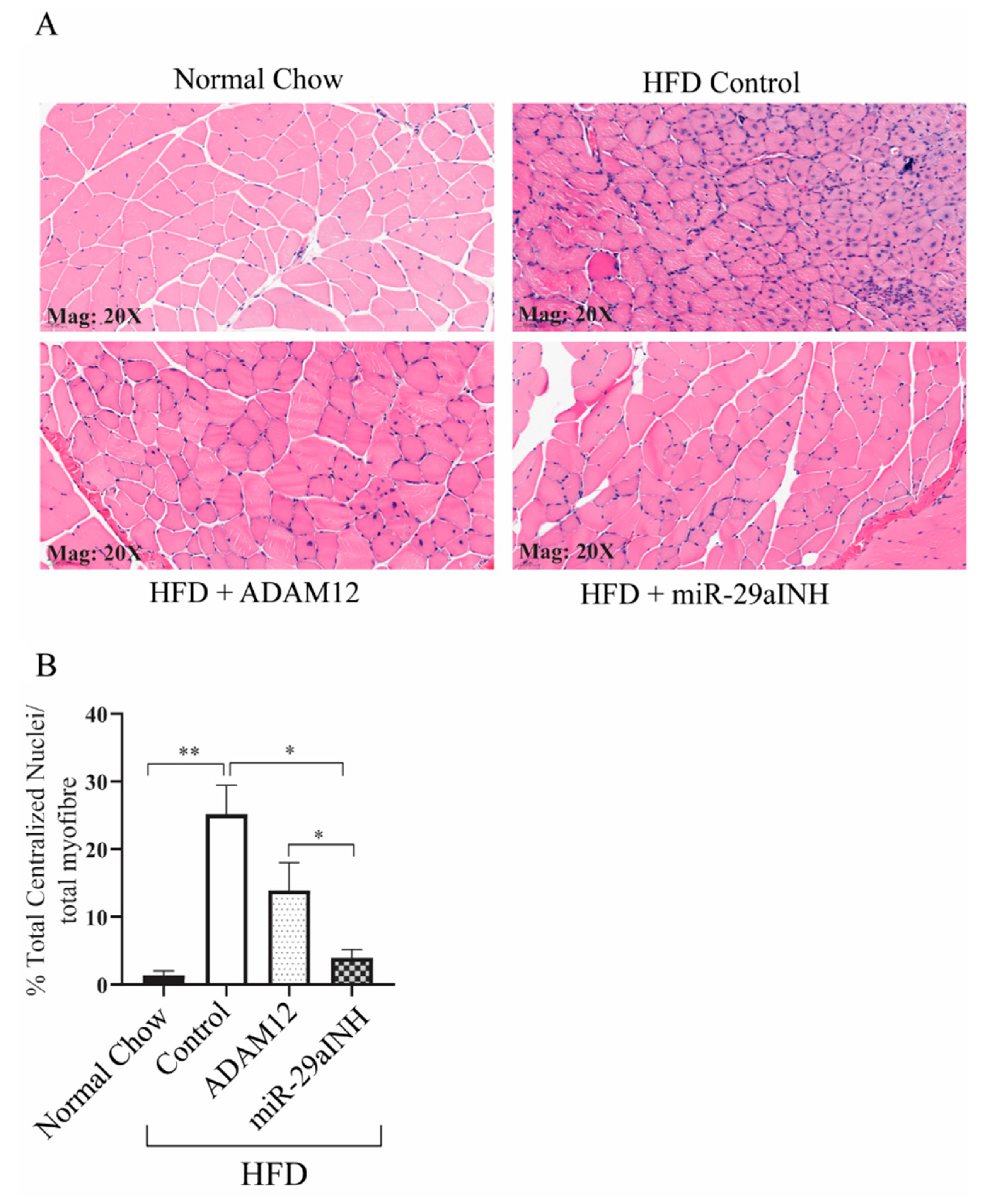
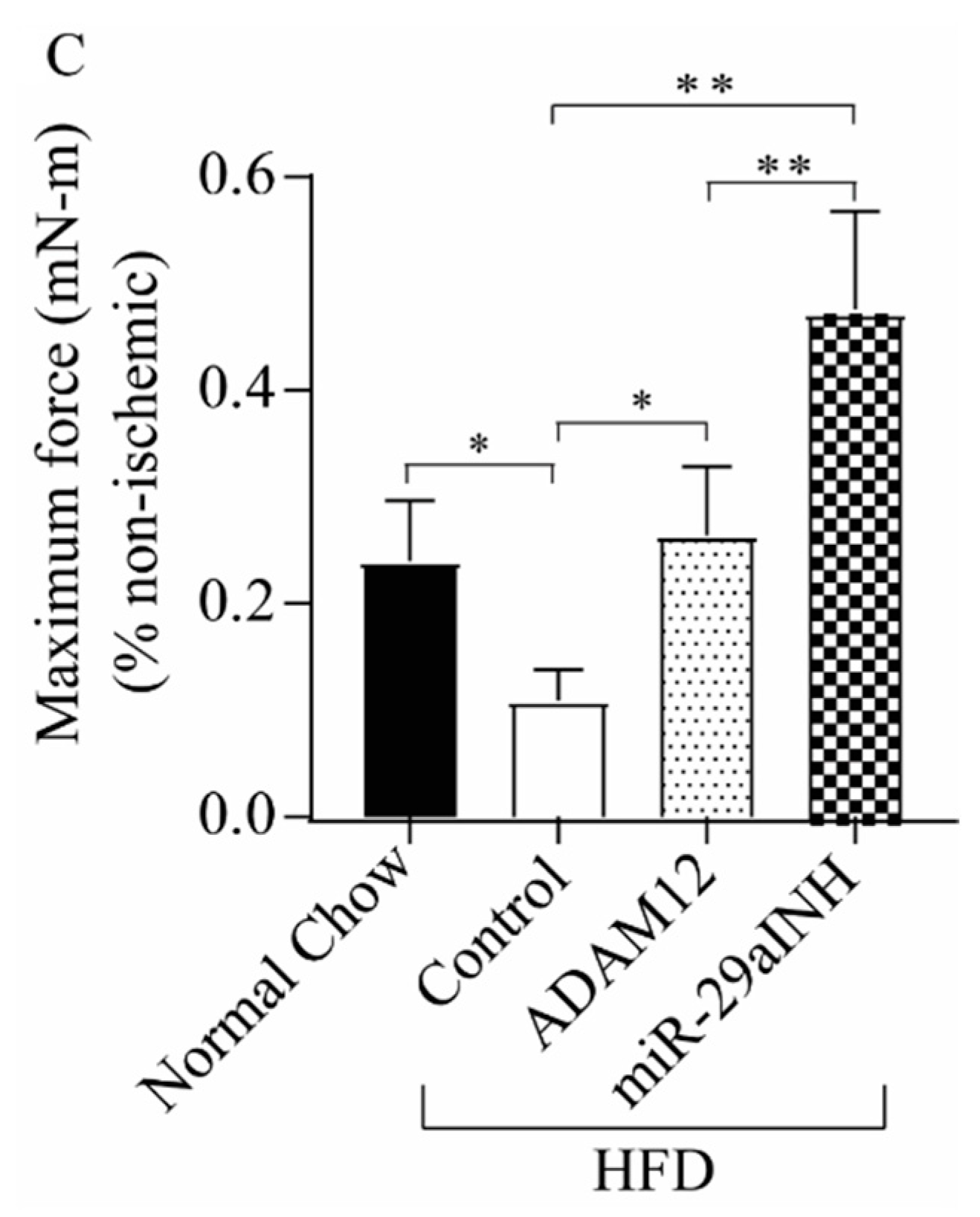
2.3. ADAM12 Augmentation and miR-29a Inhibition Reduced the Extent of Skeletal Muscle Injury and Increased Skeletal Muscle Function in Ischemic GA Muscle of HFD Mice
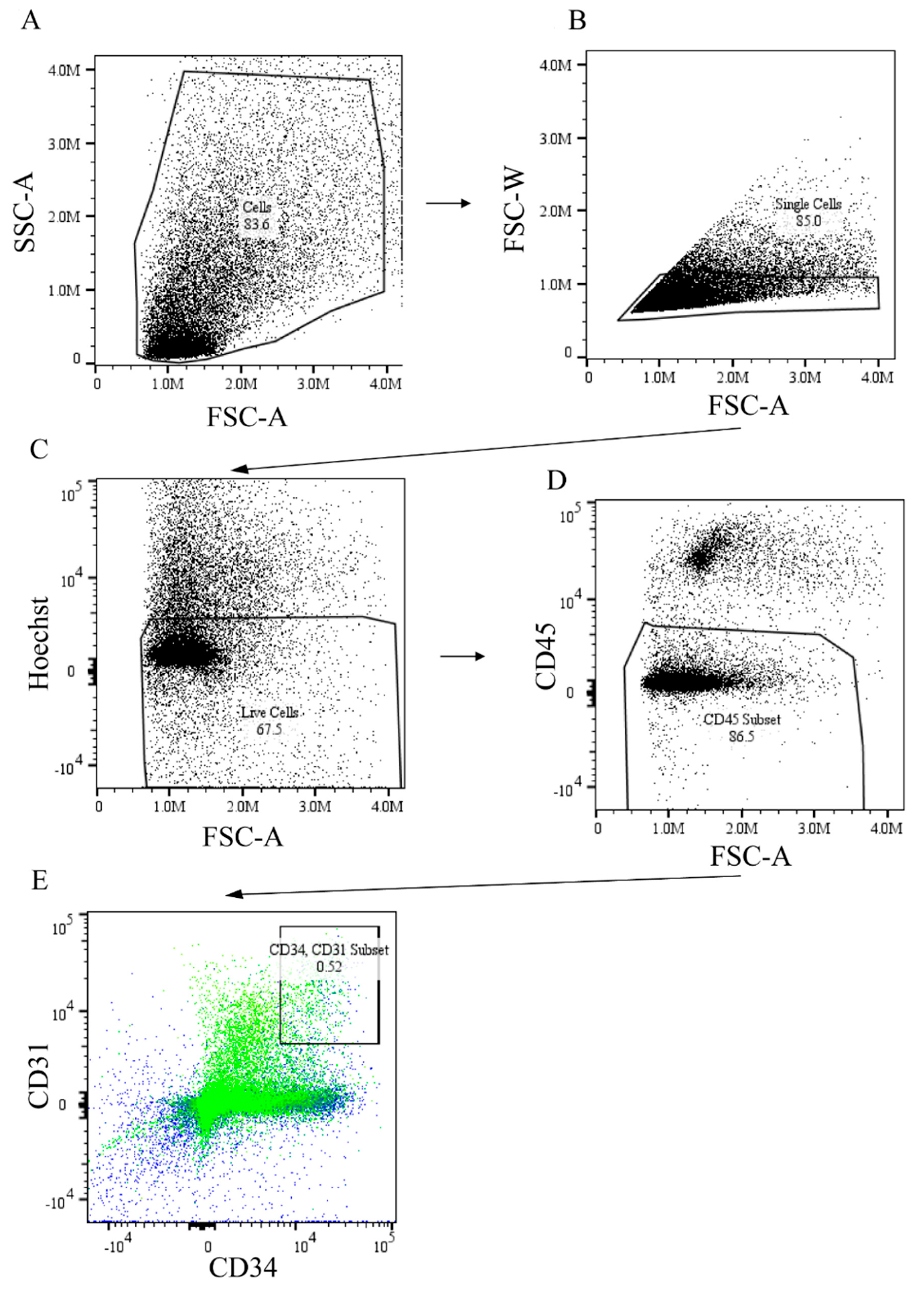
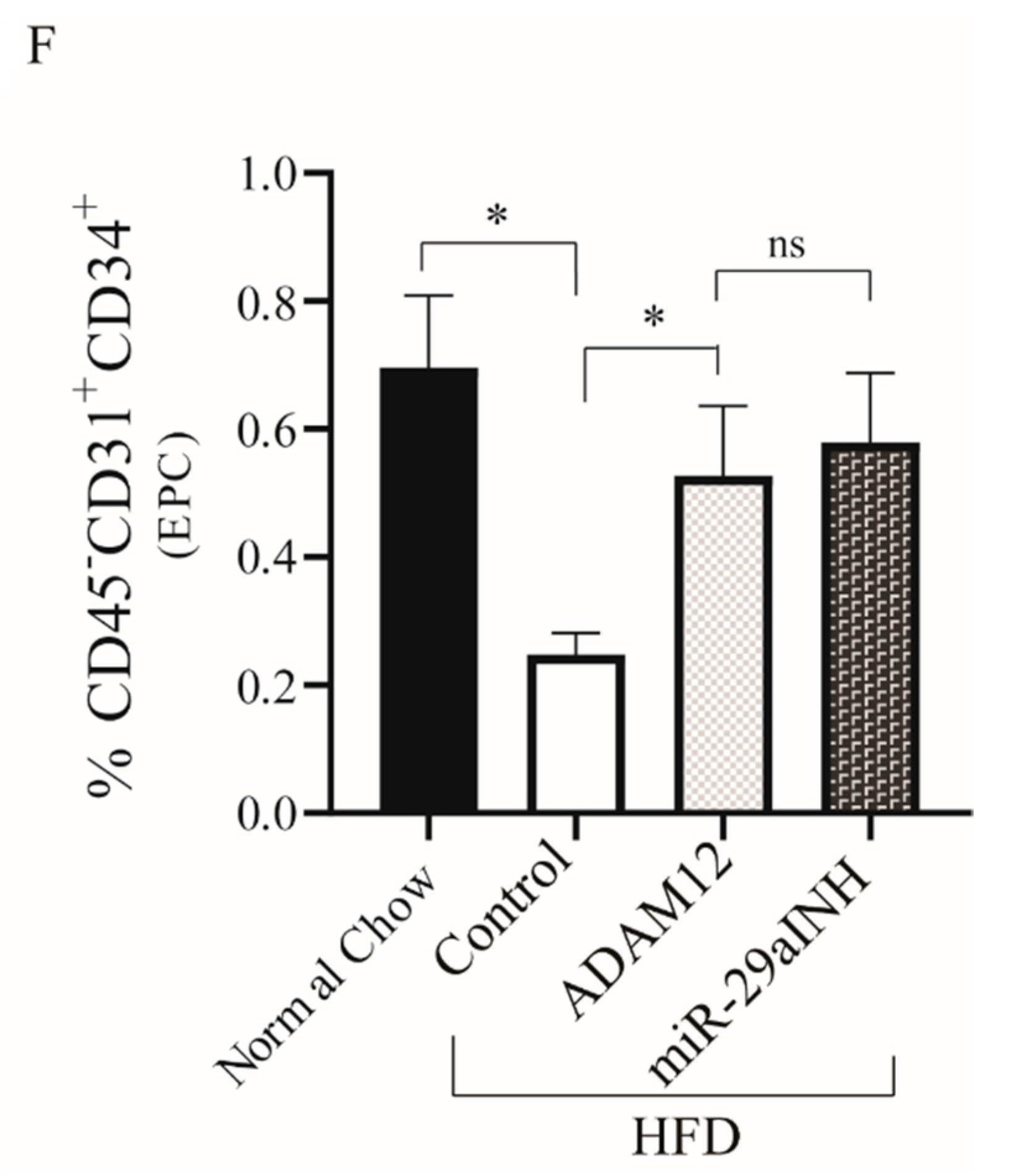
2.4. ADAM12 Gene Augmentation and miR-29a Inhibition Increased Endothelial Progenitor Cells in Post Ischemic GA Muscle of HFD Mouse
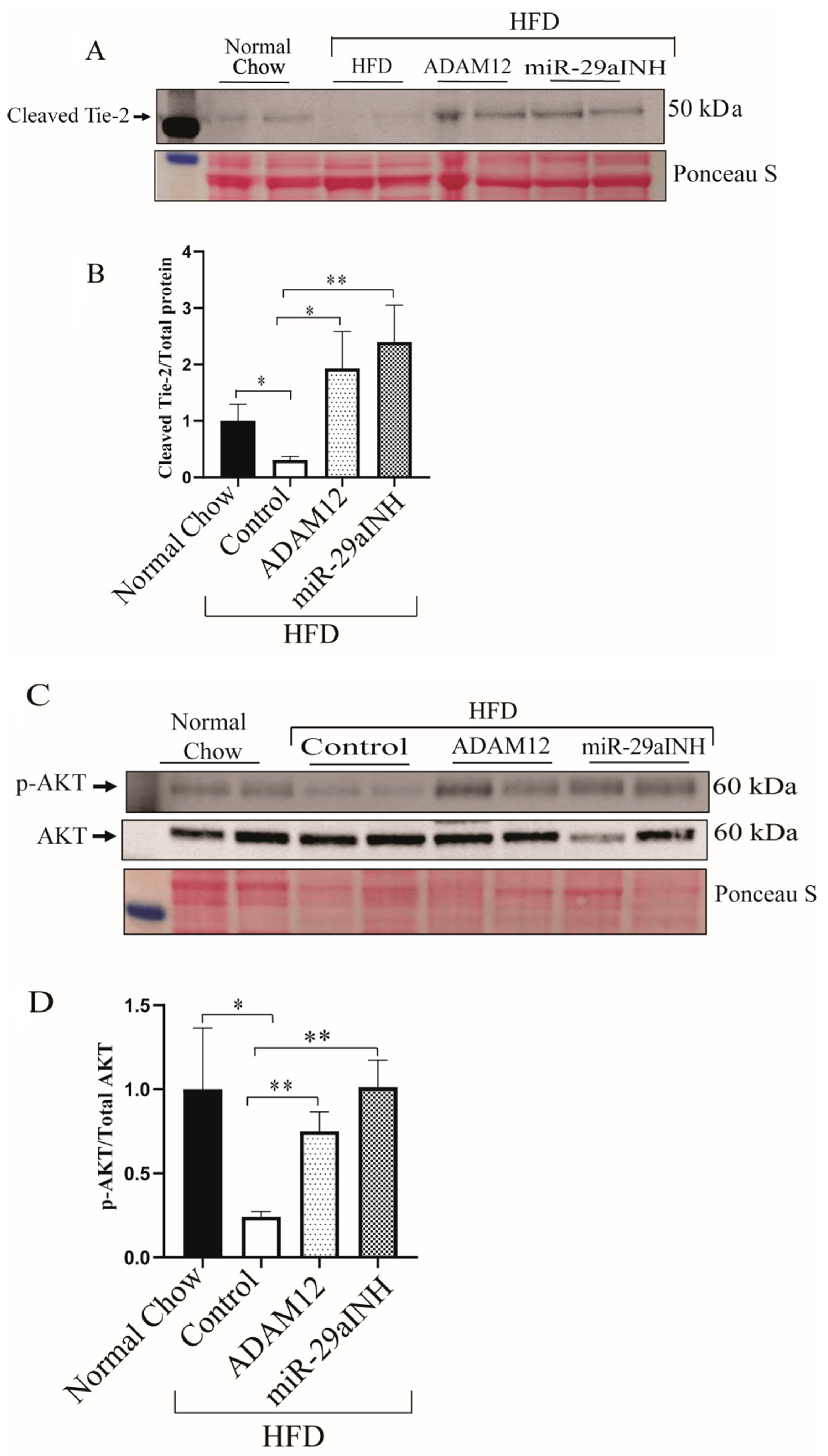
2.5. miR-29a Inhibition and ADAM12 Augmentation Modulate Tie2 Cleavage through Increased Akt Phosphorylation (P-AKT) In-Vivo
3. Discussion
4. Methods
4.1. Animals
4.2. Hind Limb Ischemia Surgery and Perfusion Imaging
4.3. In Vivo miR-29a Inhibition and ADAM12 over-Expression
4.4. Muscle Contractile Function
4.5. RNA, Quantitative PCR
4.6. Western Blot Analyses
4.7. Immunohistochemistry
4.8. Flow Cytometry
4.9. Statistical Analysis
4.10. GRANTS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhaliwal, G.; Mukherjee, D. Peripheral arterial disease: Epidemiology, natural history, diagnosis and treatment. Int. J. Angiol. 2007, 16, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Hirsch, A.T.; Duval, S. The global pandemic of peripheral artery disease. Lancet 2013, 382, 1312–1314. [Google Scholar] [CrossRef]
