
Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization

 , , and
, , and
Abstract
:1. Introduction
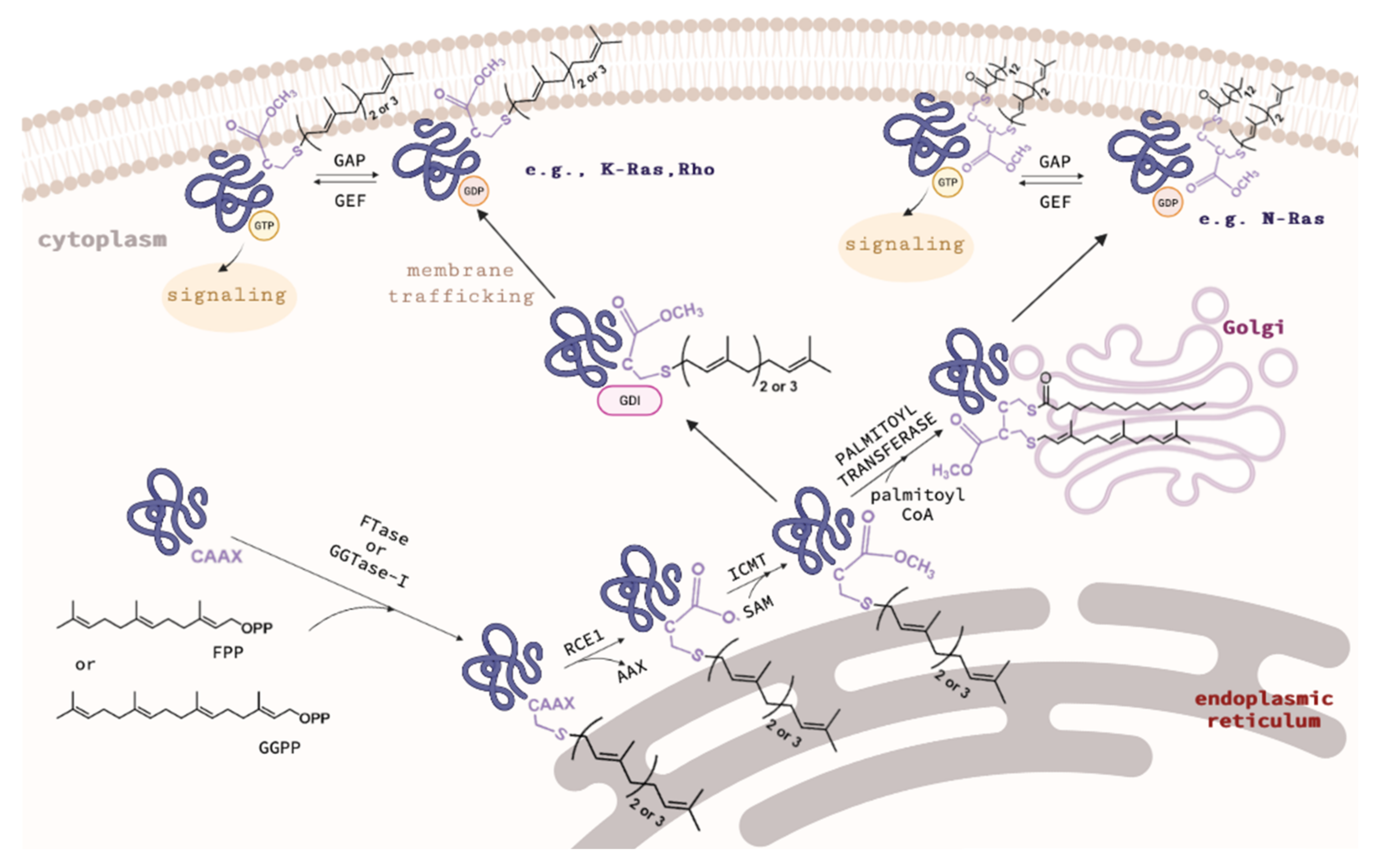
2. Overview of Protein Prenyltransferases and Their Substrates
3. Structures of CAAX Prenyltransferases: FTase and GGTase-I and Mechanism of Prenylation
4. Structure of Rab Geranylgeranyltransferase and Protein Complexes Involved in the Process of Rab Protein Prenylation and the Mechanism of Rab Prenylation
5. Structure of Geranylgeranyltransferase Type III
6. Selected Inhibitors of Prenyltransferases
6.1. FTase Infibitors (FTIs)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Inhibitor | Potency | PDB CODE | Mode of Binding * | Reference |
|---|---|---|---|---|---|
| 1 |  | IC50 0.86 nM (FTase); 90 clinical trials | 1SA4 | protein substrate competitive | [58] |
| 2 |  | EC50 0.34 μM @ EC 10 Vorinostat EC50 > 8 μM without Vorinostat | 7RNI | protein substrate competitive | [53] |
| 3 |  | IC50 1.9 nM (FTase); 40 clinical trials | 1O5M | protein substrate competitive ** | [52] |
| 4 |  | IC50 1.4 nM (FTase); IC50 1.7 μM (GGTase-I) | = | - | [54] |
6.2. GGTase-I Inhibitors (GGTIs)
| Entry | Inhibitor | Potency | PDB CODE | Reference |
|---|---|---|---|---|
| 1 |  | IC50 21 nM (GGTase-I) IC50 5.6μM (FTase) | - | [54] |
| 2 |  | IC50 2.2 μM (GGTase-I) (K562 Proliferation) | - | [65] |
| 3 |  | IC50 313 nM (GGTase-I) | - | [64] |
| 4 |  | IC50 9.5 nM (GGTase-I) IC50 53 μM (FTase) (recruiting for clinical trial) | - | [62] |
| 5 |  | IC50 8.24 nM(GGTase-I) IC50 > 2 μM(FTase) | - | [61] |
6.3. Selected Dual Inhibitors of FTase and GGTase-I
6.4. GGTase-II Inhibitors
| Entry | Inhibitor | Potency | PDB CODE | Mode of Binding * | Reference |
|---|---|---|---|---|---|
| 1 |  | IC50 2.1 μM | - | [64] | |
| 9 |  | 2: IC50 22.7 ± 1.7 μM (Rab GGTase) 3: IC50 9 ± 1 μM (Rab GGTase) | 2: 3C72 3: 3HXD | TAG tunel | [72] |
 | 4: IC50 11 ± 1.2 μM (Rab GGTase) 5: IC50 4.7 ± 0.1 μM (Rab GGTase) | 4: 3HXF 5: 3HXE | TAG tunel | [72] | |
| 2 |  | IC50 6.4 ± 4.8 nM (Rab GGTase)RA IC50 724 ± 321 nM (Rab GGTase)FL | 3PZ1 (BMS3:RGGT), 3PZ2 (BMS3:RGGT:GGPP), 3PZ4 (BMS3:FTase:FPP) | [70,71] | |
| 3 |  | IC50 1547 ± 101 nM (Rab GGTase)RA IC50 616.2 ± 415.7 nM (Rab GGTase)FL | 3PZ3 | TAG tunel and LBS | [70,71] |
| 2 |  | IC50 260 ± 18 nM (Rab GGTase)RA IC50 41.6 ± 9 nM nM (Rab GGTase)FL | - | TAG tunel and LBS | [70,71] |
| 3 |  | IC50 1.4 μM | 4EHM | Isoprenoid substrate competitive | [73] |
| 4 |  | LED 25 μM | - | [78] | |
| 5 |  | LED 10 μM | - | [81] | |
| 6 |  | LED 10 μM | - | [74] | |
| 7 |  | R = Me, LED 10 μM R = Br, LED 10 μM R = p-CHO-C6H4, LED 25 μM | - | [75,76] | |
| 8 |  | IC50 154 μM LED 25 μM | - | [79] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gendaszewska-Darmach, E.; Garstka, M.A.; Błażewska, K.M. Targeting Small GTPases and Their Prenylation in Diabetes Mellitus. J. Med. Chem. 2021, 64, 9677–9710. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Sakurai, A.; Tamura, S.; Takahashi, N.; Abe, K.; Tsuchiya, E.; Fukui, S.; Kitada, C.; Fujino, M. Structure of Rhodotorucine A, a Novel Lipopeptide, Inducing Mating Tube Formation in Rhodosporidiumtoruloides. Biochem. Biophys. Res. Commun. 1978, 83, 1077–1083. [Google Scholar] [CrossRef]
