
Cytosolic Quality Control of Mitochondrial Protein Precursors—The Early Stages of the Organelle Biogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. One Entry but Diverse Routes of Import
3. Where Are Precursor Proteins Born?
4. Molecular Chaperones Are Essential to Run the Post-Translational Import
5. Ubiquitin-Proteasome System Degrades Mitochondrial Precursor Proteins in the Cytosol
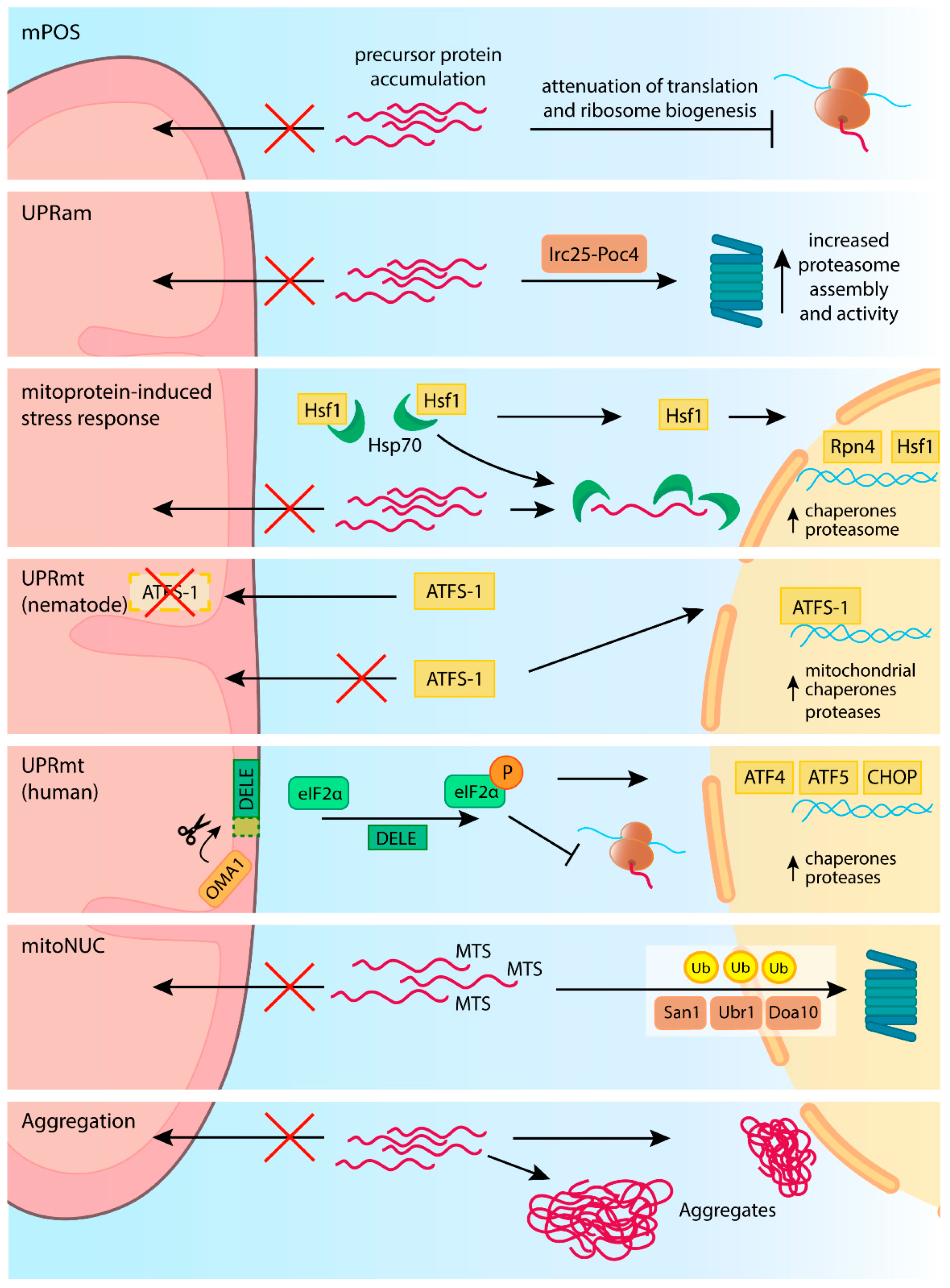
6. Disturbance and Restoration of Proteostasis in the Cytosol
7. Mitochondrial Precursor Proteins Are Prone to Aggregation
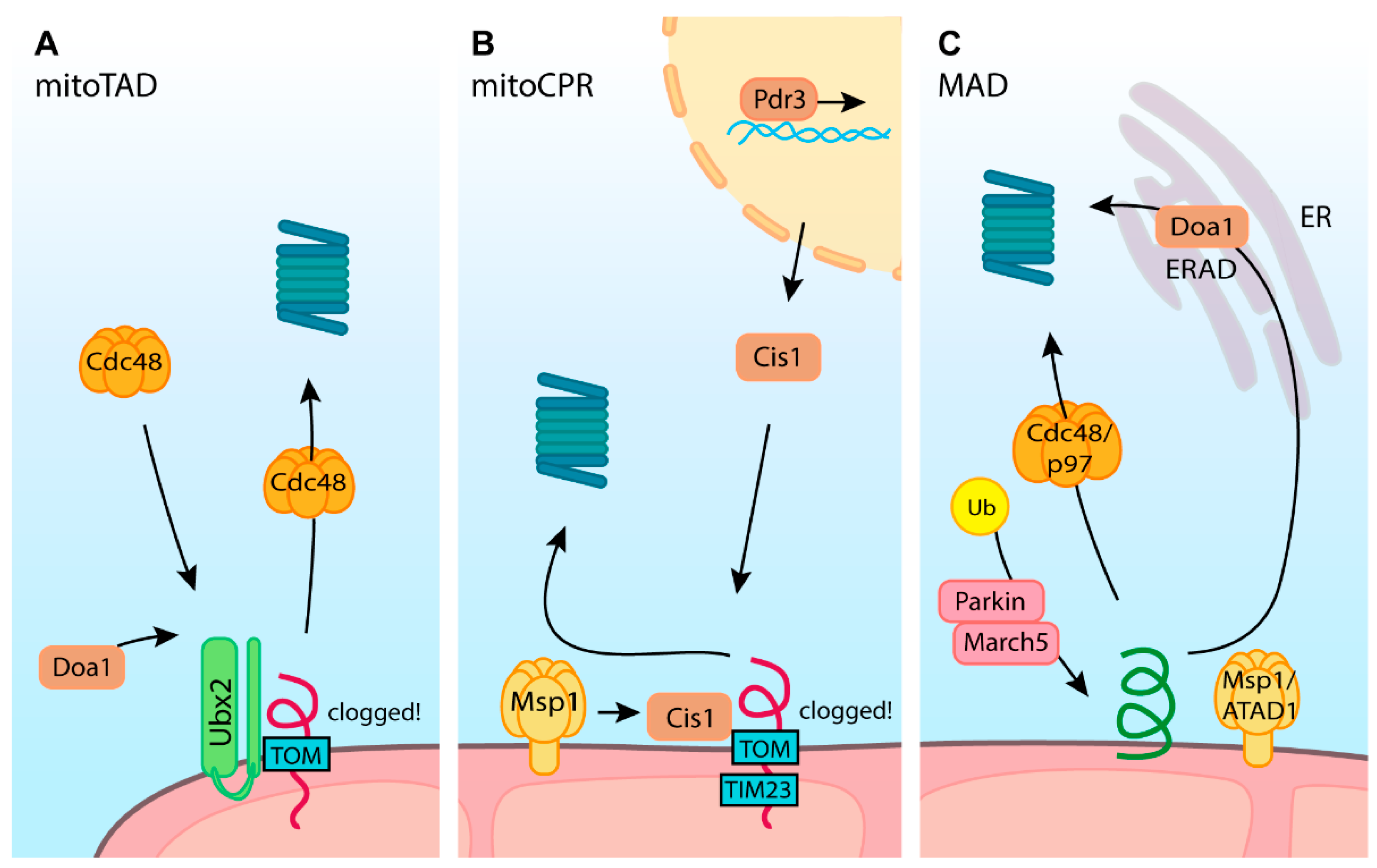
8. Restoration of Import through the Outer Mitochondrial Membrane
9. Quality Control in Co-Translational Targeting
10. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Youle, R.J. Mitochondria—Striking a balance between host and endosymbiont. Science 2019, 365. [Google Scholar] [CrossRef]
- Gabaldón, T. Relative timing of mitochondrial endosymbiosis and the “pre-mitochondrial symbioses” hypothesis. IUBMB Life 2018, 70, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.V.F.; Žárský, V.; Barlow, L.; Herman, E.K.; et al. A Eukaryote without a Mitochondrial Organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Guaragnella, N.; Coyne, L.P.; Chen, X.J.; Giannattasio, S. Mitochondria–cytosol–nucleus crosstalk: Learning from Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, 18. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Zahedi, R.; Sickmann, A.; Pfanner, N.; Meisinger, C. The Protein Import Machinery of Mitochondria—A Regulatory Hub in Metabolism, Stress, and Disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Mårtensson, C.U.; Priesnitz, C.; Song, J.; Ellenrieder, L.; Doan, K.N.; Boos, F.; Floerchinger, A.; Zufall, N.; Oeljeklaus, S.; Warscheid, B.; et al. Mitochondrial protein translocation-associated degradation. Nat. Cell Biol. 2019, 569, 679–683. [Google Scholar] [CrossRef]
- Weidberg, H.; Amon, A. MitoCPR—A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Neupert, W. A Mitochondrial Odyssey. Annu. Rev. Biochem. 2012, 81, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.; Park, E. Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2019, 26, 1158–1166. [Google Scholar] [CrossRef]
- Ahting, U.; Thieffry, M.; Engelhardt, H.; Hegerl, R.; Neupert, W.; Nussberger, S. Tom40, the Pore-Forming Component of the Protein-Conducting Tom Channel in the Outer Membrane of Mitochondria. J. Cell Biol. 2001, 153, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kühlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700.e7. [Google Scholar] [CrossRef] [PubMed]
- Araiso, Y.; Tsutsumi, A.; Qiu, J.; Imai, K.; Shiota, T.; Song, J.; Lindau, C.; Wenz, L.-S.; Sakaue, H.; Yunoki, K.; et al. Structure of the mitochondrial import gate reveals distinct preprotein paths. Nat. Cell Biol. 2019, 575, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Kater, L.; Wagener, N.; Berninghausen, O.; Becker, T.; Neupert, W.; Beckmann, R. Structure of the Bcs1 AAA-ATPase suggests an airlock-like translocation mechanism for folded proteins. Nat. Struct. Mol. Biol. 2020, 27, 142–149. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global Analysis of the Mitochondrial N-Proteome Identifies a Processing Peptidase Critical for Protein Stability. Cell 2009, 139, 428–439. [Google Scholar] [CrossRef]
- Calvo, S.E.; Julien, O.; Clauser, K.; Shen, H.; Kamer, K.J.; Wells, J.A.; Mootha, V.K. Comparative Analysis of Mitochondrial N-Termini from Mouse, Human, and Yeast. Mol. Cell. Proteom. 2017, 16, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Schendzielorz, A.B.; Schulz, C.; Lytovchenko, O.; Clancy, A.; Guiard, B.; Ieva, R.; van der Laan, M.; Rehling, P. Two distinct membrane potential–dependent steps drive mitochondrial matrix protein translocation. J. Cell Biol. 2017, 216, 83–92. [Google Scholar] [CrossRef]
- Backes, S.; Hess, S.; Boos, F.; Woellhaf, M.W.; Gödel, S.; Jung, M.; Mühlhaus, T.; Herrmann, J.M. Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J. Cell Biol. 2018, 217, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossmann, D.; Meisinger, C.; Vögtle, F.-N. Processing of mitochondrial presequences. Biochim. Biophys. Bioenerg. 2012, 1819, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Donzeau, M.; Káldi, K.; Adam, A.; Paschen, S.; Wanner, G.; Guiard, B.; Bauer, M.F.; Neupert, W.; Brunner, M. Tim23 Links the Inner and Outer Mitochondrial Membranes. Cell 2000, 101, 401–412. [Google Scholar] [CrossRef] [Green Version]
- E Jensen, R.; Dunn, C. Protein import into and across the mitochondrial inner membrane: Role of the TIM23 and TIM22 translocons. Biochim. Biophys. Acta Bioenerg. 2002, 1592, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Straub, S.P.; Stiller, S.B.; Wiedemann, N.; Pfanner, N. Dynamic organization of the mitochondrial protein import machinery. Biol. Chem. 2016, 397, 1097–1114. [Google Scholar] [CrossRef] [PubMed]
- Brix, J.; Rüdiger, S.; Bukau, B.; Schneider-Mergener, J.; Pfanner, N. Distribution of Binding Sequences for the Mitochondrial Import Receptors Tom20, Tom22, and Tom70 in a Presequence-carrying Preprotein and a Non-cleavable Preprotein. J. Biol. Chem. 1999, 274, 16522–16530. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Wang, Q.; Guan, Z.; Wu, Y.; Shen, C.; Hong, S.; Cao, J.; Zhang, X.; Yan, C.; Yin, P. Cryo-EM structure of the human mitochondrial translocase TIM22 complex. Cell Res. 2021, 31, 369–372. [Google Scholar] [CrossRef]
- Erdogan, A.; Riemer, J. Mitochondrial disulfide relay and its substrates: Mechanisms in health and disease. Cell Tissue Res. 2016, 367, 59–72. [Google Scholar] [CrossRef]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of import and assembly pathways in mitochondrial protein biogenesis. Biol. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef]
