
Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses


Abstract
:1. Introduction
2. E3 Ubiquitin Ligases and DUBs as Immune Modulators
2.1. E3 Ubiquitin Ligases (E3s)
2.2. Deubiquitinating Enzymes (DUBs)
3. RLR Signaling Modulation
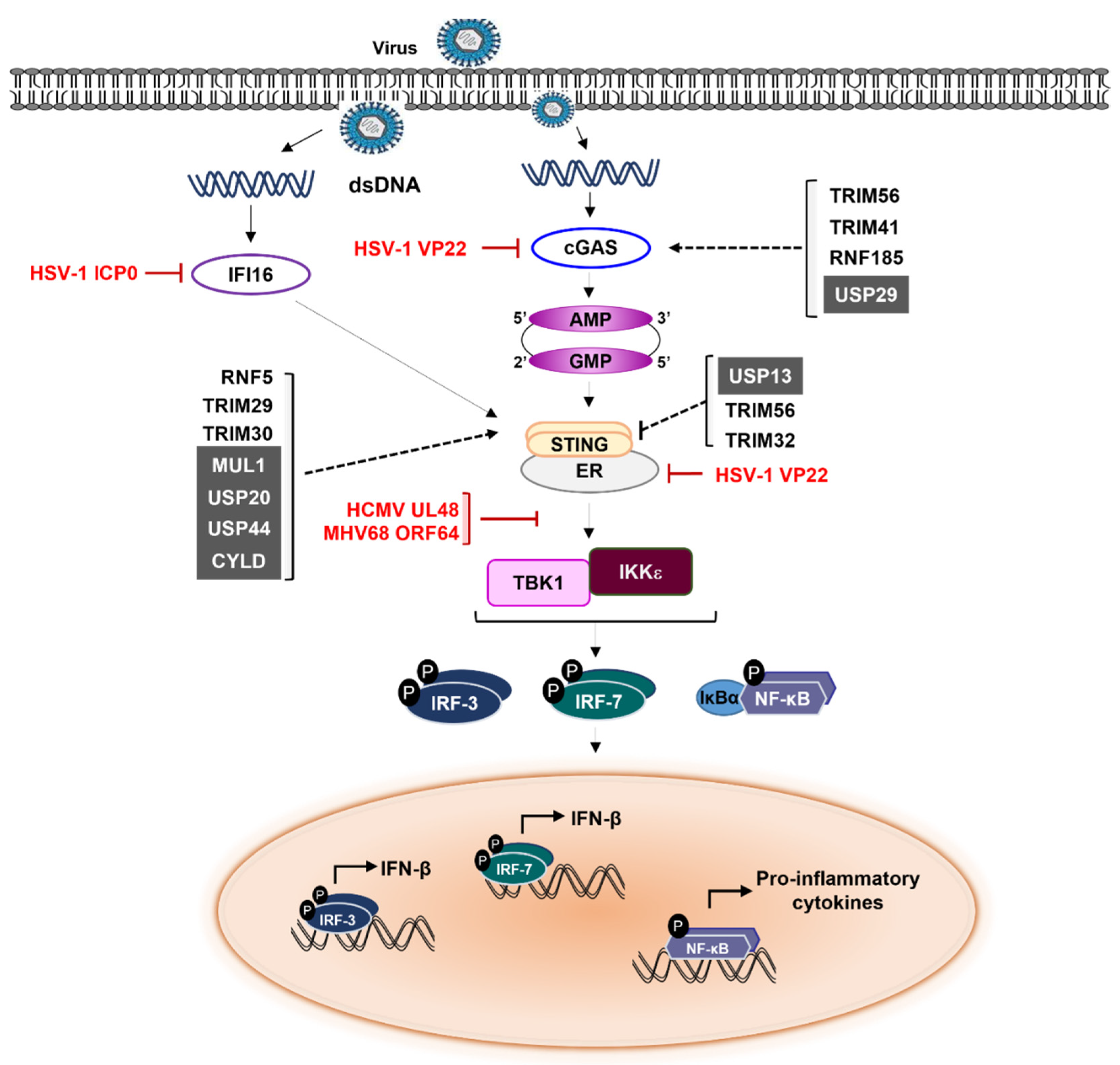
4. cGAS-STING Signaling Modulation
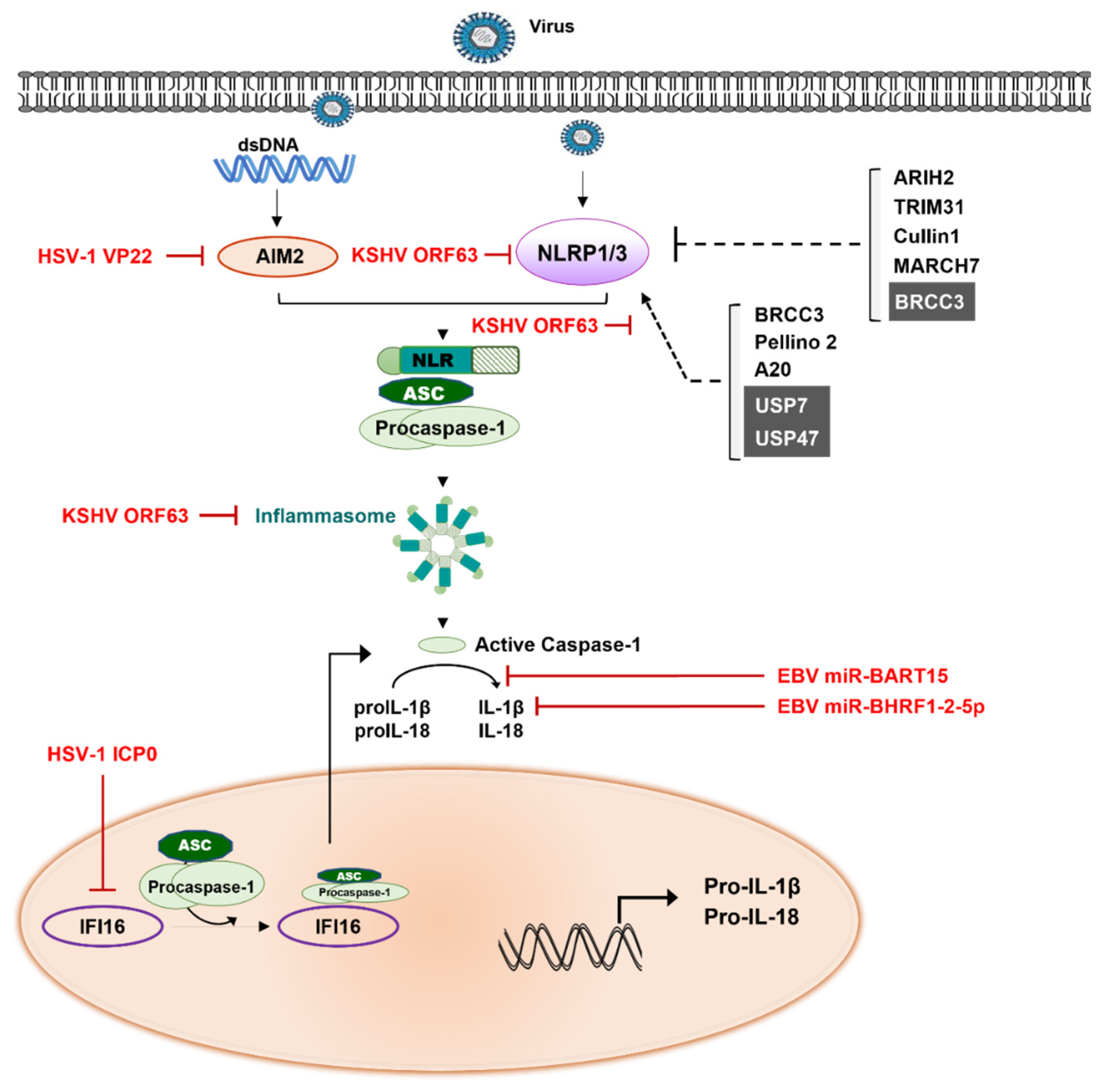
5. Inflammatory Signaling Modulation
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| UPS | Ubiquitin proteasome system |
| DUBs | Deubiquitinases |
| RIG-I | Retinoic acid-inducible gene-I |
| RLR | RIG-I-like receptors |
| GMP | Guanosine monophosphate |
| AMP | Adenine monophosphate |
| cGAS | cyclic-GMP-AMP synthase |
| IFN | Interferon |
| HSV | Herpes simplex virus |
| VZV | Varicella zoster virus |
| HCMV | Human cytomegalovirus |
| HHV | Human herpesvirus |
| EBV | Epstein-Barr virus |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| PRRs | Pathogen recognition receptors |
| PAMPs | Pathogen-associated molecular patterns |
| TLRs | Toll-like receptors |
| NOD | Nucleotide-binding oligomerization domain |
| NLR | NOD-like receptors |
| Ub | Ubiquitin |
| E6-AP | E6-associated protein |
| HECT | Homologous to E6-associated protein (E6-AP) carboxyl terminus |
| RBR | RING-between-RING |
| CRLs | Cullin-RING ligases |
| APC/C | Anaphase-promoting complex/cyclosome |
| Nedd4 | Neuronal precursor cell-expressed developmentally downregulated 4 |
| HERC | HECT and RCC1-like domain |
| RCC1 | Regulators of chromosome condensation 1 |
| RLDs | RCC-1-like domains |
| USPs | Ubiquitin-specific proteases |
| OTUs | Ovarian tumor |
| UCHs | Ubiquitin C-terminal hydrolases |
| MJDs | Machado-Joseph disease |
| MIU | Motif interacting with ubiquitin |
| MINDYs | MIU-containing novel DUB |
| JAMM | JAB1/MPN/MOV34 metalloenzyme family |
| AMSH | Associated Molecule with the SH3-domain of STAM |
| UBA | Ubiquitin associated |
| UIM | Ubiquitin interacting motif |
| ZnF | Zinc finger |
| JOSD | Josephin domain |
| ZUFSP | Zinc finger with UFM1-specific peptidase domain protein |
| HUBL | HAUSP ubiquitin-like domain |
| HAUSP | Herpesvirus-associated ubiquitin-specific protease |
| RNA | Ribonucleic acid |
| MDA | Melanoma differentiation-associated |
| LGP | Laboratory of genetics and physiology |
| CARD | Caspase activation recruitment domains |
| dsRNA | Double strand RNA |
| MAVS | Mitochondrial antiviral signaling |
| TNF | Tumor necrotic factor |
| TRAF | TNF Receptor Associated Factor |
| TANK | TRAF Family Member Associated NF-kB Activator |
| TBK1 | TANK-binding kinase |
| IKK | I-kappa B kinase |
| IRF | Interferon regulatory factor |
| TRIM | Tripartite motif |
| PCBP | Poly(C)-binding protein |
| VSV | Vesicular Stomatitis Virus |
| HOIP | HOIL-1L interacting protein |
| HOIL-1L | Heme-oxidized IRP2 ubiquitin ligase-1 |
| LUBAC | Linear ubiquitin assembly complex |
| CYLD | Cylindromatosis tumor suppressor |
| OTUD | OTU domain |
| ISG | Interferon stimulating genes |
| CHIP | Carboxy-terminus of Hsc70-interacting protein |
| LMP | Latent membrane protein |
| MHC | Major histocompatibility complex |
| ORF61 | Open reading frame61 |
| IFI16 | Interferon-Inducible Protein 16 |
| ER | Endoplasmic recticulum |
| AMFR | Autocrine motility factor receptor |
| INSIG1 | Insulin-induced gene 1 |
| MUL1 | Mitochondrial E3 Ubiquitin Ligase 1 |
| NF-kB | Nuclear Factor Kappa B |
| PEL | Primary effusion lymphoma |
| DAMPs | Damage-associated molecular pattern |
| NLRP1 | NLR-family pyrin domain (PYD)-containing 1 |
| AIM2 | Absent In Melanoma 2 |
| ACS | Apoptosis-associated speck-like protein containing a CARD domain |
| Cbl | Casitas B-cell lymphoma |
| BRCA1 | Breast cancer 1 |
| BRCC3 | BRCA1-BRCA2-containing complex |
