
Osteoimmunology in Periodontitis: Local Proteins and Compounds to Alleviate Periodontitis

 ,
,  , , ,
, , ,  and
and {kind=link}
Abstract
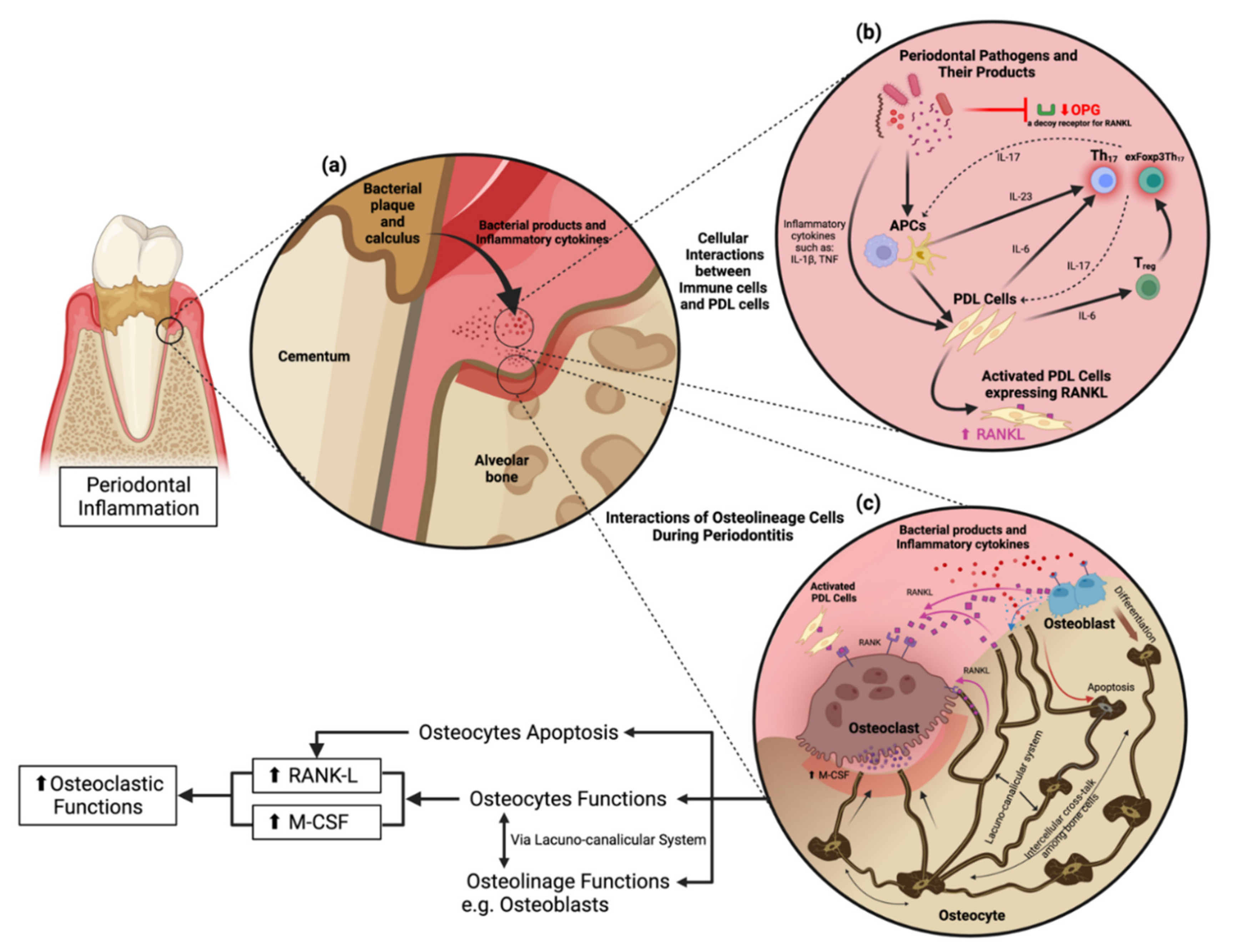
:1. Introduction
2. Periodontitis: Immune Response and Microbiota
2.1. The Novel Classification of Periodontal Diseases (the AAP 2017 Classification)
2.2. Roles of Th17 and exFoxp3Th17 Cells in Periodontitis
2.3. Cellular Interaction to Orchestrate Inflammation
2.4. Roles of Interleukin 6 and Its Receptors in the Development of Periodontitis
2.5. “Polymicrobial Synergy and Dysbiosis” Concept
2.6. Roles of Neutrophils in Periodontitis
3. Periodontitis: Osteoimmunology
3.1. RANKL and OPG
3.2. Osteocytes Are Not Just Quiescent Resident Cells
3.3. Osteocytes Induce Osteoclastogenesis via M-CSF (CSF-1) and RANKL
3.4. Periodontitis Induces Osteocyte Apoptosis Leading to Osteoclastogenesis
3.5. Tissue-Specific Immunity Concept
4. Periodontitis: Treatment
4.1. Complement
4.2. Periostin
4.3. Exendin-4 (EX-4)
4.4. Developmental Endothelial Locus-1 (DEL-1)
4.5. Synthetic and Natural Compounds That Alleviate Periodontitis
5. Closing Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, H.; Kim, S.; Jung, S. Instruction of microbiome taxonomic profiling based on 16S rRNA sequencing. J. Microbiol. 2020, 58, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobbs, A.H.; Lamont, R.J.; Jenkinson, H.F. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 2009, 73, 407–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Lenartova, M.; Tesinska, B.; Janatova, T.; Hrebicek, O.; Mysak, J.; Janata, J.; Najmanova, L. The oral microbiome in periodontal health. Front. Cell. Infect. Microbiol. 2021, 11, 119. [Google Scholar] [CrossRef]
- Van Dyke, T.E.; Bartold, P.M.; Reynolds, E.C. The nexus between periodontal inflammation and dysbiosis. Front. Immunol. 2020, 11, 511. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, R. Osteoimmunology: Inflammatory osteolysis and regeneration of the alveolar bone. J. Clin. Periodontol. 2019, 46 (Suppl. S21), 52–69. [Google Scholar] [CrossRef] [Green Version]
- Tsukasaki, M. RANKL and osteoimmunology in periodontitis. J. Bone Miner. Metab. 2021, 39, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Caton, J. Periodontal diagnosis and diagnostic aids. In Proceedings of the World Workshop in Clinical Periodontics; American Academy of Periodontology: Chicago, IL, USA, 1989; pp. 1–22. [Google Scholar]
- Armitage, G.C. Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 1999, 4, 1–6. [Google Scholar] [CrossRef]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J. Periodontol. 2018, 89, S1–S8. [Google Scholar] [CrossRef]
- Fine, D.H.; Patil, A.G.; Loos, B.G. Classification and diagnosis of aggressive periodontitis. J. Periodontol. 2018, 89 (Suppl. S1), S103–S119. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Periodontol. 2018, 89 (Suppl. S1), S159–S172. [Google Scholar] [CrossRef] [Green Version]
- Berglundh, T.; Armitage, G.; Araujo, M.G.; Avila-Ortiz, G.; Blanco, J.; Camargo, P.M.; Chen, S.; Cochran, D.; Derks, J.; Figuero, E.; et al. Peri-implant diseases and conditions: Consensus report of workgroup 4 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S313–S318. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The conceptual framework unifying the immune and skeletal systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A Dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10, eaat0797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host Defense against oral microbiota by bone-damaging T Cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef] [PubMed]