- Wasilewski, M.; Chojnacka, K.; Chacinska, A. Protein trafficking at the crossroads to mitochondria. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta Bioenerg. 2013, 1833, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Suissa, M.; Schatz, G. Import of Proteins into Mitochondria Translatable Mrnas for Imported Mitochondrial Proteins are Present in Free as Well as Mitochondria-Bound Cytoplasmic Polysomes. J. Biol. Chem. 1982, 257, 13048–13055. [Google Scholar] [CrossRef]
- Marc, P.; Margeot, A.; Devaux, F.; Blugeon, C.; Corral-Debrinski, M.; Jacq, C. Genome-wide analysis of mRNAs targeted to yeast mitochondria. EMBO Rep. 2002, 3, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Egea, G.; Izquierdo, J.M.; Ricart, J.; Martín, C.S.; Cuezva, J.M. mRNA encoding the β-subunit of the mitochondrial F1-ATPase complex is a localized mRNA in rat hepatocytes. Biochem. J. 1997, 322, 557–565. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Blugeon, C.; Jacq, C. In Yeast, the 3′ Untranslated Region or the Presequence of ATM1 Is Required for the Exclusive Localization of Its mRNA to the Vicinity of Mitochondria. Mol. Cell. Biol. 2000, 20, 7881–7892. [Google Scholar] [CrossRef] [PubMed]
- Gadir, N.; Haim-Vilmovsky, L.; Kraut-Cohen, J.; Gerst, J.E. Localization of mRNAs coding for mitochondrial proteins in the yeast Saccharomyces cerevisiae. RNA 2011, 17, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Uchiumi, T.; Saito, T.; Yagi, M.; Takazaki, S.; Kanki, T.; Kang, D. Localization of mRNAs encoding human mitochondrial oxidative phosphorylation proteins. Mitochondrion 2012, 12, 391–398. [Google Scholar] [CrossRef]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin Control Localized Translation of Respiratory Chain Component mRNAs on Mitochondria Outer Membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef]
- Zabezhinsky, D.; Slobodin, B.; Rapaport, D.; Gerst, J.E. An Essential Role for COPI in mRNA Localization to Mitochondria and Mitochondrial Function. Cell Rep. 2016, 15, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, F.; Lelandais, G.; Garcia, M.; Goussard, S.; Jacq, C. Posttranscriptional control of mitochondrial biogenesis: Spatio-temporal regulation of the protein import process. FEBS Lett. 2010, 584, 4273–4279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenault, T.; Lithgow, T.; Traven, A. PUF proteins: Repression, activation and mRNA localization. Trends Cell Biol. 2011, 21, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Gucek, M.; Xu, H. The mitochondrial outer membrane protein MDI promotes local protein synthesis and mt DNA replication. EMBO J. 2016, 35, 1045–1057. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Myers, S.A.; Carey, D.K.; Carr, S.A.; Ting, A.Y. Spatiotemporally-resolved mapping of RNA binding proteins via functional proximity labeling reveals a mitochondrial mRNA anchor promoting stress recovery. Nat. Commun. 2021, 12, 4980. [Google Scholar] [CrossRef]
- Becker, T.; Bhushan, S.; Jarasch, A.; Armache, J.-P.; Funes, S.; Jossinet, F.; Gumbart, J.; Mielke, T.; Berninghausen, O.; Schulten, K.; et al. Structure of Monomeric Yeast and Mammalian Sec61 Complexes Interacting with the Translating Ribosome. Science 2009, 326, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Rapoport, T.A. Mechanisms of Sec61/SecY-Mediated Protein Translocation Across Membranes. Annu. Rev. Biophys. 2012, 41, 21–40. [Google Scholar] [CrossRef]
- Elvekrog, M.M.; Walter, P. Dynamics of co-translational protein targeting. Curr. Opin. Chem. Biol. 2015, 29, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Foerster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. [Google Scholar] [CrossRef] [Green Version]
- Mahamid, J.; Pfeffer, S.; Schaffer, M.; Villa, E.; Danev, R.; Cuellar, L.K.; Förster, F.; Hyman, A.A.; Plitzko, J.M.; Baumeister, W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 2016, 351, 969–972. [Google Scholar] [CrossRef]
- Zhang, Y.; Berndt, U.; Gölz, H.; Tais, A.; Oellerer, S.; Wölfle, T.; Fitzke, E.; Rospert, S. NAC functions as a modulator of SRP during the early steps of protein targeting to the endoplasmic reticulum. Mol. Biol. Cell 2012, 23, 3027–3040. [Google Scholar] [CrossRef]
- Crowley, K.S.; Payne, R.M. Ribosome Binding to Mitochondria Is Regulated by GTP and the Transit Peptide. J. Biol. Chem. 1998, 273, 17278–17285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellems, R.E.; Allison, V.F.; A Butow, R. Cytoplasmic type 80S ribosomes associated with yeast mitochondria. IV. Attachment of ribosomes to the outer membrane of isolated mitochondria. J. Cell Biol. 1975, 65, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gold, V.A.; Chroscicki, P.; Bragoszewski, P.; Chacinska, A. Visualization of cytosolic ribosomes on the surface of mitochondria by electron cryo-tomography. EMBO Rep. 2017, 18, 1786–1800. [Google Scholar] [CrossRef]
- Sylvestre, J.; Vialette, S.; Debrinski, M.C.; Jacq, C. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003, 4, R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.U.; Beech, P.L.; Lay, S.T.; Gilson, P.R.; Fisher, P.R. Import-Associated Translational Inhibition: Novel In Vivo Evidence for Cotranslational Protein Import into Dictyostelium discoideum Mitochondria. Eukaryot. Cell 2006, 5, 1314–1327. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, T.; Viana, M.P.; Xu, F.; Yu, J.; Chanchani, R.; Arceo, X.G.; Tutucci, E.; Choi, J.; Chen, Y.S.; Singer, R.H.; et al. Mitochondrial volume fraction and translation duration impact mitochondrial mRNA localization and protein synthesis. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Karniely, S.; Pines, O. Translation-coupled Translocation of Yeast Fumarase into Mitochondria in Vivo. J. Biol. Chem. 2007, 282, 29222–29229. [Google Scholar] [CrossRef] [Green Version]