- Wolda, S.L.; Glomset, J.A. Evidence for Modification of Lamin B by a Product of Mevalonic Acid. J. Biol. Chem. 1988, 263, 5997–6000. [Google Scholar] [CrossRef]
- Farnsworth, C.C.; Wolda, S.L.; Gelbs, M.H.; Glomsetg, J.A. Human Lamin B Contains a Farnesylated Cysteine Residue. J. Biol. Chem. 1989, 264, 20422–20429. [Google Scholar] [CrossRef]
- Nguyen, U.T.T.; Wu, Y.; Goodall, A.; Alexandrov, K. Analysis of Protein Prenylation In Vitro and In Vivo Using Functionalized Phosphoisoprenoids. Curr. Protoc. Protein Sci. 2010, 14. [Google Scholar] [CrossRef] [PubMed]
- Brioschi, M.; Martinez Fernandez, A.; Banfi, C. Exploring the Biochemistry of the Prenylome and Its Role in Disease through Proteomics: Progress and Potential. Expert Rev. Proteom. 2017, 14, 515–528. [Google Scholar] [CrossRef]
- Lane, K.T.; Beese, L.S. Thematic Review Series: Lipid Posttranslational Modifications. Structural Biology of Protein Farnesyltransferase and Geranylgeranyltransferase Type I. J. Lipid Res. 2006, 47, 681–699. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, R.; Goto-Ito, S.; Goto, K.; Wakayama, S.; Kubo, H.; Sakata, N.; Trinh, D.A.; Yamagata, A.; Sato, Y.; Masumoto, H.; et al. A SNARE Geranylgeranyltransferase Essential for the Organization of the Golgi Apparatus. EMBO J. 2020, 39, e104120. [Google Scholar] [CrossRef]
- Kuchay, S.; Wang, H.; Marzio, A.; Jain, K.; Homer, H.; Fehrenbacher, N.; Philips, M.R.; Zheng, N.; Pagano, M. GGTase3 Is a Newly Identified Geranylgeranyltransferase Targeting a Ubiquitin Ligase. Nat. Struct. Mol. Biol. 2019, 26, 628–636. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting Protein Prenylation for Cancer Therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Jeong, A.; Suazo, K.F.; Wood, W.G.; Distefano, M.D.; Li, L. Isoprenoids and Protein Prenylation: Implications in the Pathogenesis and Therapeutic Intervention of Alzheimer’s Disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable Ras: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, Y.; Goldstein, J.L.; Seabra, M.C.; Casey, P.J.; Brown, M.S. Inhibition of Purified P21ras Farnesyl:Protein Transferase by Cys-AAX Tetrapeptides. Cell 1990, 62, 81–88. [Google Scholar] [CrossRef]
- Seabra, M.C.; Reiss, Y.; Casey, P.J.; Brown, M.S.; Goldstein, J.L. Protein Farnesyltransferase and Geranylgeranyltransferase Share a Common Alpha Subunit. Cell 1991, 65, 429–434. [Google Scholar] [CrossRef]
- Seabra, M.C.; Brown, M.S.; Slaughter, C.A.; Südhof, T.C.; Goldstein, J.L. Purification of Component A of Rab Geranylgeranyl Transferase: Possible Identity with the Choroideremia Gene Product. Cell 1992, 70, 1049–1057. [Google Scholar] [CrossRef]
- Seabra, M.C.; Goldstein, J.L.; Siidhof, T.C.; Brown, M.S. Rab Geranylgeranyl Transferase. A Multisubunit Enzyme That Prenylates GTP-Binding Proteins Terminating in Cys-X-Cys or Cys-Cys. J. Biol. Chem. 1992, 267, 14497–14503. [Google Scholar] [CrossRef]
- Ochocki, J.D.; Distefano, M.D. Prenyltransferase Inhibitors: Treating Human Ailments from Cancer to Parasitic Infections. Medchemcomm 2013, 4, 476–492. [Google Scholar] [CrossRef] [Green Version]
- Suazo, K.F.; Park, K.-Y.; Distefano, M.D. A Not-So-Ancient Grease History: Click Chemistry and Protein Lipid Modifications. Chem. Rev. 2021, 121, 7178–7248. [Google Scholar] [CrossRef]