- Jude, E.B.; Eleftheriadou, I.; Tentolouris, N. Peripheral arterial disease in diabetes—A review. Diabet. Med. 2010, 27, 4–14. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Thiruvoipati, T.; Kielhorn, C.E.; Armstrong, E.J. Peripheral artery disease in patients with diabetes: Epidemiology, mechanisms, and outcomes. World J. Diabetes 2015, 6, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Haltmayer, M.; Mueller, T.; Horvath, W.; Luft, C.; Poelz, W.; Haidinger, D. Impact of atherosclerotic risk factors on the anatomical distribution of peripheral arterial disease. Int. Angiol. 2001, 20, 200–207. [Google Scholar]
- Jude, E.B.; Oyibo, S.O.; Chalmers, N.; Boulton, A.J. Peripheral arterial disease in diabetic and nondiabetic patients: A comparison of severity and outcome. Diabetes Care 2001, 24, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Hiatt, W.R. Peripheral arterial disease in patients with diabetes. J. Am. Coll. Cardiol. 2006, 47, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton, L.J., 3rd; Macken, K.M.; Palumbo, P.J.; Elveback, L.R. Incidence and prevalence of clinical peripheral vascular disease in a population-based cohort of diabetic patients. Diabetes Care 1980, 3, 650–654. [Google Scholar] [CrossRef]
- Dokun, A.O.; Chen, L.; Lanjewar, S.S.; Lye, R.J.; Annex, B.H. Glycaemic control improves perfusion recovery and VEGFR2 protein expression in diabetic mice following experimental PAD. Cardiovasc. Res. 2014, 101, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiatt, W.R. Medical treatment of peripheral arterial disease and claudication. N. Engl. J. Med. 2001, 344, 1608–1621. [Google Scholar] [CrossRef]