- Gerber, A.P.; Herschlag, D.; O Brown, P. Extensive Association of Functionally and Cytotopically Related mRNAs with Puf Family RNA-Binding Proteins in Yeast. PLoS Biol. 2004, 2, e79. [Google Scholar] [CrossRef]
- Garciía-Rodriíguez, L.J.; Gay, A.C.; Pon, L.A. Puf3p, a Pumilio family RNA binding protein, localizes to mitochondria and regulates mitochondrial biogenesis and motility in budding yeast. J. Cell Biol. 2007, 176, 197–207. [Google Scholar] [CrossRef]
- Saint-Georges, Y.; Garcia, M.; Delaveau, T.; Jourdren, L.; Le Crom, S.; Lemoine, S.; Tanty, V.; Devaux, F.; Jacq, C. Yeast Mitochondrial Biogenesis: A Role for the PUF RNA-Binding Protein Puf3p in mRNA Localization. PLoS ONE 2008, 3, e2293. [Google Scholar] [CrossRef] [Green Version]
- Vardi-Oknin, D.; Arava, Y. Characterization of Factors Involved in Localized Translation Near Mitochondria by Ribosome-Proximity Labeling. Front. Cell Dev. Biol. 2019, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- George, R.; Beddoe, T.; Landl, K.; Lithgow, T. The yeast nascent polypeptide-associated complex initiates protein targeting to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 2296–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fünfschilling, U.; Rospert, S. Nascent Polypeptide–associated Complex Stimulates Protein Import into Yeast Mitochondria. Mol. Biol. Cell 1999, 10, 3289–3299. [Google Scholar] [CrossRef] [Green Version]
- George, R.; Walsh, P.; Beddoe, T.; Lithgow, T. The nascent polypeptide-associated complex (NAC) promotes interaction of ribosomes with the mitochondrial surface in vivo. FEBS Lett. 2002, 516, 213–216. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, J.A.; Payne, R.M. Ribosomes Specifically Bind to Mammalian Mitochondria via Protease-sensitive Proteins on the Outer Membrane. J. Biol. Chem. 2004, 279, 9803–9810. [Google Scholar] [CrossRef] [Green Version]
- Lesnik, C.; Cohen, Y.; Atir-Lande, A.; Schuldiner, M.; Arava, Y. OM14 is a mitochondrial receptor for cytosolic ribosomes that supports co-translational import into mitochondria. Nat. Commun. 2014, 5, 5711. [Google Scholar] [CrossRef] [Green Version]
- Jores, T.; Lawatscheck, J.; Beke, V.; Franz-Wachtel, M.; Yunoki, K.; Fitzgerald, J.C.; Macek, B.; Endo, T.; Kalbacher, H.; Buchner, J.; et al. Cytosolic Hsp70 and Hsp40 chaperones enable the biogenesis of mitochondrial β-barrel proteins. J. Cell Biol. 2018, 217, 3091–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F. Molecular Chaperones Hsp90 and Hsp70 Deliver Preproteins to the Mitochondrial Import Receptor Tom. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B.; Gaume, B.; Herrmann, J.M.; Neupert, W.; Schwarz, E. Role of the mitochondrial DnaJ homolog Mdj1p as a chaperone for mitochondrially synthesized and imported proteins. Mol. Cell. Biol. 1996, 16, 7063–7071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faou, P.; Hoogenraad, N.J. Tom34: A cytosolic cochaperone of the Hsp90/Hsp70 protein complex involved in mitochondrial protein import. Biochim. Biophys. Acta Bioenerg. 2012, 1823, 348–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trcka, F.; Durech, M.; Vankova, P.; Vandova, V.; Simoncik, O.; Kavan, D.; Vojtesek, B.; Muller, P.; Man, P. The interaction of the mitochondrial protein importer TOMM34 with HSP70 is regulated by TOMM34 phosphorylation and binding to 14-3-3 adaptors. J. Biol. Chem. 2020, 295, 8928–8944. [Google Scholar] [CrossRef] [PubMed]
- Stuart, R.A.; Cyr, D.M.; Neupert, W. Hsp70 in mitochondrial biogenesis: From chaperoning nascent polypeptide chains to facilitation of protein degradation. Cell. Mol. Life Sci. 1994, 50, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Zavodszky, E.; Shao, S.; Wohlever, M.; Keenan, R.; Hegde, R.S. Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol. Cell 2016, 63, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Clerico, E.M.; Tilitsky, J.M.; Meng, W.; Gierasch, L.M. How Hsp70 Molecular Machines Interact with Their Substrates to Mediate Diverse Physiological Functions. J. Mol. Biol. 2015, 427, 1575–1588. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Mol. Biol. 1997, 4, 342–349. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Koch, B.D.; Werner-Washburne, M.; Craig, E.A.; Schekman, R. A subfamily of stress proteins facilitates translocation of secretory and mitochondrial precursor polypeptides. Nat. Cell Biol. 1988, 332, 800–805. [Google Scholar] [CrossRef]
- Horton, L.E.; James, P.; Craig, E.A.; Hensold, J.O. The Yeast hsp70 Homologue Ssa Is Required for Translation and Interacts with Sis1 and Pab1 on Translating Ribosomes. J. Biol. Chem. 2001, 276, 14426–14433. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Walter, W.; Yan, W.; Craig, E.A. Functional Interaction of Cytosolic Hsp70 and a DnaJ-Related Protein, Ydj1p, in Protein Translocation In Vivo. Mol. Cell. Biol. 1996, 16, 4378–4386. [Google Scholar] [CrossRef] [Green Version]
- Döring, K.; Ahmed, N.; Riemer, T.; Suresh, H.G.; Vainshtein, Y.; Habich, M.; Riemer, J.; Mayer, M.; O’Brien, E.P.; Kramer, G.; et al. Profiling Ssb-Nascent Chain Interactions Reveals Principles of Hsp70-Assisted Folding. Cell 2017, 170, 298–311.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, K.C.; Kriel, A.; Frydman, J. Nascent Polypeptide Domain Topology and Elongation Rate Direct the Cotranslational Hierarchy of Hsp70 and TRiC/CCT. Mol. Cell 2019, 75, 1117–1130.e5. [Google Scholar] [CrossRef]