- Okui, T.; Aoki, Y.; Ito, H.; Honda, T.; Yamazaki, K. The presence of IL-17+/FOXP3+ double-positive cells in periodontitis. J. Dent. Res. 2012, 91, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. New developments in neutrophil biology and periodontitis. Periodontology 2000 2020, 82, 78–92. [Google Scholar] [CrossRef]
- Dutzan, N.; Konkel, J.E.; Greenwell-Wild, T.; Moutsopoulos, N.M. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Ruddy, M.J.; Wong, G.C.; Liu, X.K.; Yamamoto, H.; Kasayama, S.; Kirkwood, K.L.; Gaffen, S.L. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 2004, 279, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Graves, D. Cytokines that promote periodontal tissue destruction. J. Periodontol. 2008, 79, 1585–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and specificity: Insights from the interleukin 6 family of cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting interleukin-6 signaling in clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Düsterhöft, S.; Zhu, Y.; Grötzinger, J.; et al. Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Guo, B.; Fu, M.; Guo, W.; Yuan, Y.; Yuan, H.; Zhang, S.; Yu, H. Interleukin-6-174G/C polymorphism contributes to periodontitis susceptibility: An updated meta-analysis of 21 case-control studies. Dis. Markers. 2016, 2016, 9612421. [Google Scholar] [CrossRef]
- Stadler, A.F.; Angst, P.D.; Arce, R.M.; Gomes, S.C.; Oppermann, R.V.; Susin, C. Gingival crevicular fluid levels of cytokines/chemokines in chronic periodontitis: A meta-analysis. J. Clin. Periodontol. 2016, 43, 727–745. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Kirakodu, S.; Novak, M.J.; Stromberg, A.J.; Shen, S.; Orraca, L.; Gonzalez-Martinez, J.; Burgos, A.; Gonzalez, O.A. Cytokine gene expression profiles during initiation, progression and resolution of periodontitis. J. Clin. Periodontol. 2014, 41, 853–861. [Google Scholar] [CrossRef]
- Lee, H.J.; Kang, I.K.; Chung, C.P.; Choi, S.M. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J. Clin. Periodontol. 1995, 22, 885–890. [Google Scholar] [CrossRef]
- Vidal, F.; Figueredo, C.M.; Cordovil, I.; Fischer, R.G. Periodontal therapy reduces plasma levels of interleukin-6, C-reactive protein, and fibrinogen in patients with severe periodontitis and refractory arterial hypertension. J. Periodontol. 2009, 80, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Apolinário Vieira, G.H.; Aparecida Rivas, A.C.; Figueiredo Costa, K.; Ferreira Oliveira, L.F.; Tanaka Suzuki, K.; Reis Messora, M.; Sprone Ricoldi, M.; Gonçalves de Almeida, A.L.; Taba, M., Jr. Specific inhibition of IL-6 receptor attenuates inflammatory bone loss in experimental periodontitis. J. Periodontol. 2021, 92, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Irwin, C.R.; Myrillas, T.T.; Traynor, P.; Leadbetter, N.; Cawston, T.E. The role of soluble interleukin (IL)-6 receptor in mediating the effects of IL-6 on matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 expression by gingival fibroblasts. J. Periodontol. 