| BRISC | BRCC36 isopeptidase complex |
| ABRO1 | Abraxas brother protein 1 |
| RIPK3 | Receptor-interacting serine/threonine-protein kinase 3 |
References
- Grinde, B. Herpesviruses: Latency and reactivation—viral strategies and host response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, R.J. Herpesviruses. In Medical Microbiology; Baron, S., Ed.; The University of Texas Medical Branch at Galveston.: Galveston, TX, USA, 1996. [Google Scholar]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Genet. 2020, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Henao-Mejia, J.; Flavell, R.A. Regulation of the Antimicrobial Response by NLR Proteins. Immunity 2011, 34, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.-M.; Gale, M., Jr. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Ismail, H.; Li, B.; Jin, T. Molecular and Structural Basis of DNA Sensors in Antiviral Innate Immunity. Front. Immunol. 2020, 11, 613039. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Schwesinger, C. The ubiquitin–proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 2019, 15, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Lee, M.K.; Kim, C.W.; Kim, M. TRIM Proteins and Their Roles in the Influenza Virus Life Cycle. Microorganisms 2020, 8, 1424. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, C. E3 ubiquitin ligases, the powerful modulator of innate antiviral immunity. Cell. Immunol. 2019, 340, 103915. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Jin, L.; Williamson, A.; Banerjee, S.; Philipp, I.; Rape, M. Mechanism of Ubiquitin-Chain Formation by the Human Anaphase-Promoting Complex. Cell 2008, 133, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Monia, B.P.; Ecker, D.J.; Jonnalagadda, S.; Marsh, J.; Gotlib, L.; Butt, T.R.; Crooke, S.T. Gene Synthesis, Expression, and Processing of Human Ubiquitin Carboxyl Extension Proteins. J. Biol. Chem. 1989, 264, 4093–4103. [Google Scholar] [CrossRef]
- Liu, Q.; Rao, Y.; Tian, M.; Zhang, S.; Feng, P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses 2019, 11, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bie, P.; Ciechanover, A. Ubiquitination of E3 ligases: Self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011, 18, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S.; Yada, M.; Matsumoto, M.; Ishida, N.; Nakayama, K.-I. U Box Proteins as a New Family of Ubiquitin-Protein Ligases. J. Biol. Chem. 2001, 276, 33111–33120. [Google Scholar] [CrossRef] [Green Version]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef] [Green Version]
- Ohi, M.D.; Kooi, C.W.V.; Rosenberg, J.A.; Chazin, W.J.; Gould, K.L. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Mol. Biol. 2003, 10, 250–255. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta BBA -Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Talis, A.L.; Huibregtse, J.M.; Howley, P.M. The Role of E6AP in the Regulation of p53 Protein Levels in Human Papillomavirus (HPV)-positive and HPV-negative Cells. J. Biol. Chem. 1998, 273, 6439–6445. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, H.; Xu, W.; Li, X.; Dempsey, D.R.; Zhang, X.; Devreotes, P.; Wolberger, C.; Amzel, L.M.; Gabelli, S.B.; et al. A Tunable Brake for HECT Ubiquitin Ligases. Mol. Cell 2017, 66, 345–357.e6. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Rosa, J.L. The HERC proteins: Functional and evolutionary insights. Cell. Mol. Life Sci. CMLS 2005, 62, 1826–1838. [Google Scholar] [CrossRef]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef]