- Hirsch, A.T. Treatment of peripheral arterial disease--extending “intervention” to “therapeutic choice”. N. Engl. J. Med. 2006, 354, 1944–1947. [Google Scholar] [CrossRef]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.; Group, T.I.W. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67. [Google Scholar] [CrossRef] [Green Version]
- Annex, B.H. Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 2013, 10, 387–396. [Google Scholar] [CrossRef]
- Annex, B.H.; Beller, G.A. Towards the Development of Novel Therapeutics for Peripheral Artery Disease. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 224–234. [Google Scholar]
- Peacock, J.M.; Keo, H.H.; Duval, S.; Baumgartner, I.; Oldenburg, N.C.; Jaff, M.R.; Henry, T.D.; Yu, X.; Hirsch, A.T. The incidence and health economic burden of ischemic amputation in Minnesota, 2005–2008. Prev. Chronic. Dis. 2011, 8, A141. [Google Scholar]
- Wennberg, P.W. Approach to the patient with peripheral arterial disease. Circulation 2013, 128, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Kusumanto, Y.H.; van Weel, V.; Mulder, N.H.; Smit, A.J.; van den Dungen, J.J.; Hooymans, J.M.; Sluiter, W.J.; Tio, R.A.; Quax, P.H.; Gans, R.O.; et al. