
Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers?

Abstract
1. Introduction
2. Carbamylation, a Physiological Process Increased in Chronic Kidney Disease
2.1. Carbamylation Is a Physiological Process
2.2. Chronic Kidney Disease Exacerbates Carbamylation
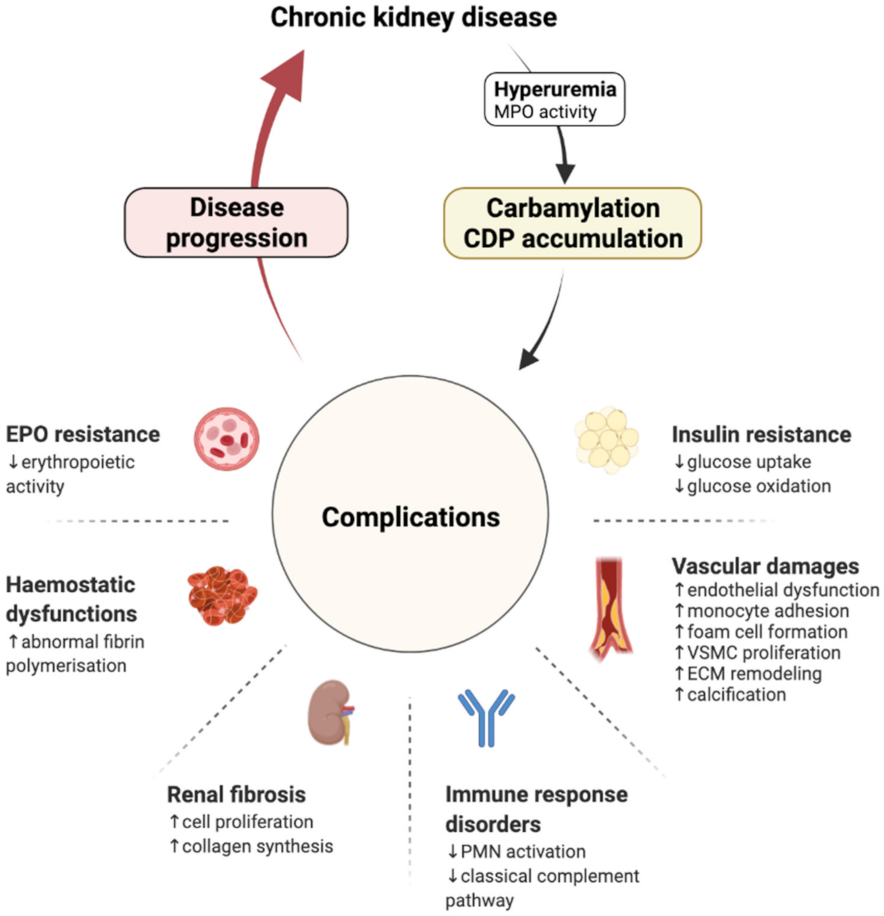
3. Carbamylation and Renal Disease Progression: The Consequence Becomes the Cause
3.1. Cardiovascular Damages
3.1.1. Lipoprotein Metabolism
3.1.2. Cellular Effects
3.1.3. ECM Remodelling
3.1.4. Calcification
3.2. Renal Fibrosis
3.3. Haemostasis Dysfunctions
3.4. Immune Response Disorders
3.5. Erythropoietin Resistance
3.6. Insulin Resistance
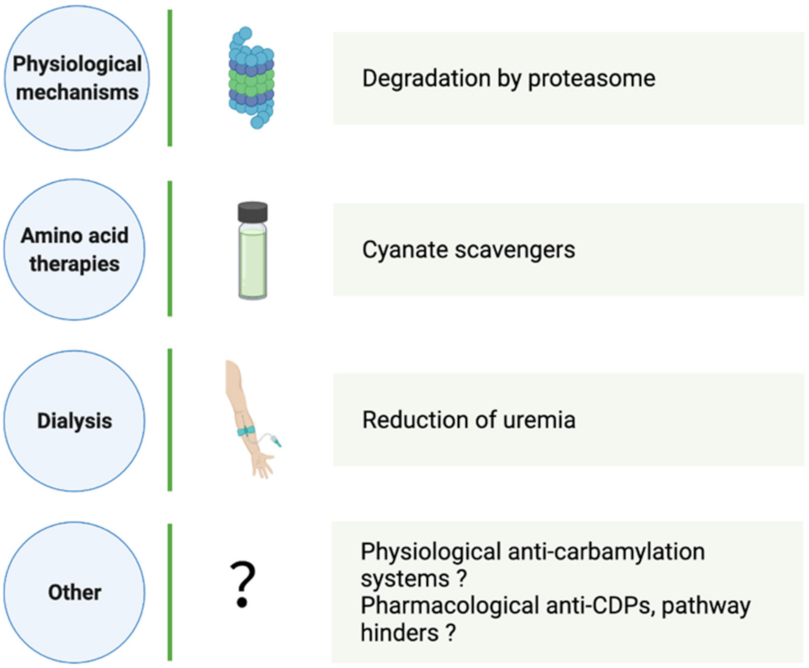
4. How Can CDP Formation and Accumulation Be Reduced?
4.1. Physiological CDP Elimination
4.2. Aid in Carbamylation Limitation
4.2.1. Dialysis
4.2.2. Amino Acid Therapies
4.2.3. Other Strategies
5. CDPs: Biomarkers, Predictors of CKD Progression
5.1. Homocitrulline
5.2. Carbamylated Haemoglobin
5.3. Carbamylated Albumin
5.4. Carbamylated Lipoproteins
6. Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. GBD Chronic Kidney Disease Collaboration Global, Regional, and National Burden of Chronic Kidney Disease, 1990-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Mehta, R.L.; Cerdá, J.; Burdmann, E.A.; Tonelli, M.; García-García, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 Initiative for Acute Kidney Injury (Zero Preventable Deaths by 2025): A Human Rights Case for Nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on Uremic Toxins: Classification, Concentration, and Interindividual Variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Falconi, C.A.; Junho, C.V.d.C.; Fogaça-Ruiz, F.; Vernier, I.C.S.; da Cunha, R.S.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front. Physiol. 2021, 12, 686249. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.J.; Sidor, N.A.; Tonial, N.C.; Che, A.; Urquhart, B.L. Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets. Toxins 2021, 13, 142. [Google Scholar] [CrossRef]
- Lau, W.L.; Vaziri, N.D. Urea, a True Uremic Toxin: The Empire Strikes Back. Clin. Sci. 2017, 131, 3–12. [Google Scholar] [CrossRef]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the Lack of Urea Toxicity in Dialysis Patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef]
- Stark, G.R. [53] Modification of Proteins with Cyanate. Methods Enzymol. 1972, 25, 579–584. [Google Scholar] [CrossRef]
- Dirnhuber, P.; Schütz, F. The Isomeric Transformation of Urea into Ammonium Cyanate in Aqueous Solutions. Biochem. J. 1948, 42, 628–632. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein Carbamylation Links Inflammation, Smoking, Uremia and Atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Veres, P.R.; Cochran, A.K.; Warneke, C.; Burling, I.R.; Yokelson, R.J.; Lerner, B.; Gilman, J.B.; Kuster, W.C.; Fall, R.; et al. Isocyanic Acid in the Atmosphere and Its Possible Link to Smoke-Related Health Effects. Proc. Natl. Acad. Sci. USA 2011, 108, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Kassa, R.M.; Kasensa, N.L.; Monterroso, V.H.; Kayton, R.J.; Klimek, J.E.; David, L.L.; Lunganza, K.R.; Kayembe, K.T.; Bentivoglio, M.; Juliano, S.L.; et al. On the Biomarkers and Mechanisms of Konzo, a Distinct Upper Motor Neuron Disease Associated with Food (Cassava) Cyanogenic Exposure. Food Chem. Toxicol. 2011, 49, 571–578. [Google Scholar] [CrossRef]
- Desmons, A.; Jaisson, S.; Pietrement, C.; Rieu, P.; Wynckel, A.; Gillery, P. Homocitrulline: A New Marker for Differentiating Acute from Chronic Renal Failure. Clin. Chem. Lab. Med. 2016, 54, 73–79. [Google Scholar] [CrossRef]
- Pietrement, C.; Gorisse, L.; Jaisson, S.; Gillery, P. Chronic Increase of Urea Leads to Carbamylated Proteins Accumulation in Tissues in a Mouse Model of CKD. PLoS ONE 2013, 8, e82506. [Google Scholar] [CrossRef]
- Gorisse, L.; Pietrement, C.; Vuiblet, V.; Schmelzer, C.E.H.; Köhler, M.; Duca, L.; Debelle, L.; Fornès, P.; Jaisson, S.; Gillery, P. Protein Carbamylation Is a Hallmark of Aging. Proc. Natl. Acad. Sci. USA 2016, 113, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.; Jaisson, S.; Gorisse, L.; Tessier, F.J.; Niquet-Léridon, C.; Jacolot, P.; Pietrement, C.; Gillery, P. Carbamylation Is a Competitor of Glycation for Protein Modification in Vivo. Diabetes Metab. 2018, 44, 160–167. [Google Scholar] [CrossRef]
