
Impact of Atorvastatin on Skeletal Muscle Mitochondrial Activity, Locomotion and Axonal Excitability—Evidence from ApoE-/- Mice

 ,
,  , , , ,
, , , , 
 ,
, 
Abstract
:1. Introduction
2. Results
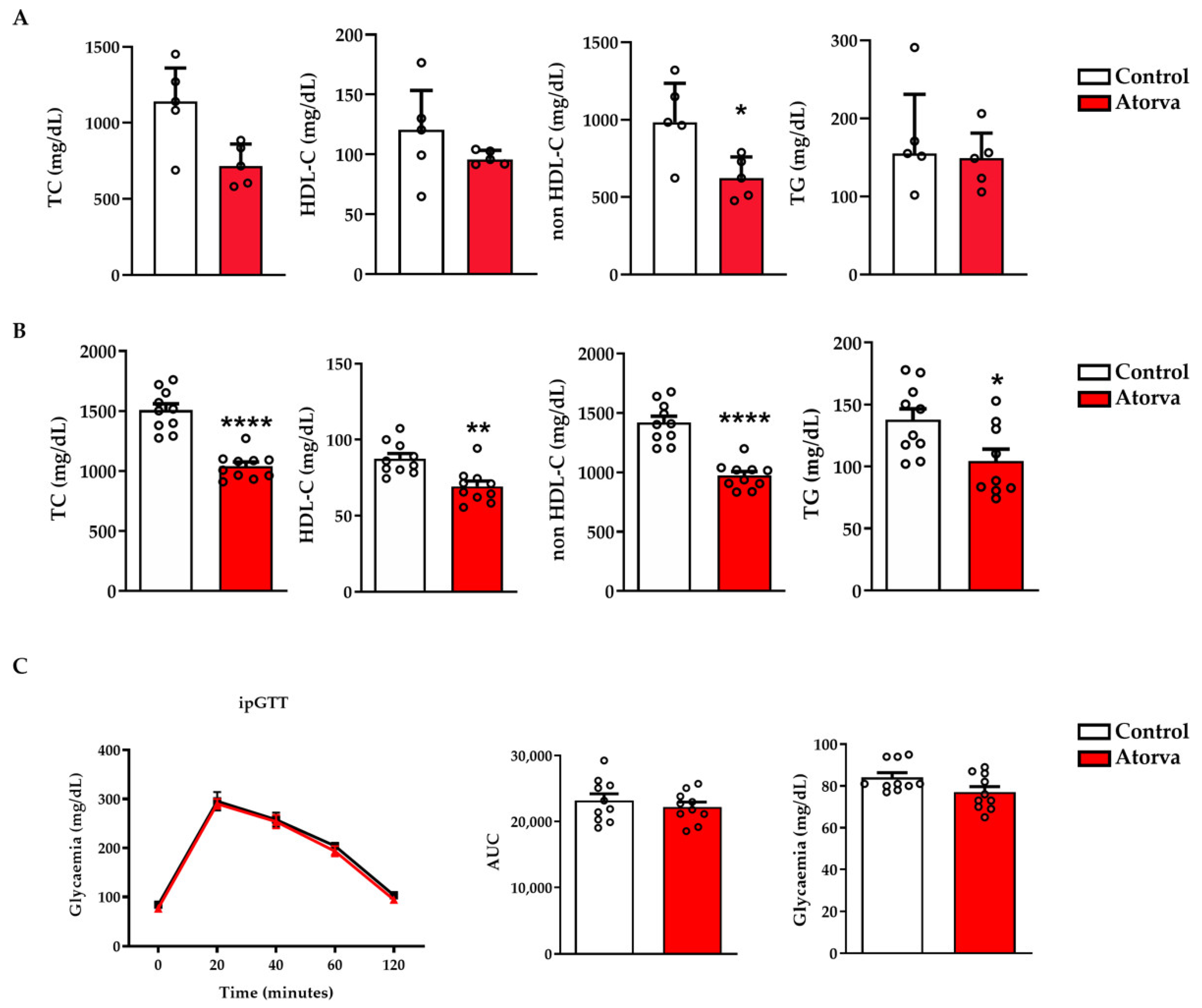
2.1. Impact of Atorvastatin on Lipid and Glycaemic Profiles
2.2. Impact of Atorvastatin on Body Weight and Organs’ Weight
2.3. Impact of Atorvastatin on Mitochondrial Biogenesis and Functionality
2.4. Impact of Atorvastatin on Skeletal Muscle Strength and Locomotion
2.5. Impact of Atorvastatin on Nerve Conduction
3. Discussion
4. Materials and Methods
4.1. Animals and Dietary Regimen
4.2. Chemicals
4.3. Biochemical Evaluations
4.4. Glucose Tolerance Test
4.5. Mitochondria Isolation and Mitochondrial Respiration Analysis
4.6. MRNA Extraction and QPCR Analysis
4.7. Western Blot
4.8. Grip and Locomotion Tests
4.9. Compound Action Potential (CAP) Recording
4.10. Statistical Analyses
5. Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cholesterol Treatment Trialists’ (CTT) Collaboratio; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathawala, F.G. HMG-CoA reductase inhibitors: An exciting development in the treatment of hyperlipoproteinemia. Med. Res. Rev. 1991, 11, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Braunwald, E. Cholesterol: The race to the bottom. Eur. Heart J. 2021, 42, 4612–4613. [Google Scholar] [CrossRef]
- Rodriguez, F.; Maron, D.J.; Knowles, J.W.; Virani, S.S.; Lin, S.; Heidenreich, P.A. Association of Statin Adherence With Mortality in Patients With Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2019, 4, 206–213. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgozoglu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, N.A.E.; Schirris, T.J.J.; Verheggen, R.J.; Russel, F.G.M.; Rodenburg, R.J.; Smeitink, J.A.M.; Thompson, P.D.; Hopman, M.T.E.; Timmers, S. Statins Affect Skeletal Muscle Performance: Evidence for Disturbances in Energy Metabolism. J. Clin. Endocrinol. Metab. 2018, 103, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, P.S.; Haas, R.H.; Bannykh, S.; Hathaway, S.; Gray, N.L.; Kimura, B.J.; Vladutiu, G.D.; England, J.D.; Scripps Mercy Clinical Research, C. Statin-associated myopathy with normal creatine kinase levels. Ann. Intern. Med. 2002, 137, 581–585. [Google Scholar] [CrossRef]
- Krishnan, G.M.; Thompson, P.D. The effects of statins on skeletal muscle strength and exercise performance. Curr. Opin. Lipidol. 2010, 21, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A. Peripheral neuropathy. BMJ 2002, 324, 466–469. [Google Scholar] [CrossRef] [PubMed]
- England, J.D.; Asbury, A.K. Peripheral neuropathy. Lancet 2004, 363, 2151–2161. [Google Scholar] [CrossRef]