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: A double-blind randomized trial. Hum. Gene Ther. 2006, 17, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinen, K.; Manninen, H.; Hedman, M.; Matsi, P.; Mussalo, H.; Alhava, E.; Yla-Herttuala, S. Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: A randomized, placebo-controlled, double-blinded phase II study. Mol. Ther. 2002, 6, 127–133. [Google Scholar] [CrossRef]
- Muona, K.; Makinen, K.; Hedman, M.; Manninen, H.; Yla-Herttuala, S. 10-year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther. 2012, 19, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikol, S.; Baumgartner, I.; Van Belle, E.; Diehm, C.; Visona, A.; Capogrossi, M.C.; Ferreira-Maldent, N.; Gallino, A.; Graham Wyatt, M.; Dinesh Wijesinghe, L.; et al. Therapeutic Angiogenesis With Intramuscular NV1FGF Improves Amputation-free Survival in Patients With Critical Limb Ischemia. Mol. Ther. 2008, 16, 972–978. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Mohler, E.R., 3rd; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: A phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 2003, 108, 1933–1938. [Google Scholar]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of miRNA-Mediated Gene Regulation from Common Downregulation to mRNA-Specific Upregulation. Int. J. Genom. 2014, 2014, 970607. [Google Scholar] [CrossRef] [Green Version]