- Uchida, C.; Kitagawa, M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr. Cancer Drug Targets 2016, 16, 157–174. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Coulson, J.M.; Urbé, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125 Pt 2, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.-K.; Saindane, M.; Baek, K.-H. p53 stability is regulated by diverse deubiquitinating enzymes. Biochim. Biophys. Acta (BBA)—Bioenerg. 2017, 1868, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.-H.; Song, M.-H.; Baek, K.-H. Decision for cell fate: Deubiquitinating enzymes in cell cycle checkpoint. Cell. Mol. Life Sci. 2016, 73, 1439–1455. [Google Scholar] [CrossRef]
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Haahr, P.; Borgermann, N.; Guo, X.; Typas, D.; Achuthankutty, D.; Hoffmann, S.; Shearer, R.; Sixma, T.K.; Mailand, N. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol. Cell 2018, 70, 165–174.e6. [Google Scholar] [CrossRef] [Green Version]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, D.J.; Hess, S.; Knobeloch, K.-P. Strategies to Target ISG15 and USP18 Toward Therapeutic Applications. Front. Chem. 2020, 7, 923. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.J.; Shin, H.M.; Nam, E.; Kim, W.S.; Kim, J.; Oh, B.; Yun, Y. DeSUMOylating isopeptidase: A second class of SUMO protease. EMBO Rep. 2012, 13, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.-W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell 2002, 111, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Aravind, L.; Koonin, E.V. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem. Sci. 2000, 25, 50–52. [Google Scholar] [CrossRef]
- Du, J.; Fu, L.; Sui, Y.; Zhang, L. The function and regulation of OTU deubiquitinases. Front. Med. 2019, 14, 542–563. [Google Scholar] [CrossRef] [Green Version]
- Que, L.T.; Morrow, M.E.; Wolberger, C. Comparison of Cross-Regulation by Different OTUB1:E2 Complexes. Biochemistry 2020, 59, 921–932. [Google Scholar] [CrossRef]
- Gong, B.; Leznik, E. The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect. 2007, 20, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.N.; Krantz, A.B.A.; Wilkinson, K.D. Substrate Specificity of Deubiquitinating Enzymes: Ubiquitin C-Terminal Hydrolases. Biochemistry 1998, 37, 3358–3368. [Google Scholar] [CrossRef]
- Zeng, C.; Zhao, C.; Ge, F.; Li, Y.; Cao, J.; Ying, M.; Lu, J.; He, Q.; Yang, B.; Dai, X.; et al. Machado-Joseph Deubiquitinases: From Cellular Functions to Potential Therapy Targets. Front. Pharmacol. 2020, 11, 1311. [Google Scholar] [CrossRef]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Gong, L.; Williams, A.; Sakai, N.; Todi, S.V.; Paulson, H.L. JosD1, a Membrane-targeted Deubiquitinating Enzyme, Is Activated by Ubiquitination and Regulates Membrane Dynamics, Cell Motility, and Endocytosis. J. Biol. Chem. 2013, 288, 17145–17155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.D.; Grasty, K.C.; Hernandez-Cuebas, L.; Loll, P.J. Crystal structure of a Josephin-ubiquitin complex: Evolutionary restraints on ataxin-3 deubiquitinating activity. J. Biol. Chem. 2011, 286, 4555–4565. [Google Scholar] [CrossRef] [Green Version]
- Winborn, B.J.; Travis, S.M.; Todi, S.V.; Scaglione, K.M.; Xu, P.; Williams, A.J.; Cohen, R.E.; Peng, J.; Paulson, H.L. The Deubiquitinating Enzyme Ataxin-3, a Polyglutamine Disease Protein, Edits Lys63 Linkages in Mixed Linkage Ubiquitin Chains. J. Biol. Chem. 2008, 283, 26436–26443. [Google Scholar] [CrossRef] [Green Version]
- De Cesare, V.; Lopez, D.C.; Mabbitt, P.D.; Fletcher, A.J.; Soetens, M.; Antico, O.; Wood, N.T.; Virdee, S. Deubiquitinating enzyme amino acid profiling reveals a class of ubiquitin esterases. Proc. Natl. Acad. Sci. USA 2021, 118, e2006947118. [Google Scholar] [CrossRef]