- Nicolas, C.; Jaisson, S.; Gorisse, L.; Tessier, F.J.; Niquet-Léridon, C.; Jacolot, P.; Pietrement, C.; Gillery, P. Carbamylation and Glycation Compete for Collagen Molecular Aging in Vivo. Sci. Rep. 2019, 9, 18291. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Pietrement, C.; Gillery, P. Protein Carbamylation: Chemistry, Pathophysiological Involvement, and Biomarkers. Adv. Clin. Chem. 2018, 84, 1–38. [Google Scholar] [CrossRef]
- Berg, A.H.; Drechsler, C.; Wenger, J.; Buccafusca, R.; Hod, T.; Kalim, S.; Ramma, W.; Parikh, S.M.; Steen, H.; Friedman, D.J.; et al. Carbamylation of Serum Albumin as a Risk Factor for Mortality in Patients with Kidney Failure. Sci. Transl. Med. 2013, 5, 175ra29. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Trottier, C.A.; Wenger, J.B.; Wibecan, J.; Ahmed, R.; Ankers, E.; Karumanchi, S.A.; Thadhani, R.; Berg, A.H. Longitudinal Changes in Protein Carbamylation and Mortality Risk after Initiation of Hemodialysis. Clin. J. Am. Soc. Nephrol. 2016, 11, 1809–1816. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; Cobo, G.; Dai, L.; Lindholm, B.; Stenvinkel, P. Role of Uremic Toxins in Early Vascular Ageing and Calcification. Toxins 2021, 13, 26. [Google Scholar] [CrossRef]
- Bankir, L.T.; Trinh-Trang-Tan, M.M. Renal Urea Transporters. Direct and Indirect Regulation by Vasopressin. Exp. Physiol. 2000, 85, 243S–252S. [Google Scholar] [CrossRef] [PubMed]
- Desmons, A.; Okwieka, A.; Doué, M.; Gorisse, L.; Vuiblet, V.; Pietrement, C.; Gillery, P.; Jaisson, S. Proteasome-Dependent Degradation of Intracellular Carbamylated Proteins. Aging 2019, 11, 3624–3638. [Google Scholar] [CrossRef]
- Claxton, J.S.; Sandoval, P.C.; Liu, G.; Chou, C.-L.; Hoffert, J.D.; Knepper, M.A. Endogenous Carbamylation of Renal Medullary Proteins. PLoS ONE 2013, 8, e82655. [Google Scholar] [CrossRef][Green Version]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein Carbamylation Exacerbates Vascular Calcification. Kidney Int. 2018, 94, 72–90. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.G.; Holden, M.; Benton, M.; Brown, C.B. Glycosylated and Carbamylated Haemoglobin in Uraemia. Nephrol. Dial. Transplant. 1989, 4, 96–100. [Google Scholar] [PubMed]
- Apostolov, E.O.; Shah, S.V.; Ok, E.; Basnakian, A.G. Quantification of Carbamylated LDL in Human Sera by a New Sandwich ELISA. Clin. Chem. 2005, 51, 719–728. [Google Scholar] [CrossRef]
- Doué, M.; Okwieka, A.; Berquand, A.; Gorisse, L.; Maurice, P.; Velard, F.; Terryn, C.; Molinari, M.; Duca, L.; Piétrement, C.; et al. Carbamylation of Elastic Fibers Is a Molecular Substratum of Aortic Stiffness. Sci. Rep. 2021, 11, 17827. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.G.; Dunn, J.A.; Thorpe, S.R.; Bailie, K.E.; Lyons, T.J.; McCance, D.R.; Baynes, J.W. Accumulation of Maillard Reaction Products in Skin Collagen in Diabetes and Aging. J. Clin. Investig. 1993, 91, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Sell, D.R.; Lane, M.A.; Johnson, W.A.; Masoro, E.J.; Mock, O.B.; Reiser, K.M.; Fogarty, J.F.; Cutler, R.G.; Ingram, D.K.; Roth, G.S.; et al. Longevity and the Genetic Determination of Collagen Glycoxidation Kinetics in Mammalian Senescence. Proc. Natl. Acad. Sci. USA 1996, 93, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Ramirez-Boscá, A.; Soler, A.; Díez, A.; Carrión-Gutiérrez, M.A.; Díaz-Alperi, J.; Quintanilla-Ripoll, E.; Bernd, A.; Quintanilla-Almagro, E. Increase with Age of Serum Lipid Peroxides: Implications for the Prevention of Atherosclerosis. Mech. Ageing Dev. 1998, 100, 17–24. [Google Scholar] [CrossRef]
- Hoy, A.; Trégouët, D.; Leininger-Muller, B.; Poirier, O.; Maurice, M.; Sass, C.; Siest, G.; Tiret, L.; Visvikis, S. Serum Myeloperoxidase Concentration in a Healthy Population: Biological Variations, Familial Resemblance and New Genetic Polymorphisms. Eur. J. Hum. Genet. 2001, 9, 780–786. [Google Scholar] [CrossRef][Green Version]