- Dokun, A.O.; Chen, L.; Okutsu, M.; Farber, C.R.; Hazarika, S.; Jones, W.S.; Craig, D.; Marchuk, D.A.; Lye, R.J.; Shah, S.H.; et al. ADAM12: A genetic modifier of preclinical peripheral arterial disease. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H790–H803. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Okeke, E.; Ayalew, D.; Wang, D.; Shahid, L.; Dokun, A.O. Modulation of miR29a improves impaired post-ischemic angiogenesis in hyperglycemia. Exp. Biol. Med. 2017, 242, 1432–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazarika, S.; Dokun, A.O.; Li, Y.; Popel, A.S.; Kontos, C.D.; Annex, B.H. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: Differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ. Res. 2007, 101, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hazarika, S.; Xie, D.; Pippen, A.M.; Kontos, C.D.; Annex, B.H. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes 2007, 56, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Folker, E.S.; Baylies, M.K. Nuclear positioning in muscle development and disease. Front. Physiol. 2013, 4, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalka, C.; Masuda, H.; Takahashi, T.; Kalka-Moll, W.M.; Silver, M.; Kearney, M.; Li, T.; Isner, J.M.; Asahara, T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. USA 2000, 97, 3422–3427. [Google Scholar] [CrossRef]
- Shen, W.C.; Liang, C.J.; Wu, V.C.; Wang, S.H.; Young, G.H.; Lai, I.R.; Chien, C.L.; Wang, S.M.; Wu, K.D.; Chen, Y.L. Endothelial progenitor cells derived from Wharton’s jelly of the umbilical cord reduces ischemia-induced