2002, 73, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Naruishi, K.; Takashiba, S.; Chou, H.H.; Arai, H.; Nishimura, F.; Murayama, Y. Role of soluble interleukin-6 receptor in inflamed gingiva for binding of interleukin-6 to gingival fibroblasts. J. Periodontal Res. 1999, 34, 296–300. [Google Scholar] [CrossRef]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T.; et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 11924–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedghi, L.M.; Bacino, M.; Kapila, Y.L. Periodontal disease: The good, the bad, and the unknown. Front. Cell. Infect. Microbiol. 2021, 11, 1210. [Google Scholar] [CrossRef]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontology 2000 2020, 83, 14–25. [Google Scholar] [CrossRef]
- Lourenço, T.G.; Heller, D.; Silva-Boghossian, C.M.; Cotton, S.L.; Paster, B.J.; Colombo, A.P. Microbial signature profiles of periodontally healthy and diseased patients. J. Clin. Periodontol. 2014, 41, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Feres, M.; Teles, F.; Teles, R.; Figueiredo, L.C.; Faveri, M. The subgingival periodontal microbiota of the aging mouth. Periodontology 2000 2016, 72, 30–53. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 2014, 29, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Yost, S.; Duran-Pinedo, A.E.; Teles, R.; Krishnan, K.; Frias-Lopez, J. Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 2015, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Attar, A.; Alimova, Y.; Kirakodu, S.; Kozal, A.; Novak, M.J.; Stromberg, A.J.; Orraca, L.; Gonzalez-Martinez, J.; Martinez, M.; Ebersole, J.L.; et al. Activation of Notch-1 in oral epithelial cells by P. gingivalis triggers the expression of the antimicrobial protein PLA(2)-IIA. Mucosal Immunol. 2018, 11, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellebrekers, P.; Vrisekoop, N.; Koenderman, L. Neutrophil phenotypes in health and disease. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12943. [Google Scholar] [CrossRef] [PubMed]
- Medara, N.; Lenzo, J.C.; Walsh, K.A.; Reynolds, E.C.; O’Brien-Simpson, N.M.; Darby, I.B. Peripheral neutrophil phenotypes during management of periodontitis. J. Periodontal Res. 2021, 56, 58–68. [Google Scholar] [CrossRef]
- Movat, H.Z.; Cybulsky, M.I.; Colditz, I.G.; Chan, M.K.; Dinarello, C.A. Acute inflammation in gram-negative infection: Endotoxin, interleukin 1, tumor necrosis factor, and neutrophils. Fed. Proc. 1987, 46, 97–104. [Google Scholar]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus: Same foe different M.O. Front. Immunol. 2021, 12, 570. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef]
- Lakschevitz, F.S.; Aboodi, G.M.; Glogauer, M. Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PLoS ONE 2013, 8, e68983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popadiak, K.; Potempa, J.; Riesbeck, K.; Blom, A.M. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J. Immunol. 2007, 178, 7242–7250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Krauss, J.L.; Domon, H.; McIntosh, M.L.; Hosur, K.B.; Qu, H.; Li, F.; Tzekou, A.; Lambris, J.D.; Hajishengallis, G. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J. Immunol. 2011, 186, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; Mcintosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.I. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontology 2000 2010, 53, 124–137. [Google Scholar] [CrossRef]
- Lee, W.; Aitken, S.; Sodek, J.; McCulloch, C.A. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: Role of active enzyme in human periodontitis. J. Periodontal Res. 1995, 30, 23–33. [Google Scholar] [CrossRef]