- Rajabally, Y.A.; Varakantam, V.; Abbott, R.J. Disorder resembling Guillain-Barre syndrome on initiation of statin therapy. Muscle Nerve 2004, 30, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Gaist, D.; Jeppesen, U.; Andersen, M.; Garcia Rodriguez, L.A.; Hallas, J.; Sindrup, S.H. Statins and risk of polyneuropathy: A case-control study. Neurology 2002, 58, 1333–1337. [Google Scholar] [CrossRef]
- Weimer, L.H. Medication-induced peripheral neuropathy. Curr. Neurol. Neurosci. Rep. 2003, 3, 86–92. [Google Scholar] [CrossRef]
- Bjerre, L.M.; LeLorier, J. Expressing the magnitude of adverse effects in case-control studies: “The number of patients needed to be treated for one additional patient to be harmed”. BMJ 2000, 320, 503–506. [Google Scholar] [CrossRef]
- Leis, A.A.; Stokic, D.S.; Olivier, J. Statins and polyneuropathy: Setting the record straight. Muscle Nerve 2005, 32, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.B.; Preiss, D.; Tobert, J.A.; Jacobson, T.A.; Page, R.L., 2nd; Goldstein, L.B.; Chin, C.; Tannock, L.R.; Miller, M.; Raghuveer, G.; et al. Statin Safety and Associated Adverse Events: A Scientific Statement From the American Heart Association. Arter. Thromb. Vasc. Biol. 2019, 39, e38–e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailman, T.; Hariharan, M.; Karten, B. Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J. Neurochem. 2011, 119, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Bjorkegren, J.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Penson, P.E.; Banach, M. Nocebo/drucebo effect in statin-intolerant patients: An attempt at recommendations. Eur. Heart J. 2021, 42, 4787–4788. [Google Scholar] [CrossRef] [PubMed]
- Sinzinger, H.; O’Grady, J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br. J. Clin. Pharmacol. 2004, 57, 525–528. [Google Scholar] [CrossRef] [Green Version]
- De Vera, M.A.; Bhole, V.; Burns, L.C.; Lacaille, D. Impact of statin adherence on cardiovascular disease and mortality outcomes: A systematic review. Br. J. Clin. Pharmacol. 2014, 78, 684–689. [Google Scholar] [CrossRef]
- Ruscica, M.; Ferri, N.; Banach, M.; Sirtori, C.R.; Corsini, A. Side effects of statins—From pathophysiology and epidemiology to diagnostic and therapeutic implications. Cardiovasc. Res. 2022. [Google Scholar] [CrossRef]
- Moos, W.H.; Faller, D.V.; Glavas, I.P.; Harpp, D.N.; Kamperi, N.; Kanara, I.; Kodukula, K.; Mavrakis, A.N.; Pernokas, J.; Pernokas, M.; et al. Pathogenic mitochondrial dysfunction and metabolic abnormalities. Biochem. Pharmacol. 2021, 193, 114809. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Song, L.N.; Yang, J.K. ACE2 and energy metabolism: The connection between COVID-19 and chronic metabolic disorders. Clin. Sci. 2021, 135, 535–554. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachexia Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Bland, A.R.; Payne, F.M.; Ashton, J.C.; Jamialahmadi, T.; Sahebkar, A. The cardioprotective actions of statins in targeting mitochondrial dysfunction associated with myocardial ischaemia-reperfusion injury. Pharmacol. Res. 2022, 175, 105986. [Google Scholar] [CrossRef]