- Blanden, M.J.; Suazo, K.F.; Hildebrandt, E.R.; Hardgrove, D.S.; Patel, M.; Saunders, W.P.; Distefano, M.D.; Schmidt, W.K.; Hougland, J.L. Efficient Farnesylation of an Extended C-Terminal C(x)3X Sequence Motif Expands the Scope of the Prenylated Proteome. J. Biol. Chem. 2018, 293, 2770–2785. [Google Scholar] [CrossRef] [Green Version]
- Schey, G.L.; Buttery, P.H.; Hildebrandt, E.R.; Novak, S.X.; Schmidt, W.K.; Hougland, J.L.; Distefano, M.D. MALDI-MS Analysis of Peptide Libraries Expands the Scope of Substrates for Farnesyltransferase. Int. J. Mol. Sci. 2021, 22, 12042. [Google Scholar] [CrossRef]
- Ashok, S.; Hildebrandt, E.R.; Ruiz, C.S.; Hardgrove, D.S.; Coreno, D.W.; Schmidt, W.K.; Hougland, J.L. Protein Farnesyltransferase Catalyzes Unanticipated Farnesylation and Geranylgeranylation of Shortened Target Sequences. Biochemistry 2020, 59, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Blanden, M.J.; Ashok, S.; Hougland, J.L. Mechanisms of CaaX Protein Processing: Protein Prenylation by FTase and GGTase-I; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Mohammed, I.; Hampton, S.E.; Ashall, L.; Hildebrandt, E.R.; Kutlik, R.A.; Manandhar, S.P.; Floyd, B.J.; Smith, H.E.; Dozier, J.K.; Distefano, M.D.; et al. 8-Hydroxyquinoline-Based Inhibitors of the Rce1 Protease Disrupt Ras Membrane Localization in Human Cells. Bioorg. Med. Chem. 2016, 24, 160–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, H.Y.; Tang, J.; Casey, P.J.; Wang, M. Isoprenylcysteine Carboxylmethyltransferase Is Critical for Malignant Transformation and Tumor Maintenance by All RAS Isoforms. Oncogene 2017, 36, 3934–3942. [Google Scholar] [CrossRef] [Green Version]
- Wahlstrom, A.M.; Cutts, B.A.; Liu, M.; Lindskog, A.; Karlsson, C.; Sjogren, A.K.M.; Andersson, K.M.E.; Young, S.G.; Bergo, M.O. Inactivating Icmt Ameliorates K-RAS–Induced Myeloproliferative Disease. Blood 2008, 112, 1357–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Ramos, N.I.; Ortega-Nogales, F.J.; Torrecillas, R.; Gil-Ordo, A.; Marcos-Ramiro, B.; Aguilar-Garrido, P.; Cushman, I.; Romero, A.; Medrano, F.J.; Gajate, C.; et al. A Potent Isoprenylcysteine Carboxylmethyltransferase (ICMT) Inhibitor Improves Survival in Ras-Driven Acute Myeloid Leukemia. J. Med. Chem. 2019, 62, 6035–6046. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A Polybasic Domain or Palmitoylation Is Required in Addition to the CAAX Motif to Localize P21ras to the Plasma Membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Fukasawa, M.; Varlamov, O.; Eng, W.S.; Söllner, T.H.; Rothman, J.E. Localization and Activity of the SNARE Ykt6 Determined by Its Regulatory Domain and Palmitoylation. Proc. Natl. Acad. Sci. USA 2004, 101, 4815–4820. [Google Scholar] [CrossRef] [Green Version]
- Zverina, E.A.; Lamphear, C.L.; Wright, E.N.; Fierke, C.A. Recent Advances in Protein Prenyltransferases: Substrate Identification, Regulation, and Disease Interventions. Curr. Opin. Chem. Biol. 2012, 16, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Goody, R.S.; Abagyan, R.; Alexandrov, K. Structure of the Disordered C Terminus of Rab7 GTPase Induced by Binding to the Rab Geranylgeranyl Transferase Catalytic Complex Reveals the Mechanism of Rab Prenylation. J. Biol. Chem. 2009, 284, 13185–13192. [Google Scholar] [CrossRef] [Green Version]