- Sima, C.; Glogauer, M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: Current state of knowledge. Curr. Oral Health Rep. 2014, 1, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Chapple, I.L.; Matthews, J.B. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontology 2000 2007, 43, 160–232. [Google Scholar] [CrossRef]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Takahashi, K.; Azuma, T.; Motohira, H.; Kinane, D.F.; Kitetsu, S. The potential role of interleukin-17 in the immunopathology of periodontal disease. J. Clin. Periodontol. 2005, 32, 369–374. [Google Scholar] [CrossRef]
- Maekawa, T.; Briones, R.A.; Resuello, R.R.; Tuplano, J.V.; Hajishengallis, E.; Kajikawa, T.; Koutsogiannaki, S.; Garcia, C.A.; Ricklin, D.; Lambris, J.D.; et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J. Clin. Periodontol. 2016, 43, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- López Roldán, A.; García Giménez, J.L.; Alpiste Illueca, F. Impact of periodontal treatment on the RANKL/OPG ratio in crevicular fluid. PLoS ONE 2020, 15, e0227757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, T.; Abe, T.; Hajishengallis, E.; Hosur, K.B.; DeAngelis, R.A.; Ricklin, D.; Lambris, J.D.; Hajishengallis, G. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J. Immunol. 2014, 192, 6020–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Okamoto, K.; Nakai, Y.; Tsutsumi, M.; Muro, R.; Suematsu, A.; Hashimoto, K.; Okamura, T.; Ehata, S.; Nitta, T.; et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat. Metab. 2019, 1, 868–875. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL Biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Crotti, T.; Smith, M.D.; Hirsch, R.; Soukoulis, S.; Weedon, H.; Capone, M.; Ahern, M.J.; Haynes, D. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J. Periodontal Res. 2003, 38, 380–387. [Google Scholar] [CrossRef]
- Ochiai, N.; Nakachi, Y.; Yokoo, T.; Ichihara, T.; Eriksson, T.; Yonemoto, Y.; Kato, T.; Ogata, H.; Fujimoto, N.; Kobayashi, Y.; et al. Murine osteoclasts secrete serine protease HtrA1 capable of degrading osteoprotegerin in the bone microenvironment. Commun. Biol. 2019, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Miyamoto, Y.; Yoshimura, K.; Yamada, A.; Takami, M.; Suzawa, T.; Hoshino, M.; Imamura, T.; Akiyama, C.; Yasuhara, R.; et al. Porphyromonas gingivalis-derived lysine gingipain enhances osteoclast differentiation induced by tumor necrosis factor-alpha and interleukin-1 beta but suppresses that by interleukin-17A: Importance of proteolytic degradation of osteoprotegerin by lysine gingipain. J. Biol. Chem. 2014, 289, 15621–15630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Xie, M.; Xie, Y.; Mei, F.; Lu, X.; Li, X.; Chen, L. The roles of osteocytes in alveolar bone destruction in periodontitis. J. Transl. Med. 2020, 18, 479. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Bonewald, L.F. Osteocytes as dynamic multifunctional cells. Ann. N. Y. Acad. Sci. 2007, 1116, 281–290. [Google Scholar] [CrossRef]
- Bellido, T. Osteocyte-driven bone remodeling. Calcif. Tissue Int. 2014, 94, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Miner. Metab. 2019, 37, 9–17. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef] [Green Version]