- Goodman, C.A.; Pol, D.; Zacharewicz, E.; Lee-Young, R.S.; Snow, R.J.; Russell, A.P.; McConell, G.K. Statin-Induced Increases in Atrophy Gene Expression Occur Independently of Changes in PGC1alpha Protein and Mitochondrial Content. PLoS ONE 2015, 10, e0128398. [Google Scholar] [CrossRef] [Green Version]
- Bouitbir, J.; Charles, A.L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef]
- Lantier, L.; Fentz, J.; Mounier, R.; Leclerc, J.; Treebak, J.T.; Pehmoller, C.; Sanz, N.; Sakakibara, I.; Saint-Amand, E.; Rimbaud, S.; et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 2014, 28, 3211–3224. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Southern, W.M.; Nichenko, A.S.; Shill, D.D.; Spencer, C.C.; Jenkins, N.T.; McCully, K.K.; Call, J.A. Skeletal muscle metabolic adaptations to endurance exercise training are attainable in mice with simvastatin treatment. PLoS ONE 2017, 12, e0172551. [Google Scholar] [CrossRef] [Green Version]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Wu, Q.; Guo, J.; Ares, I.; Rodriguez, J.L.; Martinez-Larranaga, M.R.; Yuan, Z.; Anadon, A.; Wang, X.; Martinez, M.A. Statins: Adverse reactions, oxidative stress and metabolic interactions. Pharmacol. Ther. 2019, 195, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, P.; Fabre, O.; Bordenave, S.; Hillaire-Buys, D.; Raynaud De Mauverger, E.; Lacampagne, A.; Mercier, J. Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol. Appl. Pharmacol. 2012, 259, 263–268. [Google Scholar] [CrossRef]
- Pierno, S.; Didonna, M.P.; Cippone, V.; De Luca, A.; Pisoni, M.; Frigeri, A.; Nicchia, G.P.; Svelto, M.; Chiesa, G.; Sirtori, C.; et al. Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: A biochemical, histological and electrophysiological study. Br. J. Pharmacol. 2006, 149, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Sirtori, C.R.; Mombelli, G.; Triolo, M.; Laaksonen, R. Clinical response to statins: Mechanism(s) of variable activity and adverse effects. Ann. Med. 2012, 44, 419–432. [Google Scholar] [CrossRef]
- Parker, B.A.; Capizzi, J.A.; Grimaldi, A.S.; Clarkson, P.M.; Cole, S.M.; Keadle, J.; Chipkin, S.; Pescatello, L.S.; Simpson, K.; White, C.M.; et al. Effect of statins on skeletal muscle function. Circulation 2013, 127, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Mikus, C.R.; Boyle, L.J.; Borengasser, S.J.; Oberlin, D.J.; Naples, S.P.; Fletcher, J.; Meers, G.M.; Ruebel, M.; Laughlin, M.H.; Dellsperger, K.C.; et al. Simvastatin impairs exercise training adaptations. J. Am. Coll. Cardiol. 2013, 62, 709–714. [Google Scholar] [CrossRef] [Green Version]