- Sakata, N.; Shirakawa, R.; Goto, K.; Trinh, D.A.; Horiuchi, H. Double Prenylation of SNARE Protein Ykt6 Is Required for Lysosomal Hydrolase Trafficking. J. Biochem. 2021, 169, 363–370. [Google Scholar] [CrossRef]
- Park, H.W.; Boduluri, S.R.; Moomaw, J.F.; Casey, P.J.; Beese, L.S. Crystal Structure of Protein Farnesyltransferase at 2.25 Angstrom Resolution. Science 1997, 275, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; Reid, T.S.; Terry, K.L.; Casey, P.J.; Beese, L.S. Structure of Mammalian Protein Geranylgeranyltransferase Type-I. EMBO J. 2003, 22, 5963–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolence, J.M.; Rozema, D.B.; Poulter, C.D. Yeast Protein Farnesyltransferase. Site-Directed Mutagenesis of Conserved Residues in the Beta-Subunit. Biochemistry 1997, 36, 9246–9252. [Google Scholar] [CrossRef] [PubMed]
- Kral, A.M.; Diehl, R.E.; DeSolms, S.J.; Williams, T.M.; Kohl, N.E.; Omer, C.A. Mutational Analysis of Conserved Residues of the Beta-Subunit of Human Farnesyl:Protein Transferase. J. Biol. Chem. 1997, 272, 27319–27323. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Casey, P.J.; Beese, L.S. The Basis for K-Ras4B Binding Specificity to Protein Farnesyl-Transferase Revealed by 2 Å Resolution Ternary Complex Structures. Structure 2000, 8, 209–222. [Google Scholar] [CrossRef]
- Huang, C.C.; Hightower, K.E.; Fierke, C.A. Mechanistic Studies of Rat Protein Farnesyltransferase Indicate an Associative Transition State. Biochemistry 2000, 39, 2593–2602. [Google Scholar] [CrossRef]
- Pickett, J.S.; Bowers, K.E.; Fierke, C.A. Mutagenesis Studies of Protein Farnesyltransferase Implicate Aspartate Beta 352 as a Magnesium Ligand. J. Biol. Chem. 2003, 278, 51243–51250. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Casey, P.J.; Beese, L.S. Reaction Path of Protein Farnesyltransferase at Atomic Resolution. Nature 2002, 419, 645–650. [Google Scholar] [CrossRef]
- Long, S.B.; Casey, P.J.; Beese, L.S. Cocrystal Structure of Protein Farnesyltransferase Complexed with a Farnesyl Diphosphate Substrate. Biochemistry 1998, 37, 9612–9618. [Google Scholar] [CrossRef]
- Strickland, C.L.; Windsor, W.T.; Syto, R.; Wang, L.; Bond, R.; Wu, Z.; Schwartz, J.; Le, H.V.; Beese, L.S.; Weber, P.C. Crystal Structure of Farnesyl Protein Transferase Complexed with a CaaX Peptide and Farnesyl Diphosphate Analogue. Biochemistry 1998, 37, 16601–16611. [Google Scholar] [CrossRef]
- Zhang, H.; Seabra, M.C.; Deisenhofer, J. Crystal Structure of Rab Geranylgeranyltransferase at 2.0 Å Resolution. Structure 2000, 8, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Wu, Y.W.; Das, D.; Delon, C.; Cramer, J.; Yu, S.; Thuns, S.; Lupilova, N.; Waldmann, H.; Brunsveld, L.; et al. Structures of RabGGTase–Substrate/Product Complexes Provide Insights into the Evolution of Protein Prenylation. EMBO J. 2008, 27, 2444–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylypenko, O.; Rak, A.; Reents, R.; Niculae, A.; Sidorovitch, V.; Cioaca, M.D.; Bessolitsyna, E.; Thomä, N.H.; Waldmann, H.; Schlichting, I.; et al. Structure of Rab Escort Protein-1 in Complex with Rab Geranylgeranyltransferase. Mol. Cell 2003, 11, 483–494. [Google Scholar] [CrossRef]