- Cassuto, J.; Folestad, A.; Göthlin, J.; Malchau, H.; Kärrholm, J. The key role of proinflammatory cytokines, matrix proteins, RANKL/OPG and Wnt/beta-catenin in bone healing of hip arthroplasty patients. Bone 2018, 107, 66–77. [Google Scholar] [CrossRef]
- Harris, S.E.; MacDougall, M.; Horn, D.; Woodruff, K.; Zimmer, S.N.; Rebel, V.I.; Fajardo, R.; Feng, J.Q.; Gluhak-Heinrich, J.; Harris, M.A.; et al. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone 2012, 50, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.L.; Sharma, R.; Woodruff, K.; Horn, D.; Harris, S.E.; Gorin, Y.; Lee, D.Y.; Hua, R.; Gu, S.; Fajardo, R.J.; et al. CSF-1 in osteocytes inhibits Nox4-mediated oxidative stress and promotes normal bone homeostasis. JBMR Plus 2020, 4, e10080. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, J.K.; Figliomeni, L.; Huang, L.; Pavlos, N.J.; Rogers, M.; Tan, A.; Price, P.; Zheng, M.H. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int. J. Mol. Med. 2003, 11, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [Green Version]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, E.; Kido, J.I.; Takagi, R.; Inagaki, Y.; Naruishi, K.; Nagata, T.; Yumoto, H. Advanced glycation end-product 2 and Porphyromonas gingivalis lipopolysaccharide increase sclerostin expression in mouse osteocyte-like cells. Bone 2019, 122, 22–30. [Google Scholar] [CrossRef]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 Receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to upregulate RANKL gene expression in MLO-Y4 cells. Cell Biol. Int. 2017, 41, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-alpha directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Hayashi, M.; Hayashi, N.; Aoki, S.; Suzuki, H. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J. Bone Miner. Res. 2013, 28, 1936–1949. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Matsunaga, A.; Ono, T.; Hayashi, M.; Takayanagi, H.; Moriyama, K.; Nakashima, T. Osteocyte regulation of orthodontic force-mediated tooth movement via RANKL expression. Sci. Rep. 2017, 7, 8753. [Google Scholar] [CrossRef]
- Li, W.; Zhao, J.; Sun, W.; Wang, H.; Pan, Y.; Wang, L.; Zhang, W.B. Osteocytes promote osteoclastogenesis via autophagy-mediated RANKL secretion under mechanical compressive force. Arch. Biochem. Biophys. 2020, 694, 108594. [Google Scholar] [CrossRef]
- Graves, D.T.; Alshabab, A.; Albiero, M.L.; Mattos, M.; Corrêa, J.D.; Chen, S.; Yang, Y. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J. Clin. Periodontol. 2018, 45, 285–292. [Google Scholar] [CrossRef]
- Jun, H.K.; Jung, Y.J.; Choi, B.K. Treponema denticola, Porphyromonas gingivalis, and Tannerella forsythia induce cell death and release of endogenous danger signals. Arch. Oral Biol. 2017, 73, 72–78. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features [Review]. World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Jilka, R.L.; Noble, B.; Weinstein, R.S. Osteocyte apoptosis. Bone 2013, 54, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Zhang, F.; Wu, J.; Xu, N.; Liang, M. Gingipains disrupt F-actin and cause osteoblast apoptosis via integrin beta1. J. Periodontal Res. 2018, 53, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, S.; Pathak, J.L. Pro-inflammatory cytokines and osteocytes. Curr. Osteoporos. Rep. 2019, 17, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell … and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose-John, S. The soluble interleukin 6 receptor: Advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.Y.; Simmons, C.A.; You, L. Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone 2012, 50, 104–110. [Google Scholar] [CrossRef]