- Carracedo, J.; Ramírez-Carracedo, R.; Martínez de Toda, I.; Vida, C.; Alique, M.; De la Fuente, M.; Ramírez-Chamond, R. Protein Carbamylation: A Marker Reflecting Increased Age-Related Cell Oxidation. Int. J. Mol. Sci. 2018, 19, E1495. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Lundquist, P.; Kågedal, B.; Larsson, R. Plasma Cyanate Concentrations in Chronic Renal Failure. Clin. Chem. 1996, 42, 482–483. [Google Scholar] [CrossRef]
- Kwan, J.T.; Carr, E.C.; Barron, J.L.; Bending, M.R. Carbamylated Haemoglobin in Normal, Diabetic and Uraemic Patients. Ann. Clin. Biochem. 1992, 29 Pt 2, 206–209. [Google Scholar] [CrossRef]
- Kraus, L.M.; Gaber, L.; Handorf, C.R.; Marti, H.P.; Kraus, A.P. Carbamoylation of Glomerular and Tubular Proteins in Patients with Kidney Failure: A Potential Mechanism of Ongoing Renal Damage. Swiss. Med. Wkly. 2001, 131, 139–144. [Google Scholar] [CrossRef]
- Hasuike, Y.; Nakanishi, T.; Moriguchi, R.; Otaki, Y.; Nanami, M.; Hama, Y.; Naka, M.; Miyagawa, K.; Izumi, M.; Takamitsu, Y. Accumulation of Cyanide and Thiocyanate in Haemodialysis Patients. Nephrol. Dial. Transplant. 2004, 19, 1474–1479. [Google Scholar] [CrossRef]
- Hörkkö, S.; Huttunen, K.; Kervinen, K.; Kesäniemi, Y.A. Decreased Clearance of Uraemic and Mildly Carbamylated Low-Density Lipoprotein. Eur. J. Clin. Investig. 1994, 24, 105–113. [Google Scholar] [CrossRef]
- Ok, E.; Basnakian, A.G.; Apostolov, E.O.; Barri, Y.M.; Shah, S.V. Carbamylated Low-Density Lipoprotein Induces Death of Endothelial Cells: A Link to Atherosclerosis in Patients with Kidney Disease. Kidney Int. 2005, 68, 173–178. [Google Scholar] [CrossRef]
- Speer, T.; Owala, F.O.; Holy, E.W.; Zewinger, S.; Frenzel, F.L.; Stähli, B.E.; Razavi, M.; Triem, S.; Cvija, H.; Rohrer, L.; et al. Carbamylated Low-Density Lipoprotein Induces Endothelial Dysfunction. Eur. Heart J. 2014, 35, 3021–3032. [Google Scholar] [CrossRef]
- Apostolov, E.O.; Basnakian, A.G.; Yin, X.; Ok, E.; Shah, S.V. Modified LDLs Induce Proliferation-Mediated Death of Human Vascular Endothelial Cells through MAPK Pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1836–H1846. [Google Scholar] [CrossRef]
- Apostolov, E.O.; Shah, S.V.; Ray, D.; Basnakian, A.G. Scavenger Receptors of Endothelial Cells Mediate the Uptake and Cellular Pro-Atherogenic Effects of Carbamylated LDL. Arter. Thromb. Vasc. Biol. 2009, 29, 1622–1630. [Google Scholar] [CrossRef]
- Asci, G.; Basci, A.; Shah, S.V.; Basnakian, A.; Toz, H.; Ozkahya, M.; Duman, S.; Ok, E. Carbamylated Low-Density Lipoprotein Induces Proliferation and Increases Adhesion Molecule Expression of Human Coronary Artery Smooth Muscle Cells. Nephrology 2008, 13, 480–486. [Google Scholar] [CrossRef]
- Sun, J.T.; Yang, K.; Lu, L.; Zhu, Z.B.; Zhu, J.Z.; Ni, J.W.; Han, H.; Chen, N.; Zhang, R.Y. Increased Carbamylation Level of HDL in End-Stage Renal Disease: Carbamylated-HDL Attenuated Endothelial Cell Function. Am. J. Physiol. Renal Physiol. 2016, 310, F511–F517. [Google Scholar] [CrossRef]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia Alters HDL Composition and Function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein Carbamylation Renders High-Density Lipoprotein Dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef]
- Garnotel, R.; Sabbah, N.; Jaisson, S.; Gillery, P. Enhanced Activation of and Increased Production of Matrix Metalloproteinase-9 by Human Blood Monocytes upon Adhering to Carbamylated Collagen. FEBS Lett. 2004, 563, 13–16. [Google Scholar] [CrossRef]
- Jaisson, S.; Lorimier, S.; Ricard-Blum, S.; Sockalingum, G.D.; Delevallée-Forte, C.; Kegelaer, G.; Manfait, M.; Garnotel, R.; Gillery, P. Impact of Carbamylation on Type I Collagen Conformational Structure and Its Ability to Activate Human Polymorphonuclear Neutrophils. Chem. Biol. 2006, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Alesutan, I.; Luong, T.T.D.; Schelski, N.; Masyout, J.; Hille, S.; Schneider, M.P.; Graham, D.; Zickler, D.; Verheyen, N.; Estepa, M.; et al. Circulating Uromodulin Inhibits Vascular Calcification by Interfering with Pro-Inflammatory Cytokine Signalling. Cardiovasc. Res. 2021, 117, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; Saritas, T.; Kjolby, M.; Hermann, J.; Speer, T.; Himmelsbach, A.; Mahr, K.; Heuschkel, M.A.; Schunk, S.J.; Thirup, S.; et al. Carbamylated Sortilin Associates with Cardiovascular Calcification in Patients with Chronic Kidney Disease. Kidney Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.-L.; Piecha, G.; Bierhaus, A.; Hanke, W.; Henle, T.; Schirmacher, P.; Ritz, E. Glycated and Carbamylated Albumin Are More “Nephrotoxic” than Unmodified Albumin in the Amphibian Kidney. Am. J. Physiol. Renal Physiol. 2011, 301, F476–F485. [Google Scholar] [CrossRef] [PubMed]