- Rak, A.; Pylypenko, O.; Niculae, A.; Pyatkov, K.; Goody, R.S.; Alexandrov, K. Structure of the Rab7:REP-1 Complex: Insights into the Mechanism of Rab Prenylation and Choroideremia Disease. Cell 2004, 117, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Pradhipa Karuna, M.; Witte, L.; Linnemannstoens, K.; Choezom, D.; Danieli-Mackay, A.; Honemann-Capito, M.; Gross, J.C. Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity. Biomolecules 2020, 10, 1560. [Google Scholar] [CrossRef]
- Wen, W.; Yu, J.; Pan, L.; Wei, Z.; Weng, J.; Wang, W.; Ong, Y.S.; Tran, T.H.T.; Hong, W.; Zhang, M. Lipid-Induced Conformational Switch Controls Fusion Activity of Longin Domain SNARE Ykt6. Mol. Cell 2010, 37, 383–395. [Google Scholar] [CrossRef]
- Shen, M.; Pan, P.; Li, Y.; Li, D.; Yu, H.; Hou, T. Farnesyltransferase and Geranylgeranyltransferase I: Structures, Mechanism, Inhibitors and Molecular Modeling. Drug Discov. Today 2015, 20, 267–276. [Google Scholar] [CrossRef]
- Hunt, J.T.; Ding, C.Z.; Batorsky, R.; Bednarz, M.; Bhide, R.; Cho, Y.; Chong, S.; Chao, S.; Gullo-Brown, J.; Guo, P.; et al. Discovery of (R)-7-Cyano-2,3,4, 5-Tetrahydro-1-(1H-Imidazol-4-Ylmethyl)-3- (Phenylmethyl)-4-(2-Thienylsulfonyl)-1H-1,4-Benzodiazepine (BMS-214662), a Farnesyltransferase Inhibitor with Potent Preclinical Antitumor Activity. J. Med. Chem. 2000, 43, 3587–3595. [Google Scholar] [CrossRef]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- Reid, T.S.; Beese, L.S. Crystal Crystal Structures of the Anticancer Clinical Candidates R115777 (Tipifarnib) and BMS-214662 Complexed with Protein Farnesyltransferase Suggest a Mechanism of FTI Selectivity. Biochemistry 2004, 43, 6877–6884. [Google Scholar] [CrossRef]
- Strickland, C.L.; Weber, P.C.; Windsor, W.T.; Wu, Z.; Le, H.v.; Albanese, M.M.; Alvarez, C.S.; Cesarz, D.; del Rosario, J.; Deskus, J.; et al. Tricyclic Farnesyl Protein Transferase Inhibitors: Crystallographic and Calorimetric Studies of Structure-Activity Relationships. J. Med. Chem. 1999, 42, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Bukhtiyarova, M.; Cook, E.M.; Hancock, P.J.; Hruza, A.W.; Shaw, A.W.; Adam, G.C.; Barnard, R.J.O.; McKenna, P.M.; Holloway, M.K.; Bell, I.M.; et al. Discovery of an Anion-Dependent Farnesyltransferase Inhibitor from a Phenotypic Screen. ACS Med. Chem. Lett. 2021, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Blaskovich, M.A.; Knowles, D.; Qian, Y.; Ohkanda, J.; Bailey, R.D.; Hamilton, A.D.; Sebti, S.M. Antitumor Efficacy of a Novel Class of Non-Thiol-Containing Peptidomimetic Inhibitors of Farnesyltransferase and Geranylgeranyltransferase I: Combination Therapy with the Cytotoxic Agents Cisplatin, Taxol, and Gemcitabine. Cancer Res. 1999, 59, 4919–4926. [Google Scholar] [PubMed]
- Ohkanda, J.; Strickland, C.L.; Blaskovich, M.A.; Carrico, D.; Lockman, J.W.; Vogt, A.; Bucher, C.J.; Sun, J.; Qian, Y.; Knowles, D.; et al. Structure-Based Design of Imidazole-Containing Peptidomimetic Inhibitors of Protein Farnesyltransferase. Org. Biomol. Chem. 2006, 4, 482–492. [Google Scholar] [CrossRef]
- Scholten, J.D.; Zimmerman, K.K.; Oxender, M.G.; Leonard, D.; Sebolt-Leopold, J.; Gowan, R.; Hupe, D.J. Synergy between Anions and Farnesyldiphosphate Competitive Inhibitors of Farnesyl:Protein Transferase. J. Biol. Chem. 1997, 272, 18077–18081. [Google Scholar] [CrossRef] [Green Version]