- McCutcheon, S.; Majeska, R.J.; Spray, D.C.; Schaffler, M.B.; Vazquez, M. Apoptotic osteocytes induce RANKL production in bystanders via purinergic signaling and activation of pannexin channels. J. Bone Miner. Res. 2020, 35, 966–977. [Google Scholar] [CrossRef]
- Komori, T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. [Google Scholar] [CrossRef] [Green Version]
- Al-Dujaili, S.A.; Lau, E.; Al-Dujaili, H.; Tsang, K.; Guenther, A.; You, L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 2011, 112, 2412–2423. [Google Scholar] [CrossRef]
- Algate, K.; Haynes, D.R.; Bartold, P.M.; Crotti, T.N.; Cantley, M.D. The effects of tumour necrosis factor-alpha on bone cells involved in periodontal alveolar bone loss; Osteoclasts, osteoblasts and osteocytes. J. Periodontal Res. 2016, 51, 549–566. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. DEL-1-regulated immune plasticity and inflammatory disorders. Trends Mol. Med. 2019, 25, 444–459. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyler-Yaniv, A.; Oyler-Yaniv, J.; Whitlock, B.M.; Liu, Z.; Germain, R.N.; Huse, M.; Altan-Bonnet, G.; Krichevsky, O. A tunable diffusion-consumption mechanism of cytokine propagation enables plasticity in cell-to-cell communication in the immune system. Immunity 2017, 46, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Hasturk, H.; Lambris, J.D. C3-targeted therapy in periodontal disease: Moving closer to the clinic. Trends Immunol. 2021, 42, 856–864. [Google Scholar] [CrossRef]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Du, J.; Li, M. Functions of periostin in dental tissues and its role in periodontal tissues’ regeneration. Cell. Mol. Life Sci. 2017, 74, 4279–4286. [Google Scholar] [CrossRef]
- Matsuzawa, M.; Arai, C.; Nomura, Y.; Murata, T.; Yamakoshi, Y.; Oida, S.; Hanada, N.; Nakamura, Y. Periostin of human periodontal ligament fibroblasts promotes migration of human mesenchymal stem cell through the alphavbeta3 integrin/FAK/PI3K/Akt pathway. J. Periodontal Res. 2015, 50, 855–863. [Google Scholar] [CrossRef]
- Padial-Molina, M.; Volk, S.L.; Rios, H.F. Periostin increases migration and proliferation of human periodontal ligament fibroblasts challenged by tumor necrosis factor-alpha and Porphyromonas gingivalis lipopolysaccharides. J. Periodontal Res. 2014, 49, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Rani, S.; Barbe, M.F.; Barr, A.E.; Litivn, J. Role of TNF alpha and PLF in bone remodeling in a rat model of repetitive reaching and grasping. J. Cell. Physiol. 2010, 225, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.L. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 2012, 59, 464–471. [Google Scholar] [CrossRef]
- Sun, H.X.; Lu, N.; Liu, D.M.; Zhao, L.; Sun, L.H.; Zhao, H.Y.; Liu, J.M.; Tao, B. The bone-preserving effects of exendin-4 in ovariectomized rats. Endocrine 2016, 51, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, R.; Zhang, F.; Ding, M.; Wang, P. Exendin-4 relieves the inhibitory effects of high glucose on the proliferation and osteoblastic differentiation of periodontal ligament stem cells. Arch. Oral Biol. 2018, 91, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zheng, J.; Zheng, T.; Wang, P. Exendin-4 regulates Wnt and NF-κB signaling in lipopolysaccharide-induced human periodontal ligament stem cells to promote osteogenic differentiation. Int. Immunopharmacol. 