- McCullough, J.; Row, P.E.; Lorenzo, O.; Doherty, M.; Beynon, R.; Clague, M.J.; Urbé, S. Activation of the Endosome-Associated Ubiquitin Isopeptidase AMSH by STAM, a Component of the Multivesicular Body-Sorting Machinery. Curr. Biol. 2006, 16, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.K.; Ronau, J.A.; Davies, C.W.; Guenette, R.G.; Strieter, E.R.; Paul, L.N.; Das, C. Insights into the Mechanism of Deubiquitination by JAMM Deubiquitinases from Cocrystal Structures of the Enzyme with the Substrate and Product. Biochemistry 2014, 53, 3199–3217. [Google Scholar] [CrossRef]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Reverter, D. Molecular Mechanisms of DUBs Regulation in Signaling and Disease. Int. J. Mol. Sci. 2021, 22, 986. [Google Scholar] [CrossRef]
- Kristariyanto, Y.A.; Abdul Rehman, S.A.; Weidlich, S.; Knebel, A.; Kulathu, Y. A single MIU motif of MINDY-1 recognizes K48-linked polyubiquitin chains. EMBO Rep. 2017, 18, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Heride, C.; Urbe, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, Z.; An, R.; Ye, L.; Zhong, B. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 2020, 30, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashtalir, N.; Daou, S.; Barbour, H.; Sen, N.N.; Gagnon, J.; Hammond-Martel, I.; Dar, H.H.; Therrien, M.; Affar, E.B. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol. Cell 2014, 54, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 Regulates p53 Localization and Stability by Deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faesen, A.C.; Dirac, A.M.; Shanmugham, A.; Ovaa, H.; Perrakis, A.; Sixma, T.K. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 2011, 44, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The Innate Immune Sensor LGP2 Activates Antiviral Signaling by Regulating MDA5-RNA Interaction and Filament Assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Kinch, L.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-Induced Oligomerization of the RNA Sensors RIG-I and MDA5 Activates Antiviral Innate Immune Response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.-Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xu, M.; Liu, S.; Sun, L.; Chen, Z.J. Key Role of Ubc5 and Lysine-63 Polyubiquitination in Viral Activation of IRF3. Mol. Cell 2009, 36, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I Pathway Reveals a Signaling Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The Ubiquitin Ligase Riplet Is Essential for RIG-I-Dependent Innate Immune Responses to RNA Virus Infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2016, 18, 214–224. [Google Scholar] [CrossRef]
- del Toro Duany, Y.; Wu, B.; Hur, S. MDA5-filament, dynamics and disease. Curr. Opin. Virol 2015, 12, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef]
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92, e00321-18. [Google Scholar] [CrossRef] [Green Version]
- Arimoto, K.-I.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jiang, M.; Liu, S.; Zhang, S.; Liu, W.; Ma, Y.; Zhang, L.; Zhang, J.; Cao, X. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 9581–9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; You, F.; Chen, H.; Jiang, Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res. 2012, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.B.; Shembade, N.; Parvatiyar, K.; Balachandran, S.; Harhaj, E.W. TAX1BP1 Restrains Virus-Induced Apoptosis by Facilitating Itch-Mediated Degradation of the Mitochondrial Adaptor MAVS. Mol. Cell. Biol. 2017, 37, e00422-16. [Google Scholar] [CrossRef] [Green Version]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef]