- Samuel Metibemu, D. VHTS and 3D-QSAR for the Identification of Novel Phyto-Inhibitors of Farnesyltransferase: Validation of Ascorbic Acid Inhibition of Farnesyltransferase in an Animal Model of Breast Cancer. Drug Res. 2021, 71, 341–347. [Google Scholar] [CrossRef]
- Reid, T.S.; Long, S.B.; Beese, L.S. Crystallographic Analysis Reveals That Anticancer Clinical Candidate L-778,123 Inhibits Protein Farnesyltransferase and Geranylgeranyltransferase-I by Different Binding Modes. Biochemistry 2004, 43, 9000–9008. [Google Scholar] [CrossRef]
- Qian, Y.; Vogt, A.; Vasudevan, A.; Sebti, S.M.; Hamilton, A.D. Selective Inhibition of Type-I Geranylgeranyltransferase in Vitro and in Whole Cells by CAAL Peptidomimetics. Bioorg. Med. Chem. 1998, 6, 293–299. [Google Scholar] [CrossRef]
- Vasudevan, A.; Qian, Y.; Vogt, A.; Blaskovich, M.A.; Ohkanda, J.; Sebti, S.M.; Hamilton, A.D. Potent, Highly Selective, and Non-Thiol Inhibitors of Protein Geranylgeranyltransferase-I. J. Med. Chem. 1999, 42, 1333–1340. [Google Scholar] [CrossRef]
- Peng, H.; Carrico, D.; Thai, V.; Blaskovich, M.; Bucher, C.; Pusateri, E.E.; Sebti, S.M.; Hamilton, A.D. Synthesis and Evaluation of Potent, Highly-Selective, 3-Aryl-Piperazinone Inhibitors of Protein Geranylgeranyltransferase-I. Org. Biomol. Chem. 2006, 4, 1768–1784. [Google Scholar] [CrossRef]
- Karasic, T.B.; Chiorean, E.G.; Sebti, S.M.; O’Dwyer, P.J. A Phase I Study of GGTI-2418 (Geranylgeranyl Transferase I Inhibitor) in Patients with Advanced Solid Tumors. Target. Oncol. 2019, 14, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Kelly, P.; Weinbaum, C.A.; Casey, P.J. A Novel Protein Geranylgeranyltransferase-I Inhibitor with High Potency, Selectivity, and Cellular Activity. J. Biol. Chem. 2006, 281, 12445–12450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Fiji, H.D.G.; Guo, L.; Chan, L.; Kinderman, S.S.; Slamon, D.J.; Kwon, O.; Tamanoi, F. Inhibitors of Protein Geranylgeranyltransferase I and Rab Geranylgeranyltransferase Identified from a Library of Allenoate-Derived Compounds. J. Biol. Chem. 2008, 283, 9571–9579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Chan, L.; Fiji, H.D.G.; Dahl, R.; Kwon, O.; Tamanoi, F. In Vivo Antitumor Effect of a Novel Inhibitor of Protein Geranylgeranyltransferase-I. Mol. Cancer Ther. 2009, 8, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimonjic, D.B.; Chan, L.N.; Tripathi, V.; Lu, J.; Kwon, O.; Popescu, N.C.; Lowy, D.R.; Tamanoi, F. In Vitro and in Vivo Effects of Geranylgeranyltransferase I Inhibitor P61A6 on Non-Small Cell Lung Cancer Cells. BMC Cancer 2013, 13, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.N.; Fiji, H.D.G.; Watanabe, M.; Kwon, O.; Tamanoi, F. Identification and Characterization of Mechanism of Action of P61-E7, a Novel Phosphine Catalysis-Based Inhibitor of Geranylgeranyltransferase-I. PLoS ONE 2011, 6, e26135. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Xiang, S.; Yang, H.; Chen, L.; Kennedy, P.; Ayaz, M.; Fletcher, S.; Cummings, C.; Lawrence, H.R.; Beato, F.; et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin. Cancer Res. 2019, 25, 5984–5996. [Google Scholar] [CrossRef]