- Gregg, E.W.; Sorlie, P.; Paulose-Ram, R.; Gu, Q.; Eberhardt, M.S.; Wolz, M.; Burt, V.; Curtin, L.; Engelgau, M.; Geiss, L.; et al. Prevalence of lower-extremity disease in the US adult population ≥ 40 years of age with and without diabetes: 1999–2000 national health and nutrition examination survey. Diabetes Care 2004, 27, 1591–1597. [Google Scholar] [CrossRef] [Green Version]
- Jeppesen, U.; Gaist, D.; Smith, T.; Sindrup, S.H. Statins and peripheral neuropathy. Eur. J. Clin. Pharmacol. 1999, 54, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Tierney, E.F.; Thurman, D.J.; Beckles, G.L.; Cadwell, B.L. Association of statin use with peripheral neuropathy in the U.S. population 40 years of age or older. J. Diabetes 2013, 5, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, T.K.; Norregaard Hansen, P.; Garcia Rodriguez, L.A.; Andersen, L.; Hallas, J.; Sindrup, S.H.; Gaist, D. Statins and polyneuropathy revisited: Case-control study in Denmark, 1999–2013. Br. J. Clin. Pharmacol. 2017, 83, 2087–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisgaier, C.L.; Essenburg, A.D.; Auerbach, B.J.; Pape, M.E.; Sekerke, C.S.; Gee, A.; Wolle, S.; Newton, R.S. Attenuation of plasma low density lipoprotein cholesterol by select 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors in mice devoid of low density lipoprotein receptors. J. Lipid Res. 1997, 38, 2502–2515. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnaghi, V.; Ballabio, M.; Camozzi, F.; Colleoni, M.; Consoli, A.; Gassmann, M.; Lauria, G.; Motta, M.; Procacci, P.; Trovato, A.E.; et al. Altered peripheral myelination in mice lacking GABAB receptors. Mol. Cell Neurosci. 2008, 37, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Lakes, E.H.; Allen, K.D. Gait analysis methods for rodent models of arthritic disorders: Reviews and recommendations. Osteoarthr. Cartil. 2016, 24, 1837–1849. [Google Scholar] [CrossRef] [Green Version]
- Hetze, S.; Romer, C.; Teufelhart, C.; Meisel, A.; Engel, O. Gait analysis as a method for assessing neurological outcome in a mouse model of stroke. J. Neurosci. Methods 2012, 206, 7–14. [Google Scholar] [CrossRef]
- Shepherd, A.J.; Mohapatra, D.P. Pharmacological validation of voluntary gait and mechanical sensitivity assays associated with inflammatory and neuropathic pain in mice. Neuropharmacology 2018, 130, 18–29. [Google Scholar] [CrossRef]
- Bonalume, V.; Caffino, L.; Castelnovo, L.F.; Faroni, A.; Liu, S.; Hu, J.; Milanese, M.; Bonanno, G.; Sohns, K.; Hoffmann, T.; et al. Axonal GABAA stabilizes excitability in unmyelinated sensory axons secondary to NKCC1 activity. J. Physiol. 2021, 599, 4065–4084. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Burke, D.; Andersen, K.V.; Bostock, H. Multiple measures of axonal excitability: A new approach in clinical testing. Muscle Nerve 2000, 23, 399–409. [Google Scholar] [CrossRef]