- Pfund, C.; Lopez-Hoyo, N.; Ziegelhoffer, T.; Schilke, B.A.; Lopez-Buesa, P.; Walter, W.A.; Wiedmann, M.; Craig, E.A. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 1998, 17, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Raue, U.; Oellerer, S.; Rospert, S. Association of Protein Biogenesis Factors at the Yeast Ribosomal Tunnel Exit Is Affected by the Translational Status and Nascent Polypeptide Sequence. J. Biol. Chem. 2007, 282, 7809–7816. [Google Scholar] [CrossRef] [Green Version]
- Willmund, F.; del Alamo, M.; Pechmann, S.; Chen, T.; Albanèse, V.; Dammer, E.; Peng, J.; Frydman, J. The Cotranslational Function of Ribosome-Associated Hsp70 in Eukaryotic Protein Homeostasis. Cell 2013, 152, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nat. Cell Biol. 2015, 524, 481–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hageman, J.; Kampinga, H.H. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperon 2009, 14, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.-B.; Shao, Y.-M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Shiber, A.; Ravid, T. Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules 2014, 4, 704–724. [Google Scholar] [CrossRef] [Green Version]
- Cyr, D.M.; Ramos, C.H. Specification of Hsp70 Function by Type I and Type II Hsp. Subcell. Biochem. 2015, 78, 91–102. [Google Scholar] [CrossRef]
- Gaur, D.; Singh, P.; Guleria, J.; Gupta, A.; Kaur, S.; Sharma, D. The Yeast Hsp70 Cochaperone Ydj1 Regulates Functional Distinction of Ssa Hsp70s in the Hsp90 Chaperoning Pathway. Genet 2020, 215, 683–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahi, C.; Craig, E.A. Network of general and specialty J protein chaperones of the yeast cytosol. Proc. Natl. Acad. Sci. USA 2007, 104, 7163–7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.J.; Cyr, D.M.; Douglas, M.G. YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell 1992, 71, 1143–1155. [Google Scholar] [CrossRef]
- Hoseini, H.; Pandey, S.; Jores, T.; Schmitt, A.; Franz-Wachtel, M.; Macek, B.; Buchner, J.; Dimmer, K.S.; Rapaport, R. The cytosolic cochaperone Sti1 is relevant for mitochondrial biogenesis and morphology. FEBS J. 2016, 283, 3338–3352. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.G.; Aviram, N.; Laborenz, J.; Bibi, C.; Meyer, M.; Spang, A.; Schuldiner, M.; Herrmann, J.M. An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science 2018, 361, 1118–1122. [Google Scholar] [CrossRef] [Green Version]
- Papić, D.; Elbaz-Alon, Y.; Koerdt, S.N.; Leopold, K.; Worm, D.; Jung, M.; Schuldiner, M.; Rapaport, D. The Role of Djp1 in Import of the Mitochondrial Protein Mim1 Demonstrates Specificity between a Cochaperone and Its Substrate Protein. Mol. Cell. Biol. 2013, 33, 4083–4094. [Google Scholar] [CrossRef] [Green Version]
- Fan, A.C.Y.; Bhangoo, M.K.; Young, J.C. Hsp90 Functions in the Targeting and Outer Membrane Translocation Steps of Tom70-mediated Mitochondrial Import. J. Biol. Chem. 2006, 281, 33313–33324. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.; Toft, D.O. Hop Modulates hsp70/hsp90 Interactions in Protein Folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Smith, D.F. Hop as an Adaptor in the Heat Shock Protein 70 (Hsp70) and Hsp90 Chaperone Machinery. J. Biol. Chem. 1998, 273, 35194–35200. [Google Scholar] [CrossRef] [Green Version]
- Southworth, D.R.; Agard, D.A. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Mol. Cell 2011, 42, 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, A.; Avramova, L.V.; Weiner, H. Tom34 Unlike Tom20 Does Not Interact with the Leader Sequences of Mitochondrial Precursor Proteins. Arch. Biochem. Biophys. 2002, 400, 97–104. [Google Scholar] [CrossRef]
- Opaliński, Ł.; Song, J.; Priesnitz, C.; Wenz, L.-S.; Oeljeklaus, S.; Warscheid, B.; Pfanner, N.; Becker, T. Recruitment of Cytosolic J-Proteins by TOM Receptors Promotes Mitochondrial Protein Biogenesis. Cell Rep. 2018, 25, 2036–2043.e5. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.C.; Likić, V.A.; Waller, R.F.; Mulhern, T.D.; Lithgow, T. The C-terminal TPR Domain of Tom70 Defines a Family of Mitochondrial Protein Import Receptors Found only in Animals and Fungi. J. Mol. Biol. 2006, 358, 1010–1022. [Google Scholar] [CrossRef]
- Zanphorlin, L.M.; de Lima, T.B.; Wong, M.J.; Balbuena, T.S.; Minetti, C.; Remeta, D.; Young, J.C.; Barbosa, L.; Gozzo, F.C.; Ramos, C.H.I. Heat Shock Protein 90 kDa (Hsp90) Has a Second Functional Interaction Site with the Mitochondrial Import Receptor Tom. J. Biol. Chem. 2016, 291, 18620–18631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backes, S.; Bykov, Y.S.; Flohr, T.; Räschle, M.; Zhou, J.; Lenhard, S.; Krämer, L.; Mühlhaus, T.; Bibi, C.; Jann, C.; et al. The chaperone-binding activity of the mitochondrial surface receptor Tom70 protects the cytosol against mitoprotein-induced stress. Cell Rep. 2021, 35, 108936. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, A.M.; A Prado, M.; Peng, I.; Abbas, A.R.; Haley, B.; A Paulo, J.; Reichelt, M.; Katakam, A.; Sagolla, M.; Modrusan, Z.; et al. Ubiquilin1 promotes antigen-receptor mediated proliferation by eliminating mislocalized mitochondrial proteins. eLife 2017, 6, e26435. [Google Scholar] [CrossRef]
- Hessa, T.; Sharma, A.; Mariappan, M.; Eshleman, H.D.; Gutierrez, E.; Hegde, R.S. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nat. Cell Biol. 2011, 475, 394–397. [Google Scholar] [CrossRef]
- Rodrigo-Brenni, M.C.; Gutierrez, E.; Hegde, R.S. Cytosolic Quality Control of Mislocalized Proteins Requires RNF126 Recruitment to Bag. Mol. Cell 2014, 55, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Dederer, V.; Khmelinskii, A.; Huhn, A.G.; Okreglak, V.; Knop, M.; Lemberg, M.K. Cooperation of mitochondrial and ER factors in quality control of tail-anchored proteins. eLife 2019, 8, e45506. [Google Scholar] [CrossRef]