2019, 75, 105801. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, S.K.; Jo, K.J.; Song, D.Y.; Lim, D.M.; Park, K.Y.; Bonewald, L.F.; Kim, B.J. Exendin-4 increases bone mineral density in type 2 diabetic OLETF rats potentially through the down-regulation of SOST/Sclerostin in osteocytes. Life Sci. 2013, 92, 533–540. [Google Scholar] [CrossRef]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.H.; Liang, S.; Ciero, P.A.; Krauss, J.L.; Li, F.; Rauner, M.; et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.Y.; Chavakis, E.; Czabanka, M.A.; Langer, H.F.; Fraemohs, L.; Economopoulou, M.; Kundu, R.K.; Orlandi, A.; Zheng, Y.Y.; Prieto, D.A.; et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 2008, 322, 1101–1104. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Colamatteo, A.; Kalafati, L.; Kajikawa, T.; Wang, H.; Lim, J.H.; Bdeir, K.; Chung, K.J.; Yu, X.; Fusco, C.; et al. The DEL-1/β3 integrin axis promotes regulatory T cell responses during inflammation resolution. J. Clin. Investig. 2020, 130, 6261–6277. [Google Scholar] [CrossRef]
- Yuh, D.Y.; Maekawa, T.; Li, X.; Kajikawa, T.; Bdeir, K.; Chavakis, T.; Hajishengallis, G. The secreted protein DEL-1 activates a β3 integrin-FAK-ERK1/2-RUNX2 pathway and promotes osteogenic differentiation and bone regeneration. J. Biol. Chem. 2020, 295, 7261–7273. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Chen, L.S.; Singh, R.P.; Kourtzelis, I.; Economopoulou, M.; Kajikawa, T.; Troullinaki, M.; Ziogas, A.; Ruppova, K.; Hosur, K.; et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J. Clin. Investig. 2017, 127, 3624–3639. [Google Scholar] [CrossRef]
- Tamura, H.; Maekawa, T.; Domon, H.; Hiyoshi, T.; Hirayama, S.; Isono, T.; Sasagawa, K.; Yonezawa, D.; Takahashi, N.; Oda, M.; et al. Effects of erythromycin on osteoclasts and bone resorption via DEL-1 induction in mice. Antibiotics 2021, 10, 312. [Google Scholar] [CrossRef]
- Maekawa, T.; Tamura, H.; Domon, H.; Hiyoshi, T.; Isono, T.; Yonezawa, D.; Hayashi, N.; Takahashi, N.; Tabeta, K.; Maeda, T.; et al. Erythromycin inhibits neutrophilic inflammation and mucosal disease by upregulating DEL-1. JCI Insight 2020, 5, e136706. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Hosur, K.; Abe, T.; Kantarci, A.; Ziogas, A.; Wang, B.; Van Dyke, T.E.; Chavakis, T.; Hajishengallis, G. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3Beta-C/EBPbeta pathway. Nat. Commun. 2015, 6, 8272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Maekawa, T.; Abe, T.; Hajishengallis, E.; Hosur, K.; Pyaram, K.; Mitroulis, I.; Chavakis, T.; Hajishengallis, G. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci. Transl. Med. 2015, 7, 307ra155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Chavakis, T. DEL-1: A potential therapeutic target in inflammatory and autoimmune disease? Expert Rev. Clin. Immunol. 2021, 17, 549–552. [Google Scholar] [CrossRef]
- Tamura, H.; Maekawa, T.; Domon, H.; Hiyoshi, T.; Yonezawa, D.; Nagai, K.; Ochiai, A.; Taniguchi, M.; Tabeta, K.; Maeda, T.; et al. Peptides from rice endosperm protein restrain periodontal bone loss in mouse model of periodontitis. Arch. Oral Biol. 2019, 98, 132–139. [Google Scholar] [CrossRef]