- Tsubamoto, M.; Le, T.K.; Li, M.; Watanabe, T.; Matsumi, C.; Parvatkar, P.; Fujii, H.; Kato, N.; Sun, J.; Ohkanda, J. A Guanidyl-Based Bivalent Peptidomimetic Inhibits K-Ras Prenylation and Association with c-Raf. Chem. Eur. J. 2019, 25, 13531–13536. [Google Scholar] [CrossRef]
- Bon, R.S.; Guo, Z.; Stigter, E.A.; Wetzel, S.; Menninger, S.; Wolf, A.; Choidas, A.; Alexandrov, K.; Blankenfeldt, W.; Goody, R.S.; et al. Structure-Guided Development of Selective RabGGTase Inhibitors. Angew. Chem. Int. Ed. Engl. 2011, 50, 4957–4961. [Google Scholar] [CrossRef]
- Stigter, E.A.; Guo, Z.; Bon, R.S.; Wu, Y.W.; Choidas, A.; Wolf, A.; Menninger, S.; Waldmann, H.; Blankenfeldt, W.; Goody, R.S. Development of Selective, Potent RabGGTase Inhibitors. J. Med. Chem. 2012, 55, 8330–8340. [Google Scholar] [CrossRef]
- Tan, K.T.; Guiu-Rozas, E.; Bon, R.S.; Guo, Z.; Delon, C.; Wetzel, S.; Arndt, S.; Alexandrov, K.; Waldmann, H.; Goody, R.S.; et al. Design, Synthesis, and Characterization of Peptide-Based Rab Geranylgeranyl Transferase Inhibitors. J. Med. Chem. 2009, 52, 8025–8037. [Google Scholar] [CrossRef] [PubMed]
- Deraeve, C.; Guo, Z.; Bon, R.S.; Blankenfeldt, W.; DiLucrezia, R.; Wolf, A.; Menninger, S.; Stigter, E.A.; Wetzel, S.; Choidas, A.; et al. Psoromic Acid Is a Selective and Covalent Rab-Prenylation Inhibitor Targeting Autoinhibited Rabggtase. J. Am. Chem. Soc. 2012, 134, 7384–7391. [Google Scholar] [CrossRef] [PubMed]
- Joachimiak, Ł.; Marchwicka, A.; Gendaszewska-Darmach, E.; Błażewska, K.M. Synthesis and Biological Evaluation of Imidazole-Bearing α-Phosphonocarboxylates as Inhibitors of Rab Geranylgeranyl Transferase (RGGT). ChemMedChem 2018, 13, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, A.; Kusy, D.; Niinivehmas, S.P.; Gmach, J.; Joachimiak, Ł.; Pentikäinen, O.T.; Gendaszewska-Darmach, E.; Błazewska, K.M. Identification of the Privileged Position in the Imidazo[1,2-a]Pyridine Ring of Phosphonocarboxylates for Development of Rab Geranylgeranyl Transferase (RGGT) Inhibitors. J. Med. Chem. 2017, 60, 8781–8800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusy, D.; Marchwicka, A.; Małolepsza, J.; Justyna, K.; Gendaszewska-Darmach, E.; Błażewska, K.M. Synthesis of the 6-Substituted Imidazo[1,2-a]Pyridine-3-Yl-2- Phosphonopropionic Acids as Potential Inhibitors of Rab Geranylgeranyl Transferase. Front. Chem. 2021, 8, 596162. [Google Scholar] [CrossRef]
- Baron, R.A.; Tavaré, R.; Figueiredo, A.C.; Błazewska, K.M.; Kashemirov, B.A.; McKenna, C.E.; Ebetino, F.H.; Taylor, A.; Rogers, M.J.; Coxon, F.P.; et al. Phosphonocarboxylates Inhibit the Second Geranylgeranyl Addition by Rab Geranylgeranyl Transferase. J. Biol. Chem. 2009, 284, 6861. [Google Scholar] [CrossRef] [Green Version]
- McKenna, C.E.; Kashemirov, B.A.; Błazewska, K.M.; Mallard-Favier, I.; Stewart, C.A.; Rojas, J.; Lundy, M.W.; Ebetino, F.H.; Baron, R.A.; Dunford, J.E.; et al. Synthesis, Chiral High Performance Liquid Chromatographic Resolution and Enantiospecific Activity of a Potent New Geranylgeranyl Transferase Inhibitor, 2-Hydroxy-3-Imidazo[1,2-a]Pyridin-3-Yl-2-Phosphonopropionic Acid. J. Med. Chem. 2010, 53, 3454–3464. [Google Scholar] [CrossRef]