- Schuldiner, M.; Metz, J.; Schmid, V.; Denic, V.; Rakwalska, M.; Schmitt, H.D.; Schwappach, B.; Weissman, J.S. The GET Complex Mediates Insertion of Tail-Anchored Proteins into the ER Membrane. Cell 2008, 134, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Vitali, D.G.; Sinzel, M.; Bulthuis, E.P.; Kolb, A.; Zabel, S.; Mehlhorn, D.G.; Costa, B.F.; Farkas, Á.; Clancy, A.; Schuldiner, M.; et al. The GET pathway can increase the risk of mitochondrial outer membrane proteins to be mistargeted to the ER. J. Cell Sci. 2018, 131, jcs.211110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laborenz, J.; Bykov, Y.S.; Knöringer, K.; Räschle, M.; Filker, S.; Prescianotto-Baschong, C.; Spang, A.; Tatsuta, T.; Langer, T.; Storchová, Z.; et al. The ER protein Ema19 facilitates the degradation of nonimported mitochondrial precursor proteins. Mol. Biol. Cell 2021, 32, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Laborenz, J.; Hansen, K.; Prescianotto-Baschong, C.; Spang, A.; Herrmann, J.M. In vitro import experiments with semi-intact cells suggest a role of the Sec61 paralog Ssh1 in mitochondrial biogenesis. Biol. Chem. 2019, 400, 1229–1240. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nat. Cell Biol. 2003, 425, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Schwartz, D.; E Elias, J.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hatakeyama, S.; Oyamada, K.; Oda, Y.; Nishimura, T.; Nakayama, K.I. Large-scale analysis of the human ubiquitin-related proteome. Proteom. 2005, 5, 4145–4151. [Google Scholar] [CrossRef]
- Jeon, H.B.; Choi, E.S.; Yoon, J.H.; Hwang, J.H.; Chang, J.W.; Lee, E.K.; Choi, H.W.; Park, Z.-Y.; Yoo, Y.J. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 2007, 357, 731–736. [Google Scholar] [CrossRef]
- Sarraf, S.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nat. Cell Biol. 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Rose, C.; Isasa, M.; Ordureau, A.; Prado, M.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.; Gygi, S.P. Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3, 395–403.e4. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.S.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nat. Cell Biol. 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Duttler, S.; Pechmann, S.; Frydman, J. Principles of Cotranslational Ubiquitination and Quality Control at the Ribosome. Mol. Cell 2013, 50, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Durfee, L.A.; Huibregtse, J.M. A Cotranslational Ubiquitination Pathway for Quality Control of Misfolded Proteins. Mol. Cell 2013, 50, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margineantu, D.H.; Emerson, C.B.; Diaz, D.; Hockenbery, D.M. Hsp90 Inhibition Decreases Mitochondrial Protein Turnover. PLoS ONE 2007, 2, e1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial Protein Quality Control by the Proteasome Involves Ubiquitination and the Protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [Green Version]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The Ubiquitin-Proteasome System Regulates Mitochondrial Intermembrane Space Proteins. Mol. Cell. Biol. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, L.; Bragoszewski, P.; Khmelinskii, A.; Glow, E.; Knop, M.; Chacinska, A. Determinants of the cytosolic turnover of mitochondrial intermembrane space proteins. BMC Biol. 2018, 16, 66. [Google Scholar] [CrossRef]
- Habich, M.; Salscheider, S.L.; Murschall, L.M.; Hoehne, M.N.; Fischer, M.; Schorn, F.; Petrungaro, C.; Ali, M.; Erdogan, A.J.; Abou-Eid, S.; et al. Vectorial Import via a Metastable Disulfide-Linked Complex Allows for a Quality Control Step and Import by the Mitochondrial Disulfide Relay. Cell Rep. 2019, 26, 759–774.e5. [Google Scholar] [CrossRef] [Green Version]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, 1107–1123.e10. [Google Scholar] [CrossRef]
- Finger, Y.; Habich, M.; Gerlich, S.; Urbanczyk, S.; Van De Logt, E.; Koch, J.; Schu, L.; Lapacz, K.J.; Ali, M.; Petrungaro, C.; et al. Proteasomal degradation induced by DPP9-mediated processing competes with mitochondrial protein import. EMBO J. 2020, 39, e103889. [Google Scholar] [CrossRef]
- Mohanraj, K.; Wasilewski, M.; Benincá, C.; Cysewski, D.; Poznanski, J.; Sakowska, P.; Bugajska, Z.; Deckers, M.; Dennerlein, S.; Fernandez-Vizarra, E.; et al. Inhibition of proteasome rescues a pathogenic variant of respiratory chain assembly factor COA. EMBO Mol. Med. 2019, 11, e9561. [Google Scholar] [CrossRef]
- Pearce, D.A.; Sherman, F. Differential Ubiquitin-dependent Degradation of the Yeast Apo-cytochrome c Isozymes. J. Biol. Chem. 1997, 272, 31829–31836. [Google Scholar] [CrossRef] [Green Version]
- Shakya, V.P.; A Barbeau, W.; Xiao, T.; Knutson, C.S.; Schuler, M.H.; Hughes, A.L. A nuclear-based quality control pathway for non-imported mitochondrial proteins. eLife 2021, 10, e61230. [Google Scholar] [CrossRef]
- von Mikecz, A. The nuclear ubiquitin-proteasome system. J. Cell Sci. 2006, 119, 1977–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murschall, L.M.; Gerhards, A.; MacVicar, T.; Peker, E.; Hasberg, L.; Wawra, S.; Langer, T.; Riemer, J. The C-terminal region of the oxidoreductase MIA40 stabilizes its cytosolic precursor during mitochondrial import. BMC Biol. 2020, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsayyah, C.; Ozturk, O.; Cavellini, L.; Belgareh-Touzé, N.; Cohen, M.M. The regulation of mitochondrial homeostasis by the ubiquitin proteasome system. Biochim. Biophys. Bioenerg. 2020, 1861, 148302. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Kodroń, A.; Mussulini, B.H.; Pilecka, I.; Chacińska, A. The ubiquitin-proteasome system and its crosstalk with mitochondria as therapeutic targets in medicine. Pharmacol. Res. 2021, 163, 105248. [Google Scholar] [CrossRef]