hind limb injury in diabetic mice by inducing HIF-1alpha/IL-8 expression. Stem. Cells Dev. 2013, 22, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.H.; Chai, H.T.; Sun, C.K.; Yen, C.H.; Leu, S.; Chen, Y.L.; Chung, S.Y.; Ko, S.F.; Chang, H.W.; Wu, C.J.; et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J. Transl. Med. 2012, 10, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ieronimakis, N.; Balasundaram, G.; Reyes, M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS ONE 2008, 3, e0001753. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, C.; Klitgaard, M.; Noer, J.B.; Kotzsch, A.; Nehammer, C.; Kronqvist, P.; Berthelsen, J.; Blobel, C.; Kveiborg, M.; Albrechtsen, R.; et al. ADAM12 is expressed in the tumour vasculature and mediates ectodomain shedding of several membrane-anchored endothelial proteins. Biochem. J. 2013, 452, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Lekas, M.; Lekas, P.; Mei, S.H.; Deng, Y.; Dumont, D.J.; Stewart, D.J. Tie2-dependent neovascularization of the ischemic hindlimb is mediated by angiopoietin-2. PLoS ONE 2012, 7, e43568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findley, C.M.; Cudmore, M.J.; Ahmed, A.; Kontos, C.D. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Lee, H.S.; Lee, D.; Lee, S.D.; Cho, E.G.; Yang, S.J.; Kim, S.B.; Park, D.; Kim, M.G. Epithin/PRSS14 proteolytically regulates angiopoietin receptor Tie2 during transendothelial migration. Blood 2011, 117, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Klein, J.D.; Mitch, W.E.; Zhang, L.; Martinez, I.; Wang, X.H. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging 2014, 6, 160–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimov, A.; Merry, T.L.; Luca, E.; Rushing, E.J.; Mizbani, A.; Turcekova, K.; Hartung, A.; Croce, C.M.; Ristow, M.; Krutzfeldt, J. MicroRNA-29a in Adult Muscle Stem Cells Controls Skeletal Muscle Regeneration During Injury and Exercise Downstream of Fibroblast Growth Factor-2. Stem. Cells 2016, 34, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, J.; He, W.; Zhao, Y.; Zhang, A.; Liu, Y.; Hassounah, F.; Ma, F.; Klein, J.D.; Wang, X.H.; et al. Exogenous miR-29a Attenuates Muscle Atrophy and Kidney Fibrosis in Unilateral Ureteral Obstruction Mice. Hum. Gene Ther. 2020, 31, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, J.D.; Mere, C.P.; Kronemberger, A.; Blomme, J.; Bae, D.; Turner, K.D.; Harris, M.P.; Scudese, E.; Edwards, M.; Ebert, S.M.; et al. ULK2 is essential for degradation of ubiquitinated protein aggregates and homeostasis in skeletal muscle. FASEB J. 2019, 33, 11735–11745. [Google Scholar] [CrossRef] [Green Version]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamin, V.; Verry, J.; Eigner-Bybee, I.; Fuqua, J.D.; Wong, T.; Lira, V.A.; Dokun, A.O. Modulation of miR-29a and ADAM12 Reduces Post-Ischemic Skeletal Muscle Injury and Improves Perfusion Recovery and Skeletal Muscle Function in a Mouse Model of Type 2 Diabetes and Peripheral Artery Disease. Int. J. Mol. Sci. 2022, 23, 429. https://doi.org/10.3390/ijms23010429