- Małolepsza, J.; Marchwicka, A.; Serwa, R.A.; Niinivehmas, S.P.; Pentikäinen, O.T.; Gendaszewska-Darmach, E.; Błażewska, K.M. Rational Design, Optimization, and Biological Evaluation of Novel α-Phosphonopropionic Acids as Covalent Inhibitors of Rab Geranylgeranyl Transferase. J. Enzym. Inhib. Med. Chem. 2022, 37, 940–951. [Google Scholar] [CrossRef]
- Abo, M.; Li, C.; Weerapana, E. Isotopically-Labeled Iodoacetamide-Alkyne Probes for Quantitative Cysteine-Reactivity Profiling. Mol. Pharm. 2018, 15, 743–749. [Google Scholar] [CrossRef]
- Coxon, F.; Joachimiak, Ł.; Najumudeen, A.K.; Breen, G.; Gmach, J.; Oetken-Lindholm, C.; Way, R.; Dunford, J.; Abankwa, D.; Błazewska, K.M. Synthesis and Characterization of Novel Phosphonocarboxylate Inhibitors of RGGT. Eur. J. Med. Chem. 2014, 84, 77–89. [Google Scholar] [CrossRef]
- Palsuledesai, C.C.; Distefano, M.D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.L.; von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. Engl. 2020, 59, 6342. [Google Scholar] [CrossRef] [PubMed] [Green Version]









| FTase | GGTase-I | RGGT (GGTase-II) | GGTase-III | |
|---|---|---|---|---|
| Subunit composition | FNTA (PTAR2) and FNTB | FNTA (PTAR2) and PGGT1B | RabGGTA (PTAR3) and RabGGTB | PTAR1 and RabGGTB |
| Size of subunits (mammalian) | 44 kDa and 49 kDa | 44 kDa and 42 kDa | 65 kDa and 37 kDa | 46kDa and 37kDa |
| Protein recognition sequences | CAAX | CAAX | CC, CXC, CCX, CCXX, CCXXX (CAAX, CXXX for monogeranylgeranylation) | CAAX |
| Protein substrates | Ras, nuclear lamins | Rho, Rac, Rap | Rab | FBXL2, Ykt6 |
| Lipid donor substrates | Farnesyl diphosphate (FPP) | Geranylgeranyl diphosphate (GGPP) | Geranylgeranyl diphosphate (GGPP) | Geranylgeranyl diphosphate (GGPP) |
| Accessory proteins | - | - | REP (REP-1 or REP-2) | SKP1 |
| Metal requirements | Zn2+, Mg2+ | Zn2+ | Zn2+ | Zn2+ |
| Entry | Inhibitor | Potency | PDB CODE | Mode of Binding * | Reference |
|---|---|---|---|---|---|
| 1 |  | IC50 2 nM (FTase); IC50 98 nM (GGTase-I) (two completed clinical trials) | 1S63 | FTase: protein substrate; GGTase-I: protein and isoprenoid substrates competitive | [58] |
| 2 |  | IC50 250 nM (FTase) IC50 520 nM(GGTase-I) | - | - | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchwicka, A.; Kamińska, D.; Monirialamdari, M.; Błażewska, K.M.; Gendaszewska-Darmach, E. Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization. Int. J. Mol. Sci. 2022, 23, 5424. https://doi.org/10.3390/ijms23105424
Marchwicka A, Kamińska D, Monirialamdari M, Błażewska KM, Gendaszewska-Darmach E. Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization. International Journal of Molecular Sciences. 2022; 23(10):5424. https://doi.org/10.3390/ijms23105424
Chicago/Turabian StyleMarchwicka, Aleksandra, Daria Kamińska, Mohsen Monirialamdari, Katarzyna M. Błażewska, and Edyta Gendaszewska-Darmach. 2022. "Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization" International Journal of Molecular Sciences 23, no. 10: 5424. https://doi.org/10.3390/ijms23105424
APA StyleMarchwicka, A., Kamińska, D., Monirialamdari, M., Błażewska, K. M., & Gendaszewska-Darmach, E. (2022). Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization. International Journal of Molecular Sciences, 23(10), 5424. https://doi.org/10.3390/ijms23105424