- Schmidt, O.; Harbauer, A.B.; Rao, S.; Eyrich, B.; Zahedi, R.P.; Stojanovski, D.; Schönfisch, B.; Guiard, B.; Sickmann, A.; Pfanner, N.; et al. Regulation of Mitochondrial Protein Import by Cytosolic Kinases. Cell 2011, 144, 227–239. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Opalińska, M.; Gerbeth, C.; Herman, J.S.; Rao, S.; Schönfisch, B.; Guiard, B.; Schmidt, O.; Pfanner, N.; Meisinger, C. Cell cycle–dependent regulation of mitochondrial preprotein translocase. Science 2014, 346, 1109–1113. [Google Scholar] [CrossRef]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. Cell Rep. 2019, 28, 1659–1669.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, K.; Sathappa, M.; Landin, J.S.; E Johnson, A.; Alder, N. Structural changes in the mitochondrial Tim23 channel are coupled to the proton-motive force. Nat. Struct. Mol. Biol. 2013, 20, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Coyne, L.P.; Chen, X.J. mPOS is a novel mitochondrial trigger of cell death—Implications for neurodegeneration. FEBS Lett. 2017, 592, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nat. Cell Biol. 2015, 524, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Boos, F.; Krämer, L.; Groh, C.; Jung, F.; Haberkant, P.; Stein, F.; Wollweber, F.; Gackstatter, A.; Zöller, E.; Van Der Laan, M.; et al. Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat. Cell Biol. 2019, 21, 442–451. [Google Scholar] [CrossRef]
- Schneider, K.; Nelson, G.M.; Watson, J.L.; Morf, J.; Dalglish, M.; Luh, L.M.; Weber, A.; Bertolotti, A. Protein Stability Buffers the Cost of Translation Attenuation following eIF2α Phosphorylation. Cell Rep. 2020, 32, 108154. [Google Scholar] [CrossRef]
- Kallstrom, G.; Hedges, J.; Johnson, A. The Putative GTPases Nog1p and Lsg1p Are Required for 60S Ribosomal Subunit Biogenesis and Are Localized to the Nucleus and Cytoplasm, Respectively. Mol. Cell. Biol. 2003, 23, 4344–4355. [Google Scholar] [CrossRef] [Green Version]
- Boos, F.; Labbadia, J.; Herrmann, J.M. How the Mitoprotein-Induced Stress Response Safeguards the Cytosol: A Unified View. Trends Cell Biol. 2020, 30, 241–254. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Topf, U.; Suppanz, I.; Samluk, L.; Wrobel, L.; Böser, A.; Sakowska, P.; Knapp, B.; Pietrzyk, M.K.; Chacinska, A.; Warscheid, B. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Topf, U.; Uszczynska-Ratajczak, B.; Chacinska, A. Mitochondrial stress-dependent regulation of cellular protein synthesis. J. Cell Sci. 2019, 132, jcs226258. [Google Scholar] [CrossRef] [Green Version]
- Houston, R.; Sekine, S.; Sekine, Y. The coupling of translational control and stress responses. J. Biochem. 2020, 168, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Horibe, T.; Hoogenraad, N.J. The Chop Gene Contains an Element for the Positive Regulation of the Mitochondrial Unfolded Protein Response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Fiorese, C.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPRmt. Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Buschlen, S.; Amillet, J.-M.; Guiard, B.; Fournier, A.; Marcireau, C.; Bolotin-Fukuhara, M. TheS. cerevisiaeHAP Complex, a Key Regulator of Mitochondrial Function, Coordinates Nuclear and Mitochondrial Gene Expression. Comp. Funct. Genom. 2003, 4, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Bu, P.; Zeng, J.; Vancura, A. Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 2017, 292, 16942–16954. [Google Scholar] [CrossRef] [Green Version]
- Fiorese, C.; Haynes, C.M. Integrating the UPRmtinto the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nat. Cell Biol. 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nat. Cell Biol. 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Donaldson, G.; Christianson, S.; Zahayko, N.; Khalimonchuk, O. Stress-triggered Activation of the Metalloprotease Oma1 Involves Its C-terminal Region and Is Important for Mitochondrial Stress Protection in Yeast. J. Biol. Chem. 2014, 289, 13259–13272. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Bar-Ziv, R.; Bolas, T.; Dillin, A. Systemic effects of mitochondrial stress. EMBO Rep. 2020, 21, e50094. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef] [Green Version]
- Ciryam, P.; Kundra, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol. Sci. 2015, 36, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciryam, P.; Kundra, R.; Freer, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. A transcriptional signature of Alzheimer’s disease is associated with a metastable subproteome at risk for aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 4753–4758. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.-K.; Reitsma, J.M.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J.; Sommer, T. Ribosomal proteins produced in excess are degraded by the ubiquitin–proteasome system. Mol. Biol. Cell 2016, 27, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Rüb, C.; Bruderek, M.; Voos, W. Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef]
- Sung, M.-K.; Porras-Yakushi, T.R.; Reitsma, J.M.; Huber, F.M.; Sweredoski, M.J.; Hoelz, A.; Hess, S.; Deshaies, R.J. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 2016, 5. [Google Scholar] [CrossRef]