- Shaykh, M.; Pegoraro, A.A.; Mo, W.; Arruda, J.A.; Dunea, G.; Singh, A.K. Carbamylated Proteins Activate Glomerular Mesangial Cells and Stimulate Collagen Deposition. J. Lab. Clin. Med. 1999, 133, 302–308. [Google Scholar] [CrossRef]
- Jaisson, S.; Larreta-Garde, V.; Bellon, G.; Hornebeck, W.; Garnotel, R.; Gillery, P. Carbamylation Differentially Alters Type I Collagen Sensitivity to Various Collagenases. Matrix Biol. 2007, 26, 190–196. [Google Scholar] [CrossRef]
- Binder, V.; Bergum, B.; Jaisson, S.; Gillery, P.; Scavenius, C.; Spriet, E.; Nyhaug, A.K.; Roberts, H.M.; Chapple, I.L.C.; Hellvard, A.; et al. Impact of Fibrinogen Carbamylation on Fibrin Clot Formation and Stability. Thromb. Haemost. 2017, 117, 899–910. [Google Scholar] [CrossRef]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of Erythropoietin That Are Tissue Protective but Not Erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Park, K.-D.; Mun, K.-C.; Chang, E.-J.; Park, S.-B.; Kim, H.-C. Inhibition of Erythropoietin Activity by Cyanate. Scand. J. Urol. Nephrol. 2004, 38, 69–72. [Google Scholar] [CrossRef]
- Oimomi, M.; Hatanaka, H.; Yoshimura, Y.; Yokono, K.; Baba, S.; Taketomi, Y. Carbamylation of Insulin and Its Biological Activity. Nephron 1987, 46, 63–66. [Google Scholar] [CrossRef]
- Kraus, L.M.; Traxinger, R.; Kraus, A.P. Uremia and Insulin Resistance: N-Carbamoyl-Asparagine Decreases Insulin-Sensitive Glucose Uptake in Rat Adipocytes. Kidney Int. 2004, 65, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Koro, C.; Bielecka, E.; Dahl-Knudsen, A.; Enghild, J.J.; Scavenius, C.; Brun, J.G.; Binder, V.; Hellvard, A.; Bergum, B.; Jonsson, R.; et al. Carbamylation of Immunoglobulin Abrogates Activation of the Classical Complement Pathway. Eur. J. Immunol. 2014, 44, 3403–3412. [Google Scholar] [CrossRef]
- Tan, K.C.B.; Cheung, C.-L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Carbamylated Lipoproteins and Progression of Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 359–366. [Google Scholar] [CrossRef]
- Kalim, S.; Berg, A.H.; Karumanchi, S.A.; Thadhani, R.; Allegretti, A.S.; Nigwekar, S.; Zhao, S.; Srivastava, A.; Raj, D.; Deo, R.; et al. Protein Carbamylation and Chronic Kidney Disease Progression in the Chronic Renal Insufficiency Cohort Study. Nephrol. Dial. Transplant. 2020, gfaa347. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Vida, C.; Bodega, G.; Ceprián, N.; Morales, E.; Praga, M.; de Sequera, P.; Ramírez, R. Mechanisms of Cardiovascular Disorders in Patients With Chronic Kidney Disease: A Process Related to Accelerated Senescence. Front. Cell Dev. Biol. 2020, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Hörkkö, S.; Savolainen, M.J.; Kervinen, K.; Kesäniemi, Y.A. Carbamylation-Induced Alterations in Low-Density Lipoprotein Metabolism. Kidney Int. 1992, 41, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef]
- Carracedo, J.; Merino, A.; Briceño, C.; Soriano, S.; Buendía, P.; Calleros, L.; Rodriguez, M.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Carbamylated Low-Density Lipoprotein Induces Oxidative Stress and Accelerated Senescence in Human Endothelial Progenitor Cells. FASEB J. 2011, 25, 1314–1322. [Google Scholar] [CrossRef]
- Braun, M.; Pietsch, P.; Schrör, K.; Baumann, G.; Felix, S.B. Cellular Adhesion Molecules on Vascular Smooth Muscle Cells. Cardiovasc. Res. 1999, 41, 395–401. [Google Scholar] [CrossRef][Green Version]
- Mohindra, R.; Agrawal, D.K.; Thankam, F.G. Altered Vascular Extracellular Matrix in the Pathogenesis of Atherosclerosis. J. Cardiovasc. Transl. Res. 2021, 14, 647–660. [Google Scholar] [CrossRef]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage Expression of Active MMP-9 Induces Acute Plaque Disruption in ApoE-Deficient Mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef]