- Toker, H.; Balci Yuce, H.; Lektemur Alpan, A.; Gevrek, F.; Elmastas, M. Morphometric and histopathological evaluation of the effect of grape seed proanthocyanidin on alveolar bone loss in experimental diabetes and periodontitis. J. Periodontal Res. 2018, 53, 478–486. [Google Scholar] [CrossRef]
- Guzmán-Gómez, O.; García-Rodríguez, R.V.; Quevedo-Corona, L.; Pérez-Pastén-Borja, R.; Rivero-Ramírez, N.L.; Ríos-Castro, E.; Pérez-Gutiérrez, S.; Pérez-Ramos, J.; Chamorro-Cevallos, G.A. Amelioration of ethanol-induced gastric ulcers in rats pretreated with phycobiliproteins of Arthrospira (Spirulina) maxima. Nutrients 2018, 10, 763. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Moon, J.H.; Park, S.C.; Jang, Y.P.; Choung, S.Y. Spirulina maxima reduces inflammation and alveolar bone loss in Porphyromonas gingivalis-induced periodontitis. Phytomedicine 2021, 81, 153420. [Google Scholar] [CrossRef]
- Kimura, F.; Miyazawa, K.; Hamamura, K.; Tabuchi, M.; Sato, T.; Asano, Y.; Kako, S.; Aoki, Y.; Sugita, Y.; Maeda, H.; et al. Suppression of alveolar bone resorption by salubrinal in a mouse model of periodontal disease. Life Sci. 2021, 284, 119938. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, Z.; Lu, Y. Preparative separation of bioactive constitutes from Zanthoxylum planispinum using linear gradient counter-current chromatography. J. Sep. Sci. 2015, 38, 3735–3742. [Google Scholar] [CrossRef]
- Kim, M.H.; Lee, H.J.; Park, J.C.; Hong, J.; Yang, W.M. Zanthoxylum piperitum reversed alveolar bone loss of periodontitis via regulation of bone remodeling-related factors. J. Ethnopharmacol. 2017, 195, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Hajishengallis, G.; Curtis, M.A. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 2012, 91, 816–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feres, M.; Retamal-Valdes, B.; Gonçalves, C.; Cristina Figueiredo, L.; Teles, F. Did omics change periodontal therapy? Periodontology 2000 2021, 85, 182–209. [Google Scholar] [CrossRef] [PubMed]
- Surana, N.K.; Kasper, D.L. Moving beyond microbiome-wide associations to causal microbe identification. Nature 2017, 552, 244–247. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirisereephap, K.; Maekawa, T.; Tamura, H.; Hiyoshi, T.; Domon, H.; Isono, T.; Terao, Y.; Maeda, T.; Tabeta, K. Osteoimmunology in Periodontitis: Local Proteins and Compounds to Alleviate Periodontitis. Int. J. Mol. Sci. 2022, 23, 5540. https://doi.org/10.3390/ijms23105540
Sirisereephap K, Maekawa T, Tamura H, Hiyoshi T, Domon H, Isono T, Terao Y, Maeda T, Tabeta K. Osteoimmunology in Periodontitis: Local Proteins and Compounds to Alleviate Periodontitis. International Journal of Molecular Sciences. 2022; 23(10):5540. https://doi.org/10.3390/ijms23105540
Chicago/Turabian StyleSirisereephap, Kridtapat, Tomoki Maekawa, Hikaru Tamura, Takumi Hiyoshi, Hisanori Domon, Toshihito Isono, Yutaka Terao, Takeyasu Maeda, and Koichi Tabeta. 2022. "Osteoimmunology in Periodontitis: Local Proteins and Compounds to Alleviate Periodontitis" International Journal of Molecular Sciences 23, no. 10: 5540. https://doi.org/10.3390/ijms23105540
APA StyleSirisereephap, K., Maekawa, T., Tamura, H., Hiyoshi, T., Domon, H., Isono, T., Terao, Y., Maeda, T., & Tabeta, K. (2022). Osteoimmunology in Periodontitis: Local Proteins and Compounds to Alleviate Periodontitis. International Journal of Molecular Sciences, 23(10), 5540. https://doi.org/10.3390/ijms23105540