- Nowicka, U.; Chroscicki, P.; Stroobants, K.; Sladowska, M.; Turek, M.; Uszczynska-Ratajczak, B.; Kundra, R.; Goral, T.; Perni, M.; Dobson, C.M.; et al. Cytosolic aggregation of mitochondrial proteins disrupts cellular homeostasis by stimulating the aggregation of other proteins. eLife 2021, 10, e65484. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B.; Mogk, A.; Bukau, B. Spatially Organized Aggregation of Misfolded Proteins as Cellular Stress Defense Strategy. J. Mol. Biol. 2015, 427, 1564–1574. [Google Scholar] [CrossRef]
- Schlagowski, A.M.; Knöringer, K.; Morlot, S.; Vicente, A.S.; Flohr, T.; Krämer, L.; Boos, F.; Khalid, N.; Ahmed, S.; Schramm, J.; et al. Increased levels of mitochondrial import factor Mia40 prevent the aggregation of polyQ proteins in the cytosol. EMBO J. 2021, 40, e107913. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Slaughter, B.D.; Unruh, J.; Guo, F.; Yu, Z.; Mickey, K.; Narkar, A.; Ross, R.T.; McClain, M.; Li, R. Organelle-Based Aggregation and Retention of Damaged Proteins in Asymmetrically Dividing Cells. Cell 2014, 159, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Schleyer, M.; Neupert, W. Transport of Proteins into Mitochondria: Translocational Intermediates Spanning Contact Sites between Outer and Inner Membranes. Cell 1985, 43, 339–350. [Google Scholar] [CrossRef]
- Rassow, J.; Guiard, B.; Wienhues, U.; Herzog, V.; Hartl, F.-U.; Neupert, W. Translocation arrest by reversible folding of a precursor protein imported into mitochondria. A means to quantitate translocation contact sites. J. Cell Biol. 1989, 109, 1421–1428. [Google Scholar] [CrossRef]
- Wienhues, U.; Becker, K.; Schleyer, M.; Guiard, B.; Tropschug, M.; Horwich, A.L.; Pfanner, N.; Neupert, W. Protein folding causes an arrest of preprotein translocation into mitochondria in vivo. J. Cell Biol. 1991, 115, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Glick, B.S.; Wachter, C.; Reid, G.A.; Schatz, G. Import of cytochrome b2to the mitochondrial intermembrane space: The tightly folded heme-binding domain makes import dependent upon matrix ATP. Protein Sci. 1993, 2, 1901–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, T.; Nishikawa, S.-I.; Shin, I.; Schultz, P.G.; Endo, T. Probing the environment along the protein import pathways in yeast mitochondria by site-specific photocrosslinking. Proc. Natl. Acad. Sci. USA 1997, 94, 485–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulke, N.; Sepuri, N.B.V.; Pain, D. In vivo zippering of inner and outer mitochondrial membranes by a stable translocation intermediate. Proc. Natl. Acad. Sci. USA 1997, 94, 7314–7319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaume, B.; Klaus, C.; Ungermann, C.; Guiard, B.; Neupert, W.; Brunner, M. Unfolding of preproteins upon import into mitochondria. EMBO J. 1998, 17, 6497–6507. [Google Scholar] [CrossRef] [Green Version]
- Schülke, N.; Sepuri, N.B.V.; Gordon, D.M.; Saxena, S.; Dancis, A.; Pain, D. A Multisubunit Complex of Outer and Inner Mitochondrial Membrane Protein Translocases Stabilized in Vivo by Translocation Intermediates. J. Biol. Chem. 1999, 274, 22847–22854. [Google Scholar] [CrossRef] [Green Version]
- Voisine, C.; A Craig, E.; Zufall, N.; von Ahsen, O.; Pfanner, N.; Voos, W. The Protein Import Motor of Mitochondria: Unfolding and Trapping of Preproteins Are Distinct and Separable Functions of Matrix Hsp. Cell 1999, 97, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Chacinska, A.; Rehling, P.; Guiard, B.; Frazier, A.; Schulze-Specking, A.; Pfanner, N.; Voos, W.; Meisinger, C. Mitochondrial translocation contact sites: Separation of dynamic and stabilizing elements in formation of a TOM–TIM–preprotein supercomplex. EMBO J. 2003, 22, 5370–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, V.A.; Ieva, R.; Walter, A.; Pfanner, N.; Van Der Laan, M.; Kühlbrandt, W. Visualizing active membrane protein complexes by electron cryotomography. Nat. Commun. 2014, 5, 4129. [Google Scholar] [CrossRef] [Green Version]
- Sickmann, A.; Reinders, J.; Wagner, Y.; Joppich, C.; Zahedi, R.; Meyer, H.E.; Schönfisch, B.; Perschil, I.; Chacinska, A.; Guiard, B.; et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 2003, 100, 13207–13212. [Google Scholar] [CrossRef] [Green Version]
- Nahar, S.; Chowdhury, A.; Ogura, T.; Esaki, M. A AAA ATPase Cdc48 with a cofactor Ubx2 facilitates ubiquitylation of a mitochondrial fusion-promoting factor Fzo1 for proteasomal degradation. J. Biochem. 2019, 167, 279–286. [Google Scholar] [CrossRef]
- Chowdhury, A.; Ogura, T.; Esaki, M. Two Cdc48 cofactors Ubp3 and Ubx2 regulate mitochondrial morphology and protein turnover. J. Biochem. 2018, 164, 349–358. [Google Scholar] [CrossRef]
- Nahar, A.; Fu, X.; Polovin, G.; Orth, J.D.; Park, S. Two alternative mechanisms regulate the onset of chaperone-mediated assembly of the proteasomal ATPases. J. Biol. Chem. 2019, 294, 6562–6577. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Basch, M.; Wagner, M.; Rolland, S.; Carbonell, A.; Zeng, R.; Khosravi, S.; Schmidt, A.; Aftab, W.; Imhof, A.; Wagener, J.; et al. Msp1 cooperates with the proteasome for extraction of arrested mitochondrial import intermediates. Mol. Biol. Cell 2020, 31, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Scales, J.L.; Dunklebarger, M.F.; Loncarek, J.; Weissman, A.M. A protein quality control pathway at the mitochondrial outer membrane. eLife 2020, 9, e51065. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.-C.; Wolken, D.M.A.; Serrano, E.; Srivastava, P.; Pon, L.A. Mitochondria-Associated Degradation Pathway (MAD) Function beyond the Outer Membrane. Cell Rep. 2020, 32, 107902. [Google Scholar] [CrossRef]