- Stim, J.; Shaykh, M.; Anwar, F.; Ansari, A.; Arruda, J.A.; Dunea, G. Factors Determining Hemoglobin Carbamylation in Renal Failure. Kidney Int. 1995, 48, 1605–1610. [Google Scholar] [CrossRef]
- Abbate, M.; Zoja, C.; Remuzzi, G. How Does Proteinuria Cause Progressive Renal Damage? JASN 2006, 17, 2974–2984. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the Coagulation System. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Huang, M.-J.; Wei, R.; Wang, Y.; Su, T.; Di, P.; Li, Q.; Yang, X.; Li, P.; Chen, X. Blood Coagulation System in Patients with Chronic Kidney Disease: A Prospective Observational Study. BMJ Open 2017, 7, e014294. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.; Smailnejad Ganji, K.; Mirzakhani, M.; Mohammadnia-Afrouzi, M.; Akbari, R. The Role of Immune Response in Initiation and Progression of Chronic Kidney Disease. Iran J. Kidney Dis. 2019, 13, 283–299. [Google Scholar]
- Jaisson, S.; Delevallée-Forte, C.; Touré, F.; Rieu, P.; Garnotel, R.; Gillery, P. Carbamylated Albumin Is a Potent Inhibitor of Polymorphonuclear Neutrophil Respiratory Burst. FEBS Lett. 2007, 581, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Sardenberg, C.; Suassuna, P.; Andreoli, M.C.C.; Watanabe, R.; Dalboni, M.A.; Manfredi, S.R.; dos Santos, O.P.; Kallas, E.G.; Draibe, S.A.; Cendoroglo, M. Effects of Uraemia and Dialysis Modality on Polymorphonuclear Cell Apoptosis and Function. Nephrol. Dial. Transplant. 2006, 21, 160–165. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Jaber, B.L. Mortality Caused by Sepsis in Patients with End-Stage Renal Disease Compared with the General Population. Kidney Int. 2000, 58, 1758–1764. [Google Scholar] [CrossRef]
- Perez, G.O.; Glasson, P.; Favre, H.; Wauters, J.P.; Benzonana, G.; Jeannet, M.; Lambert, P.H. Circulating Immune Complexes in Regularly Dialyzed Patients with Chronic Renal Failure. Am. J. Nephrol. 1984, 4, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; van Veelen, P.A.; Mahler, M.; Janssen, G.M.C.; Drijfhout, J.W.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. Carbamylation and Antibodies against Carbamylated Proteins in Autoimmunity and Other Pathologies. Autoimmun. Rev. 2014, 13, 225–230. [Google Scholar] [CrossRef]
- Nakabo, S.; Hashimoto, M.; Ito, S.; Furu, M.; Ito, H.; Fujii, T.; Yoshifuji, H.; Imura, Y.; Nakashima, R.; Murakami, K.; et al. Carbamylated Albumin Is One of the Target Antigens of Anti-Carbamylated Protein Antibodies. Rheumatology 2017, 56, 1217–1226. [Google Scholar] [CrossRef]
- Mydel, P.; Wang, Z.; Brisslert, M.; Hellvard, A.; Dahlberg, L.E.; Hazen, S.L.; Bokarewa, M. Carbamylation-Dependent Activation of T Cells: A Novel Mechanism in the Pathogenesis of Autoimmune Arthritis. J. Immunol. 2010, 184, 6882–6890. [Google Scholar] [CrossRef]
- Turunen, S.; Koivula, M.-K.; Risteli, L.; Risteli, J. Anticitrulline Antibodies Can Be Caused by Homocitrulline-Containing Proteins in Rabbits. Arthritis Rheum. 2010, 62, 3345–3352. [Google Scholar] [CrossRef]
- Martínez, G.; Gómez, J.A.; Bang, H.; Martínez-Gamboa, L.; Roggenbuck, D.; Burmester, G.-R.; Torres, B.; Prada, D.; Feist, E. Carbamylated Vimentin Represents a Relevant Autoantigen in Latin American (Cuban) Rheumatoid Arthritis Patients. Rheumatol. Int. 2016, 36, 781–791. [Google Scholar] [CrossRef]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Krämer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of Vimentin Is Inducible by Smoking and Represents an Independent Autoantigen in Rheumatoid Arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef]
- Reed, E.; Jiang, X.; Kharlamova, N.; Ytterberg, A.J.; Catrina, A.I.; Israelsson, L.; Mathsson-Alm, L.; Hansson, M.; Alfredsson, L.; Rönnelid, J.; et al. Antibodies to Carbamylated α-Enolase Epitopes in Rheumatoid Arthritis Also Bind Citrullinated Epitopes and Are Largely Indistinct from Anti-Citrullinated Protein Antibodies. Arthritis Res. Ther. 2016, 18, 96. [Google Scholar] [CrossRef] [PubMed]