- Yoo, Y.-S.; Park, Y.-Y.; Kim, J.-H.; Cho, H.; Kim, S.-H.; Lee, H.-S.; Kim, T.-H.; Kim, Y.S.; Lee, Y.; Kim, C.-J.; et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tong, X.; Ye, X. Ndfip1 Negatively Regulates RIG-I–Dependent Immune Signaling by Enhancing E3 Ligase Smurf1-Mediated MAVS Degradation. J. Immunol. 2012, 189, 5304–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Li, R.; Meng, J.-L.; Mao, H.-T.; Zhang, Y.; Zhang, J. Smurf2 Negatively Modulates RIG-I–Dependent Antiviral Response by Targeting VISA/MAVS for Ubiquitination and Degradation. J. Immunol. 2014, 192, 4758–4764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-J.; Oanh, N.T.K.; Heo, J.; Kim, S.-G.; Lee, H.-S.; Lee, H.; Lee, J.-H.; Kang, H.C.; Lim, W.; Yoo, Y.-S.; et al. Dual targeting of RIG-I and MAVS by MARCH5 mitochondria ubiquitin ligase in innate immunity. Cell. Signal. 2019, 67, 109520. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, L.; Zhong, B.; Tan, B.; Liu, Y.; Shu, H.-B. The ubiquitin-specific protease 17 is involved in virus-triggered type I IFN signaling. Cell Res. 2010, 20, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.; Yang, S.; Gao, C. USP4 Positively Regulates RIG-I-Mediated Antiviral Response through Deubiquitination and Stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Zhang, M.; Zhang, M.-X.; Ren, Y.; Jin, J.; Zhao, Q.; Pan, Z.; Wu, M.; Shu, H.-B.; Dong, C.; et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc. Natl. Acad. Sci. USA 2015, 112, 11324–11329. [Google Scholar] [CrossRef] [Green Version]
- Inn, K.-S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.-Y.; Iwai, K.; Jung, J.U. Linear Ubiquitin Assembly Complex Negatively Regulates RIG-I- and TRIM25-Mediated Type I Interferon Induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Pauli, E.-K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The Ubiquitin-Specific Protease USP15 Promotes RIG-I–Mediated Antiviral Signaling by Deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 2014, 211, 313–328. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.-F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2013, 24, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Huang, S.; Wang, X.; Wen, M.; Zheng, J.; Wang, W.; Fu, Y.; Tian, S.; Li, L.; Li, Z.; et al. The Otubain YOD1 Suppresses Aggregation and Activation of the Signaling Adaptor MAVS through Lys63-Linked Deubiquitination. J. Immunol. 2019, 202, 2957–2970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Qian, L.; Feng, Q.; Wang, X.; Yuan, Y.; Zuo, Y.; Cheng, Q.; Miao, Y.; Guo, T.; et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog 2018, 14, e1007067. [Google Scholar] [CrossRef] [Green Version]
- Liuyu, T.; Yu, K.; Ye, L.; Zhang, Z.; Zhang, M.; Ren, Y.; Cai, Z.; Zhu, Q.; Lin, D.; Zhong, B. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 2018, 29, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Shahnazaryan, D.; Khalil, R.; Wynne, C.; Jefferies, C.A.; Gabhann-Dromgoole, J.N.; Murphy, C.C. Herpes simplex virus 1 targets IRF7 via ICP0 to limit type I IFN induction. Sci. Rep. 2020, 10, 22216. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [Green Version]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The Herpes Simplex Virus ICP0 RING Finger Domain Inhibits IRF3- and IRF7-Mediated Activation of Interferon-Stimulated Genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [Green Version]
- Früh, K.; Bartee, E.; Gouveia, K.; Mansouri, M. Immune evasion by a novel family of viral PHD/LAP-finger proteins of gamma-2 herpesviruses and poxviruses. Virus Res. 2002, 88, 55–69. [Google Scholar] [CrossRef]
- Brulois, K.; Toth, Z.; Wong, L.Y.; Feng, P.; Gao, S.J.; Ensser, A.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 2014, 88, 9335–9349. [Google Scholar] [CrossRef] [Green Version]