- Wu, X.; Li, L.; Jiang, H. Doa1 targets ubiquitinated substrates for mitochondria-associated degradation. J. Cell Biol. 2016, 213, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, J.M.; Lever, A.R.; Coody, T.; Gottschling, D.E.; Hughes, A.L. Rsp5 and Mdm30 reshape the mitochondrial network in response to age-induced vacuole stress. Mol. Biol. Cell 2019, 30, 2141–2154. [Google Scholar] [CrossRef]
- Kim, S.; Park, Y.; Yoo, Y.; Cho, H. Self-clearance mechanism of mitochondrial E3 ligase MARCH5 contributes to mitochondria quality control. FEBS J. 2015, 283, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Ye, Y. Doa1 is a MAD adaptor for Cdc. J. Cell Biol. 2016, 213, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Mullally, J.E.; Chernova, T.; Wilkinson, K.D. Doa1 Is a Cdc48 Adapter That Possesses a Novel Ubiquitin Binding Domain. Mol. Cell. Biol. 2006, 26, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.-A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Mossmann, D.; Vögtle, F.-N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, G.; Schlüter, O.M.; Südhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nat. Cell Biol. 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Izawa, T.; Tsuboi, T.; Kuroha, K.; Inada, T.; Nishikawa, S.-I.; Endo, T. Roles of Dom34:Hbs1 in Nonstop Protein Clearance from Translocators for Normal Organelle Protein Influx. Cell Rep. 2012, 2, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Pechmann, S.; Willmund, F.; Frydman, J. The Ribosome as a Hub for Protein Quality Control. Mol. Cell 2013, 49, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Inada, T. The Ribosome as a Platform for mRNA and Nascent Polypeptide Quality Control. Trends Biochem. Sci. 2017, 42, 5–15. [Google Scholar] [CrossRef]
- Sitron, C.S.; Park, J.H.; Giafaglione, J.M.; Brandman, O. Aggregation of CAT tails blocks their degradation and causes proteotoxicity in S. cerevisiae. PLoS ONE 2020, 15, e0227841. [Google Scholar] [CrossRef] [Green Version]
- Izawa, T.; Park, S.-H.; Zhao, L.; Hartl, F.U.; Neupert, W. Cytosolic Protein Vms1 Links Ribosome Quality Control to Mitochondrial and Cellular Homeostasis. Cell 2017, 171, 890–903.e18. [Google Scholar] [CrossRef] [Green Version]
- Sitron, C.S.; Brandman, O. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 2019, 26, 450–459. [Google Scholar] [CrossRef]
- Von Der Malsburg, K.; Shao, S.; Hegde, R.S. The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol. Biol. Cell 2015, 26, 2168–2180. [Google Scholar] [CrossRef] [PubMed]
- van Haaften-Visser, D.Y.; Harakalova, M.; Mocholi, E.; van Montfrans, J.M.; Elkadri, A.; Rieter, E.; Fiedler, K.; van Hasselt, P.M.; Triffaux, E.M.M.; van Haelst, M.M.; et al. Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J. Biol. Chem. 2017, 292, 7904–7920. [Google Scholar] [CrossRef] [Green Version]
- Yip, M.C.J.; Keszei, A.F.A.; Feng, Q.; Chu, V.; McKenna, M.; Shao, S. Mechanism for recycling tRNAs on stalled ribosomes. Nat. Struct. Mol. Biol. 2019, 26, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sitron, C.S.; Brandman, O. Detection and Degradation of Stalled Nascent Chains via Ribosome-Associated Quality Control. Annu. Rev. Biochem. 2020, 89, 417–442. [Google Scholar] [CrossRef]
- Su, T.; Izawa, T.; Thoms, M.; Yamashita, Y.; Cheng, J.; Berninghausen, O.; Hartl, F.U.; Inada, T.; Neupert, W.; Beckmann, R. Structure and function of Vms1 and Arb1 in RQC and mitochondrial proteome homeostasis. Nat. Cell Biol. 2019, 570, 538–542. [Google Scholar] [CrossRef]
- Wu, Z.; Tantray, I.; Lim, J.; Chen, S.; Li, Y.; Davis, Z.; Sitron, C.; Dong, J.; Gispert, S.; Auburger, G.; et al. MISTERMINATE Mechanistically Links Mitochondrial Dysfunction with Proteostasis Failure. Mol. Cell 2019, 75, 835–848.e8. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A Vesicular Transport Pathway Shuttles Cargo from Mitochondria to Lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.-L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef]
- Hughes, A.L.; E Hughes, C.; A Henderson, K.; Yazvenko, N.; E Gottschling, D. Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife 2016, 5, e13943. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Mitochondrial matrix proteases: Quality control and beyond. FEBS J. 2021, 15964. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Wasilewski, M.; Sakowska, P.; Gornicka, A.; Böttinger, L.; Qiu, J.; Wiedemann, N.; Chacinska, A. Retro-translocation of mitochondrial intermembrane space proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 7713–7718. [Google Scholar] [CrossRef] [Green Version]
- Habich, M.; Riemer, J. Stop wasting protein—Proteasome inhibition to target diseases linked to mitochondrial import. EMBO Mol. Med. 2019, 11, e10441. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenkiewicz, A.M.; Krakowczyk, M.; Bragoszewski, P. Cytosolic Quality Control of Mitochondrial Protein Precursors—The Early Stages of the Organelle Biogenesis. Int. J. Mol. Sci. 2022, 23, 7. https://doi.org/10.3390/ijms23010007
Lenkiewicz AM, Krakowczyk M, Bragoszewski P. Cytosolic Quality Control of Mitochondrial Protein Precursors—The Early Stages of the Organelle Biogenesis. International Journal of Molecular Sciences. 2022; 23(1):7. https://doi.org/10.3390/ijms23010007
Chicago/Turabian StyleLenkiewicz, Anna M., Magda Krakowczyk, and Piotr Bragoszewski. 2022. "Cytosolic Quality Control of Mitochondrial Protein Precursors—The Early Stages of the Organelle Biogenesis" International Journal of Molecular Sciences 23, no. 1: 7. https://doi.org/10.3390/ijms23010007
APA StyleLenkiewicz, A. M., Krakowczyk, M., & Bragoszewski, P. (2022). Cytosolic Quality Control of Mitochondrial Protein Precursors—The Early Stages of the Organelle Biogenesis. International Journal of Molecular Sciences, 23(1), 7. https://doi.org/10.3390/ijms23010007