- Sidiras, P.; Lechanteur, J.; Imbault, V.; Sokolova, T.; Durez, P.; Gangji, V.; Communi, D.; Rasschaert, J. Human Carbamylome Description Identifies Carbamylated A2-Macroglobulin and Hemopexin as Two Novel Autoantigens in Early Rheumatoid Arthritis. Rheumatology 2021, keab838. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Tamez, H.; Wenger, J.; Ankers, E.; Trottier, C.A.; Deferio, J.J.; Berg, A.H.; Karumanchi, S.A.; Thadhani, R.I. Carbamylation of Serum Albumin and Erythropoietin Resistance in End Stage Kidney Disease. Clin. J. Am. Soc. Nephrol. 2013, 8, 1927–1934. [Google Scholar] [CrossRef]
- Brines, M.; Grasso, G.; Fiordaliso, F.; Sfacteria, A.; Ghezzi, P.; Fratelli, M.; Latini, R.; Xie, Q.; Smart, J.; Su-Rick, C.; et al. Erythropoietin Mediates Tissue Protection through an Erythropoietin and Common β-Subunit Heteroreceptor. Proc. Natl. Acad. Sci. USA 2004, 101, 14907–14912. [Google Scholar] [CrossRef] [PubMed]
- Tögel, F.E.; Ahlstrom, J.D.; Yang, Y.; Hu, Z.; Zhang, P.; Westenfelder, C. Carbamylated Erythropoietin Outperforms Erythropoietin in the Treatment of AKI-on-CKD and Other AKI Models. J. Am. Soc. Nephrol. 2016, 27, 3394–3404. [Google Scholar] [CrossRef] [PubMed]
- Maltaneri, R.E.; Chamorro, M.E.; Schiappacasse, A.; Nesse, A.B.; Vittori, D.C. Differential Effect of Erythropoietin and Carbamylated Erythropoietin on Endothelial Cell Migration. Int. J. Biochem. Cell Biol. 2017, 85, 25–34. [Google Scholar] [CrossRef]
- Fantacci, M.; Bianciardi, P.; Caretti, A.; Coleman, T.R.; Cerami, A.; Brines, M.; Samaja, M. Carbamylated Erythropoietin Ameliorates the Metabolic Stress Induced in Vivo by Severe Chronic Hypoxia. Proc. Natl. Acad. Sci. USA 2006, 103, 17531–17536. [Google Scholar] [CrossRef]
- Abe, T.; Isaka, Y.; Imamura, R.; Kakuta, Y.; Okumi, M.; Yazawa, K.; Ichimaru, N.; Tsuda, H.; Nonomura, N.; Takahara, S.; et al. Carbamylated Erythropoietin Ameliorates Cyclosporine Nephropathy without Stimulating Erythropoiesis. Cell Transplant. 2012, 21, 571–580. [Google Scholar] [CrossRef]
- Imamura, R.; Isaka, Y.; Ichimaru, N.; Takahara, S.; Okuyama, A. Carbamylated Erythropoietin Protects the Kidneys from Ischemia-Reperfusion Injury without Stimulating Erythropoiesis. Biochem. Biophys. Res. Commun. 2007, 353, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Imamura, R.; Okumi, M.; Isaka, Y.; Ichimaru, N.; Moriyama, T.; Imai, E.; Nonomura, N.; Takahara, S.; Okuyama, A. Carbamylated Erythropoietin Improves Angiogenesis and Protects the Kidneys from Ischemia-Reperfusion Injury. Cell Transplant. 2008, 17, 135–141. [Google Scholar] [CrossRef]
- Stohlawetz, P.J.; Dzirlo, L.; Hergovich, N.; Lackner, E.; Mensik, C.; Eichler, H.G.; Kabrna, E.; Geissler, K.; Jilma, B. Effects of Erythropoietin on Platelet Reactivity and Thrombopoiesis in Humans. Blood 2000, 95, 2983–2989. [Google Scholar] [CrossRef]
- Zeng, C.; Li, Y.; Ma, J.; Niu, L.; Tay, F.R. Clinical/Translational Aspects of Advanced Glycation End-Products. Trends Endocrinol. Metab. 2019, 30, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Gillery, P. Impaired Proteostasis: Role in the Pathogenesis of Diabetes Mellitus. Diabetologia 2014, 57, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J.A. Decreased Proteolysis Caused by Protein Aggregates, Inclusion Bodies, Plaques, Lipofuscin, Ceroid, and “aggresomes” during Oxidative Stress, Aging, and Disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef]
- Trottier, C.; Perl, J.; Freeman, M.; Thadhani, R.; Berg, A.; Kalim, S. Protein Carbamylation in Peritoneal Dialysis and the Effect of Low Glucose Plus Amino Acid Solutions. Perit. Dial. Int. 2018, 38, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Perl, J.; Kalim, S.; Wald, R.; Goldstein, M.B.; Yan, A.T.; Noori, N.; Kiaii, M.; Wenger, J.; Chan, C.; Thadhani, R.I.; et al. Reduction of Carbamylated Albumin by Extended Hemodialysis. Hemodial. Int. 2016, 20, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Ortiz, G.; Trottier, C.A.; Deferio, J.J.; Karumanchi, S.A.; Thadhani, R.I.; Berg, A.H. The Effects of Parenteral Amino Acid Therapy on Protein Carbamylation in Maintenance Hemodialysis Patients. J. Ren. Nutr. 2015, 25, 388–392. [Google Scholar] [CrossRef]
- CKD Evaluation and Management—KDIGO. Available online: https://kdigo.org/guidelines/ckd-evaluation-and-management/ (accessed on 30 October 2020).