- Duncan, L.M.; Nathan, J.A.; Lehner, P.J. Stabilization of an E3 Ligase–E2–Ubiquitin Complex Increases Cell Surface MHC Class I Expression. J. Immunol. 2010, 184, 6978–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boname, J.M.; Lehner, P.J. What has the study of the K3 and K5 viral ubiquitin E3 ligases taught us about ubiquitin-mediated receptor regulation? Viruses 2011, 3, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar] [CrossRef]
- Brulois, K.F.; Chang, H.; Lee, A.S.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Gill, M.B.; May, J.S.; Stevenson, P.G. Murine Gammaherpesvirus-68 Inhibits Antigen Presentation by Dendritic Cells. PLoS ONE 2007, 2, e1048. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Sun, L.; Liu, W.; Duan, Z. Latent Membrane Protein 1 of Epstein–Barr Virus Promotes RIG-I Degradation Mediated by Proteasome Pathway. Front. Immunol. 2018, 9, 1446. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C.; Xing, J.; Wang, S.; Li, S.; Lin, R.; Mossman, K.L. Varicella-Zoster Virus Immediate-Early Protein ORF61 Abrogates the IRF3-Mediated Innate Immune Response through Degradation of Activated IRF3. J. Virol. 2011, 85, 11079–11089. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Inhibits Beta Interferon Production by Deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.T.; Oh, S.E.; Lee, Y.-O.; Gibson, W.; Ahn, J.-H. Cleavage Specificity of the UL48 Deubiquitinating Protease Activity of Human Cytomegalovirus and the Growth of an Active-Site Mutant Virus in Cultured Cells. J. Virol. 2009, 83, 12046–12056. [Google Scholar] [CrossRef] [Green Version]
- Gredmark-Russ, S.; Isaacson, M.K.; Kattenhorn, L.; Cheung, E.J.; Watson, N.; Ploegh, H.L. A Gammaherpesvirus Ubiquitin-Specific Protease Is Involved in the Establishment of Murine Gammaherpesvirus 68 Infection. J. Virol. 2009, 83, 10644–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosinski, K.; Kattenhorn, L.; Kaufer, B.B.; Ploegh, H.; Osterrieder, N. A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc. Natl. Acad. Sci. USA 2007, 104, 20025–20030. [Google Scholar] [CrossRef] [Green Version]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A Deubiquitinating Activity Is Conserved in the Large Tegument Protein of the Herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Dai, T.; He, X.; Zhang, Z.; Xie, F.; Wang, S.; Zhang, L.; Zhou, F. The interactions between cGAS-STING pathway and pathogens. Signal. Transduct Target. Ther. 2020, 5, 91. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.; Trapani, J. The Interferon Inducible Autoantigen, IFI 16: Localization to the Nucleolus and Identification of a DNA-Binding Domain. Biochem. Biophys. Res. Commun. 1995, 214, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Iqbal, J.; Kumar, B.; Roy, A.; Chikoti, L.; Singh, V.V.; Chandran, B. Herpesvirus Genome Recognition Induced Acetylation of Nuclear IFI16 Is Essential for Its Cytoplasmic Translocation, Inflammasome and IFN-β Responses. PLoS Pathog. 2015, 11, e1005019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almine, J.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Kong, X.; Li, S.; Cui, Y.; Liu, H.; et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Kim, C.; Shin, W.-J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-S.; Zhang, Z.-Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X.-M.; Li, T. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, G.; Konno, H.; Barber, G.N. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2017, 2, eaah7119. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lin, L.; Tong, Y.; Liu, Y.; Mou, J.; Wang, X.; Wang, X.; Gong, Y.; Zhao, Y.; Liu, Y.; et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov. 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Zhou, M.-T.; Hu, M.-M.; Hu, Y.