- Hakim, C.H.; Yang, H.T.; Burke, M.J.; Teixeira, J.; Jenkins, G.J.; Yang, N.N.; Yao, G.; Duan, D. Extensor carpi ulnaris muscle shows unexpected slow-to-fast fiber-type switch in Duchenne muscular dystrophy dogs. Dis. Model. Mech. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Bean, C.; Cancellara, P.; Laveder, P.; Reggiani, C.; Lanfranchi, G. Microgenomic analysis in skeletal muscle: Expression signatures of individual fast and slow myofibers. PLoS ONE 2011, 6, e16807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bytyci, I.; Penson, P.E.; Mikhailidis, D.P.; Wong, N.D.; Hernandez, A.V.; Sahebkar, A.; Thompson, P.D.; Mazidi, M.; Rysz, J.; Pella, D.; et al. Prevalence of statin intolerance: A meta-analysis. Eur. Heart J. 2022. [Google Scholar] [CrossRef]
- Nelson, A.; Haynes, K.; Shambhu, S. High-Intensity Statin Use Among Patients With Atherosclerosis in the U.S. J. Am. Coll. Cardiol. 2022, 79, 1802. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | FORWARD (5′→3′) | REVERSE (5′→3′) |
|---|---|---|
| Rpl13a | GCGCCTCAAGTGGTGTTGGAT | GAGCAGCAGGGACCACCAT |
| Tfam | CGGGCCATCATTCGTCG | AGACAAGACTGATAGACGAGGG |
| Primary Antibody (Dilution) | Secondary Antibody (Dilution) |
|---|---|
| pAMPK (1:1000) | anti-rabbit (1:10,000) |
| AMPK (1:1000) | anti-mouse (1:10,000) |
| DRP-1 (1:1000) | anti-mouse (1:10,000) |
| OPA1 (1:1000) | anti-rabbit (1:10,000) |
| Actin (1:1000) | anti-mouse (1:10,000) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macchi, C.; Bonalume, V.; Greco, M.F.; Mozzo, M.; Melfi, V.; Sirtori, C.R.; Magnaghi, V.; Corsini, A.; Ruscica, M. Impact of Atorvastatin on Skeletal Muscle Mitochondrial Activity, Locomotion and Axonal Excitability—Evidence from ApoE-/- Mice. Int. J. Mol. Sci. 2022, 23, 5415. https://doi.org/10.3390/ijms23105415
Macchi C, Bonalume V, Greco MF, Mozzo M, Melfi V, Sirtori CR, Magnaghi V, Corsini A, Ruscica M. Impact of Atorvastatin on Skeletal Muscle Mitochondrial Activity, Locomotion and Axonal Excitability—Evidence from ApoE-/- Mice. International Journal of Molecular Sciences. 2022; 23(10):5415. https://doi.org/10.3390/ijms23105415
Chicago/Turabian StyleMacchi, Chiara, Veronica Bonalume, Maria Francesca Greco, Marta Mozzo, Valentina Melfi, Cesare R. Sirtori, Valerio Magnaghi, Alberto Corsini, and Massimiliano Ruscica. 2022. "Impact of Atorvastatin on Skeletal Muscle Mitochondrial Activity, Locomotion and Axonal Excitability—Evidence from ApoE-/- Mice" International Journal of Molecular Sciences 23, no. 10: 5415. https://doi.org/10.3390/ijms23105415
APA StyleMacchi, C., Bonalume, V., Greco, M. F., Mozzo, M., Melfi, V., Sirtori, C. R., Magnaghi, V., Corsini, A., & Ruscica, M. (2022). Impact of Atorvastatin on Skeletal Muscle Mitochondrial Activity, Locomotion and Axonal Excitability—Evidence from ApoE-/- Mice. International Journal of Molecular Sciences, 23(10), 5415. https://doi.org/10.3390/ijms23105415