- Di Iorio, B.R.; Minutolo, R.; De Nicola, L.; Bellizzi, V.; Catapano, F.; Iodice, C.; Rubino, R.; Conte, G. Supplemented Very Low Protein Diet Ameliorates Responsiveness to Erythropoietin in Chronic Renal Failure. Kidney Int. 2003, 64, 1822–1828. [Google Scholar] [CrossRef]
- Bellizzi, V.; Di Iorio, B.R.; De Nicola, L.; Minutolo, R.; Zamboli, P.; Trucillo, P.; Catapano, F.; Cristofano, C.; Scalfi, L.; Conte, G.; et al. Very Low Protein Diet Supplemented with Ketoanalogs Improves Blood Pressure Control in Chronic Kidney Disease. Kidney Int. 2007, 71, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Marzocco, S.; Bellasi, A.; De Simone, E.; Dal Piaz, F.; Rocchetti, M.T.; Cosola, C.; Di Micco, L.; Gesualdo, L. Nutritional Therapy Reduces Protein Carbamylation through Urea Lowering in Chronic Kidney Disease. Nephrol. Dial. Transplant. 2018, 33, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.A.; Shanaki, M. In Vitro Inhibition of Low Density Lipoprotein Carbamylation by Vitamins, as an Ameliorating Atherosclerotic Risk in Uremic Patients. Scand. J. Clin. Lab. Investig. 2010, 70, 122–127. [Google Scholar] [CrossRef]
- Ghaffari, M.A.; Shanaki, M. Evalution of in Vitro Effect of Flavonoids on Human Low-Density Lipoprotein Carbamylation. Iran. J. Pharm. Res. 2010, 9, 67–74. [Google Scholar]
- Smyth, D.G. Carbamylation of Amino and Tyrosine Hydroxyl Groups. Preparation of an Inhibitor of Oxytocin with No Intrinsic Activity on the Isolated Uterus. J. Biol. Chem. 1967, 242, 1579–1591. [Google Scholar] [CrossRef]
- Bright, R.; Marchant, C.; Bartold, P.M. The Effect of Triclosan on Posttranslational Modification of Proteins through Citrullination and Carbamylation. Clin. Oral. Investig. 2018, 22, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Lalia, A.Z.; Dasari, S.; Pallauf, M.; Fitch, M.; Hellerstein, M.K.; Lanza, I.R. Eicosapentaenoic Acid but Not Docosahexaenoic Acid Restores Skeletal Muscle Mitochondrial Oxidative Capacity in Old Mice. Aging Cell 2015, 14, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Plater, M.L.; Goode, D.; Crabbe, M.J. Ibuprofen Protects Alpha-Crystallin against Posttranslational Modification by Preventing Protein Cross-Linking. Ophthalmic Res. 1997, 29, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.N.; Cotlier, E. Aspirin Prevents the Nonenzymatic Glycosylation and Carbamylation of the Human Eye Lens Crystallins in Vitro. Biochem. Biophys. Res. Commun. 1988, 151, 991–996. [Google Scholar] [CrossRef]
- Crompton, M.; Rixon, K.C.; Harding, J.J. Aspirin Prevents Carbamylation of Soluble Lens Proteins and Prevents Cyanate-Induced Phase Separation Opacities in Vitro: A Possible Mechanism by Which Aspirin Could Prevent Cataract. Exp. Eye Res. 1985, 40, 297–311. [Google Scholar] [CrossRef]
- Lewis, B.S.; Rixon, K.C.; Harding, J.J. Bendazac Prevents Cyanate Binding to Soluble Lens Proteins and Cyanate-Induced Phase-Separation Opacities in Vitro: A Possible Mechanism by Which Bendazac Could Delay Cataract. Exp. Eye Res. 1986, 43, 973–979. [Google Scholar] [CrossRef]
- Long, J.; Vela Parada, X.; Kalim, S. Protein Carbamylation in Chronic Kidney Disease and Dialysis. Adv. Clin. Chem. 2018, 87, 37–67. [Google Scholar] [CrossRef]
- Koeth, R.A.; Kalantar-Zadeh, K.; Wang, Z.; Fu, X.; Tang, W.H.W.; Hazen, S.L. Protein Carbamylation Predicts Mortality in ESRD. J. Am. Soc. Nephrol. 2013, 24, 853–861. [Google Scholar] [CrossRef]
- Jaisson, S.; Kazes, I.; Desmons, A.; Fadel, F.; Oudart, J.-B.; Santos-Weiss, I.C.R.D.; Millart, H.; Touré, F.; Rieu, P.; Gillery, P. Homocitrulline as Marker of Protein Carbamylation in Hemodialyzed Patients. Clin. Chim. Acta 2016, 460, 5–10. [Google Scholar] [CrossRef]
- Jaisson, S.; Kerkeni, M.; Santos-Weiss, I.C.R.; Addad, F.; Hammami, M.; Gillery, P. Increased Serum Homocitrulline Concentrations Are Associated with the Severity of Coronary Artery Disease. Clin. Chem. Lab. Med. 2015, 53, 103–110. [Google Scholar] [CrossRef]
- Flückiger, R.; Harmon, W.; Meier, W.; Loo, S.; Gabbay, K.H. Hemoglobin Carbamylation in Uremia. N. Engl. J. Med. 1981, 304, 823–827. [Google Scholar] [CrossRef]
- Kwan, J.T.; Carr, E.C.; Barron, J.L.; Bending, M.R. Carbamylated Haemoglobin--a Retrospective Index of Time-Averaged Urea Concentration. Nephrol. Dial. Transplant. 1993, 8, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Wynckel, A.; Randoux, C.; Millart, H.; Desroches, C.; Gillery, P.; Canivet, E.; Chanard, J. Kinetics of Carbamylated Haemoglobin in Acute Renal Failure. Nephrol. Dial. Transplant. 2000, 15, 1183–1188. [Google Scholar] [CrossRef]
- Davenport, A.; Jones, S.; Goel, S.; Astley, J.P.; Feest, T.G. Carbamylated Hemoglobin: A Potential Marker for the Adequacy of Hemodialysis Therapy in End-Stage Renal Failure. Kidney Int. 1996, 50, 1344–1351. [Google Scholar] [CrossRef][Green Version]
- Tarif, N.; Shaykh, M.; Stim, J.; Arruda, J.A.; Dunea, G. Carbamylated Hemoglobin in Hemodialysis Patients. Am. J. Kidney Dis. 1997, 30, 361–365. [Google Scholar] [CrossRef]
- Wang, X.; Peesapati, S.K.; Renedo, M.F.; Moktan, S. Hemoglobin A1c Levels in Non-Diabetic Patients with End-Stage Renal Disease (ESRD) Receiving Hemodialysis. J. Endocrinol. Investig. 2004, 27, 733–735. [Google Scholar] [CrossRef]