-H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.-B. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLoS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-X.; Cai, Z.; Zhang, M.; Wang, X.-M.; Wang, Y.; Zhao, F.; Zhou, J.; Luo, M.-H.; Zhu, Q.; Xu, Z.; et al. USP20 Promotes Cellular Antiviral Responses via Deconjugating K48-Linked Ubiquitination of MITA. J. Immunol. 2019, 202, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, M.-X.; Zhang, Q.; Zhu, G.-F.; Yuan, L.; Zhang, D.-E.; Zhu, Q.; Yao, J.; Shu, H.-B.; Zhong, B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016, 26, 1302–1319. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-Y.; Liao, B.-W.; Xu, Z.-S.; Ran, Y.; Wang, D.-P.; Yang, Y.; Luo, W.-W.; Wang, Y.-Y. USP44 positively regulates innate immune response to DNA viruses through deubiquitinating MITA. PLoS Pathog. 2020, 16, e1008178. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Liuyu, T.; Xu, Z.; Zhang, M.-X.; Luo, M.-H.; Zeng, W.-B.; Zhu, Q.; Lin, D.; Zhong, B. USP49 negatively regulates cellular antiviral responses via deconjugating K63-linked ubiquitination of MITA. PLoS Pathog. 2019, 15, e1007680. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Q.; Jing, Y.-Y.; Zhang, M.; Wang, H.-Y.; Cai, Z.; Liuyu, T.; Zhang, Z.-D.; Xiong, T.-C.; Wu, Y.; et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, Y.; Qin, Y.; Hu, J.; Xie, W.; Qin, F.X.-F.; Cui, J. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci. Adv. 2018, 4, eaar2824. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chen, J.; Cristea, I.M. Human Cytomegalovirus Tegument Protein pUL83 Inhibits IFI16-Mediated DNA Sensing for Immune Evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The Viral Ubiquitin Ligase ICP0 Is neither Sufficient nor Necessary for Degradation of the Cellular DNA Sensor IFI16 during Herpes Simplex Virus 1 Infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, 15. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Saha, I.; Narayanan, A.; Narayanan, S.; Takaoka, A.; Kumar, N.S.; Tailor, P.; Kumar, H. Essential role of HCMV deubiquitinase in promoting oncogenesis by targeting anti-viral innate immune signaling pathways. Cell Death Dis. 2017, 8, e3078. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Schattgen, S.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R.; et al. Evasion of Innate Cytosolic DNA Sensing by a Gammaherpesvirus Facilitates Establishment of Latent Infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 2019, 21, 17–29. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, T.; Little, M.A.; Brady, G. Targeting of the cGAS-STING system by DNA viruses. Biochem. Pharm. 2020, 174, 113831. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2017, 281, 99–114. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.; Zhang, Q.; Liu, W.; Jia, Y.; Ai, S.; Wang, T.; Wang, W.; Pan, P.; Yang, G.; Xiang, Q.; et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. 2019, 33, 5793–5807. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.; Moynagh, P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Palazon-Riquelme, P.; Worboys, J.; Green, J.; Valera, A.; Martín-Sánchez, F.; Pellegrini, C.; Brough, D.; López-Castejón, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19, e44766. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Luheshi, N.M.; Compan, V.; High, S.; Whitehead, R.; Flitsch, S.; Kirov, A.; Prudovsky, I.; Swanton, E.; Brough, D. Deubiquitinases Regulate the Activity of Caspase-1 and Interleukin-1β Secretion via Assembly of the Inflammasome. J. Biol. Chem. 2013, 288, 2721–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednash, J.S.; Johns, F.; Patel, N.; Smail, T.R.; Londino, J.D.; Mallampalli, R.K. The deubiquitinase STAMBP modulates cytokine secretion through the NLRP3 inflammasome. Cell. Signal. 2020, 79, 109859. [Google Scholar] [CrossRef]