Lamin V, Verry J, Eigner-Bybee I, Fuqua JD, Wong T, Lira VA, Dokun AO. Modulation of miR-29a and ADAM12 Reduces Post-Ischemic Skeletal Muscle Injury and Improves Perfusion Recovery and Skeletal Muscle Function in a Mouse Model of Type 2 Diabetes and Peripheral Artery Disease. International Journal of Molecular Sciences. 2022; 23(1):429. https://doi.org/10.3390/ijms23010429
Chicago/Turabian StyleLamin, Victor, Joseph Verry, Isaac Eigner-Bybee, Jordan D. Fuqua, Thomas Wong, Vitor A. Lira, and Ayotunde O. Dokun. 2022. "Modulation of miR-29a and ADAM12 Reduces Post-Ischemic Skeletal Muscle Injury and Improves Perfusion Recovery and Skeletal Muscle Function in a Mouse Model of Type 2 Diabetes and Peripheral Artery Disease" International Journal of Molecular Sciences 23, no. 1: 429. https://doi.org/10.3390/ijms23010429
APA StyleLamin, V., Verry, J., Eigner-Bybee, I., Fuqua, J. D., Wong, T., Lira, V. A., & Dokun, A. O. (2022). Modulation of miR-29a and ADAM12 Reduces Post-Ischemic Skeletal Muscle Injury and Improves Perfusion Recovery and Skeletal Muscle Function in a Mouse Model of Type 2 Diabetes and Peripheral Artery Disease. International Journal of Molecular Sciences, 23(1), 429. https://doi.org/10.3390/ijms23010429