- Nakao, T.; Matsumoto, H.; Okada, T.; Han, M.; Hidaka, H.; Yoshino, M.; Shino, T.; Yamada, C.; Nagaoka, Y. Influence of Erythropoietin Treatment on Hemoglobin A1c Levels in Patients with Chronic Renal Failure on Hemodialysis. Intern. Med. 1998, 37, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Okumiya, T.; Saibara, T.; Park, K.; Sasaki, M. Abnormally Decreased HbA1c Can Be Assessed with Erythrocyte Creatine in Patients with a Shortened Erythrocyte Age. Diabetes Care 1998, 21, 1732–1735. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, C.; Kalim, S.; Wenger, J.B.; Suntharalingam, P.; Hod, T.; Thadhani, R.I.; Karumanchi, S.A.; Wanner, C.; Berg, A.H. Protein Carbamylation Is Associated with Heart Failure and Mortality in Diabetic Patients with End Stage Renal Disease. Kidney Int. 2015, 87, 1201–1208. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CKD Complications | Carb. Compounds | Models | Key Findings | Refs. | |
| Vascular damages  | LDL * | Φ | Human leukemic T cells, human fibroblasts | Impairs cLDL binding to the hepatic LDL receptor | [11,39] |
| Σ | IV injection in healthy subjects IV injection in rabbits | Delays LDL clearance | |||
| Φ | Human EPCs, HAECs | Increases EPC senescence Increases HCAEC death via MAPK Uncouples eNOS and reduces NO production Increases ROS production Inhibits angiogenesis | [34,40,41] | ||
| Σ | IV injection in mice | Impairs aortic endothelium-dependent relaxation | |||
| Φ | HCAECs, human monocyte cell line | Increases expression of ICAM-1 and VCAM-1 on HCAECs Increases monocyte adhesion to endothelial cells via LOX-1 | [42,43] | ||
| Φ | HCAECs | Triggers LDL transcytosis via CD-36, SR-A1, SREC-1 | [43] | ||
| Σ | IV injection in mice | Induces subendothelial LDL accumulation | |||
| Φ | Peritoneal macrophages | Promotes lipid loading and foam cell formation through SR-A1 | [11] | ||
| Φ | Human CASMCs, human VSMCs | Increases expression of ICAM-1 and VCAM-1 on CASMCs Increases CASMC proliferation Increases VSMC proliferation via SR-A1 | [11,42,44] | ||
| HDL * | Φ | Bovine aortic endothelial cells | Induces cell apoptosis | [11] | |
| Φ | HAECs | Inhibits cell migration and proliferation Inhibits angiogenesis | [45] | ||
| Φ | Human monocyte cell line | Impairs cholesterol efflux Promotes cholesterol accumulation and lipid droplet formation via SR-BI | [46,47] | ||
| Type I collagen | Φ | Human monocytes | Increases monocyte adhesion Increases MMP-9 production and activation | [48] | |
| Φ | Biochemical assay | Induces local conformational changes in the triple helixImpairs fibrillogenesis | [49] | ||
| Elastin | Σ | ApoE-/- mice fed with cyanate-supplemented water | Increases aortic elastic fibre stiffness Increases aortic pulse wave velocity | [29] | |
| Mitochondrial proteins * | Φ | Human VSMCs | Promotes mitochondrial dysfunctions and oxidative stress Inhibits ENPP1 and reduces PPi production Increases cell calcification | [26] | |
| Σ | Nephrectomised rats fed with urea-supplemented diet | Increases aorta calcification by suppressing PPi production | |||
| Uromodulin | Φ | Human VSMCs | Impairs interaction with and trapping of TNF-α and IL-1β Loses inhibitory effects on osteo-/chondrogenic transdifferentiation | [50] | |
| Sortilin * | Φ | Human CASMCs | Promotes cell calcification by increasing ALPL and RUNX2 expression and TNAP activity Increases the binding of IL-6, amplifying cell calcification | [51] | |
| Ψ | Rat aorta | Increases calcification of aortic rings | |||
Renal Fibrosis | Albumin * | Σ | IP injection in Axolotl | Induces expression of fibronectin and pro-fibrogenic factors (NF-κB, TGF-β1, PDGF-AB, IL-8, ET-1) in tubular cells | [52] |
| FBS proteins | Φ | Mesangial cells | Increases cell proliferation Increases synthesis of collagen I and IV | [53] | |
| Type I Collagen | Φ | Biochemical assay | Increases resistance to MMP-1, MMP-8 and MMP-13 | [54] | |
Haemostasis dysfunctions | Fibrinogen * | Φ | Biochemical assay | Alters fibrinogen structure Interferes with factor XIIIa-mediated fibrin cross-linking and impairs fibrin polymerization Induces clot resistance to fibrinolysis | [55] |
| Fibrino-peptide A | Φ | PMNs | Increases neutrophil chemotaxis | ||
| EPO resistance  | EPO | Φ | Human leukemic cell line | Impairs binding to EPO receptor | [56] |
| Σ | SC injections in mice SC injections in rats | Impairs EPO effect on haemoglobin concentrations and haematocrit | [56,57] | ||
| Insulin resistance  | Insulin | Φ | Rat hepatocytes, rat adipocytes | Decreases binding activity Decreases glucose oxidation | [58] |
| Free L-Asn * | Φ | Rat adipocytes | Reduces insulin-sensitive glucose uptake | [59] | |
| Immune response disorders  | IgG | Φ | Biochemical assay | Impairs C1q binding to IgG Inhibits formation of C4b and C3b | [60] |
| Φ | Lymphoma cell line | Decreases cell lysis | |||
| Type I collagen | Φ | PMNs | Inhibits degranulation and ROS release | [49] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorisse, L.; Jaisson, S.; Piétrement, C.; Gillery, P. Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers? Int. J. Mol. Sci. 2022, 23, 574. https://doi.org/10.3390/ijms23010574
Gorisse L, Jaisson S, Piétrement C, Gillery P. Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers? International Journal of Molecular Sciences. 2022; 23(1):574. https://doi.org/10.3390/ijms23010574
Chicago/Turabian StyleGorisse, Laëtitia, Stéphane Jaisson, Christine Piétrement, and Philippe Gillery. 2022. "Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers?" International Journal of Molecular Sciences 23, no. 1: 574. https://doi.org/10.3390/ijms23010574
APA StyleGorisse, L., Jaisson, S., Piétrement, C., & Gillery, P. (2022). Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers? International Journal of Molecular Sciences, 23(1), 574. https://doi.org/10.3390/ijms23010574