- Py, B.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Dixit, V.M. Regulation of NF-κB by deubiquitinases. Immunol. Rev. 2012, 246, 107–124. [Google Scholar] [CrossRef] [Green Version]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 Restricts Ubiquitination of Pro-Interleukin-1β Protein Complexes and Suppresses NLRP3 Inflammasome Activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.-I.; Reed, J.C.; Ting, J.P.Y.; Damania, B. Discovery of a Viral NLR Homolog that Inhibits the Inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.J.; Masters, S.L. Cutting Edge: miR-223 and EBV miR-BART15 Regulate the NLRP3 Inflammasome and IL-1β Production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [Green Version]
- Skinner, C.M.; Ivanov, N.S.; Barr, S.A.; Chen, Y.; Skalsky, R.L. An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1. J. Virol. 2017, 91, e00530-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 2018, 23, 254–265.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulon, P.-G.; Dhanushkodi, N.; Prakash, S.; Srivastava, R.; Roy, S.; Alomari, N.I.; Nguyen, A.M.; Warsi, W.R.; Ye, C.; Carlos-Cruz, E.A.; et al. NLRP3, NLRP12, and IFI16 Inflammasomes Induction and Caspase-1 Activation Triggered by Virulent HSV-1 Strains Are Associated With Severe Corneal Inflammatory Herpetic Disease. Front. Immunol. 2019, 10, 1631. [Google Scholar] [CrossRef] [Green Version]
- Karaba, A.H.; Figueroa, A.; Massaccesi, G.; Botto, S.; DeFilippis, V.R.; Cox, A.L. Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLoS ONE 2020, 15, e0229570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Virus | Viral Protein | Mimicking Function | Role in Host Cell |
|---|---|---|---|
| KSHV | K3, K5 | E3 ubiquitin ligase | Downregulates surface expression of MHC-I molecules via lysosomal degradation [115,116,117,118] |
| ORF64 | DUB | Suppresses RIG-I-mediated IFN signaling by reducing the ubiquitination of RIG-I [126] | |
| ORF63 | DUB | Prevents NLRP1 oligomerization via interaction with NLRP1 [175] | |
| HSV-1 | ICP0 | E3 ubiquitin ligase | Dampens and Terminate TNF-α-mediated and TLR-mediated NF-kB activation. Blocks DNA sensing via IFI16 degradation [111,182] |
| UL36 | DUB | Deubiquitinates TRAF3 to prevent recruitment of TBK1 [123] | |
| HCMV | UL48 | DUB | Inhibits type I IFN synthesis via deubiquitination of TRAF6, IRF7, and STING [157] |
| VZV | ORF61 | E3 ubiquitin ligase | Antagonizes the IFN-β pathway [122] |
| MHV68 | K3, K5 | E3 ubiquitin ligase | Downregulates surface expression of MHC-I molecules via proteasomal degradation [113] |
| ORF64 | DUB | Impedes STING signalosome signaling via deubiquitination of STING [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soh, S.-M.; Kim, Y.-J.; Kim, H.-H.; Lee, H.-R. Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses. Int. J. Mol. Sci. 2022, 23, 492. https://doi.org/10.3390/ijms23010492
Soh S-M, Kim Y-J, Kim H-H, Lee H-R. Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses. International Journal of Molecular Sciences. 2022; 23(1):492. https://doi.org/10.3390/ijms23010492
Chicago/Turabian StyleSoh, Sandrine-M., Yeong-Jun Kim, Hong-Hee Kim, and Hye-Ra Lee. 2022. "Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses" International Journal of Molecular Sciences 23, no. 1: 492. https://doi.org/10.3390/ijms23010492
APA StyleSoh, S.-M., Kim, Y.-J., Kim, H.-H., & Lee, H.-R. (2022). Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses. International Journal of Molecular Sciences, 23(1), 492. https://doi.org/10.3390/ijms23010492