
Regulation of Translation, Translocation, and Degradation of Proteins at the Membrane of the Endoplasmic Reticulum


Abstract
:1. Introduction
2. Control of Protein Synthesis by ERdj1, ERdj2, and ERdj6
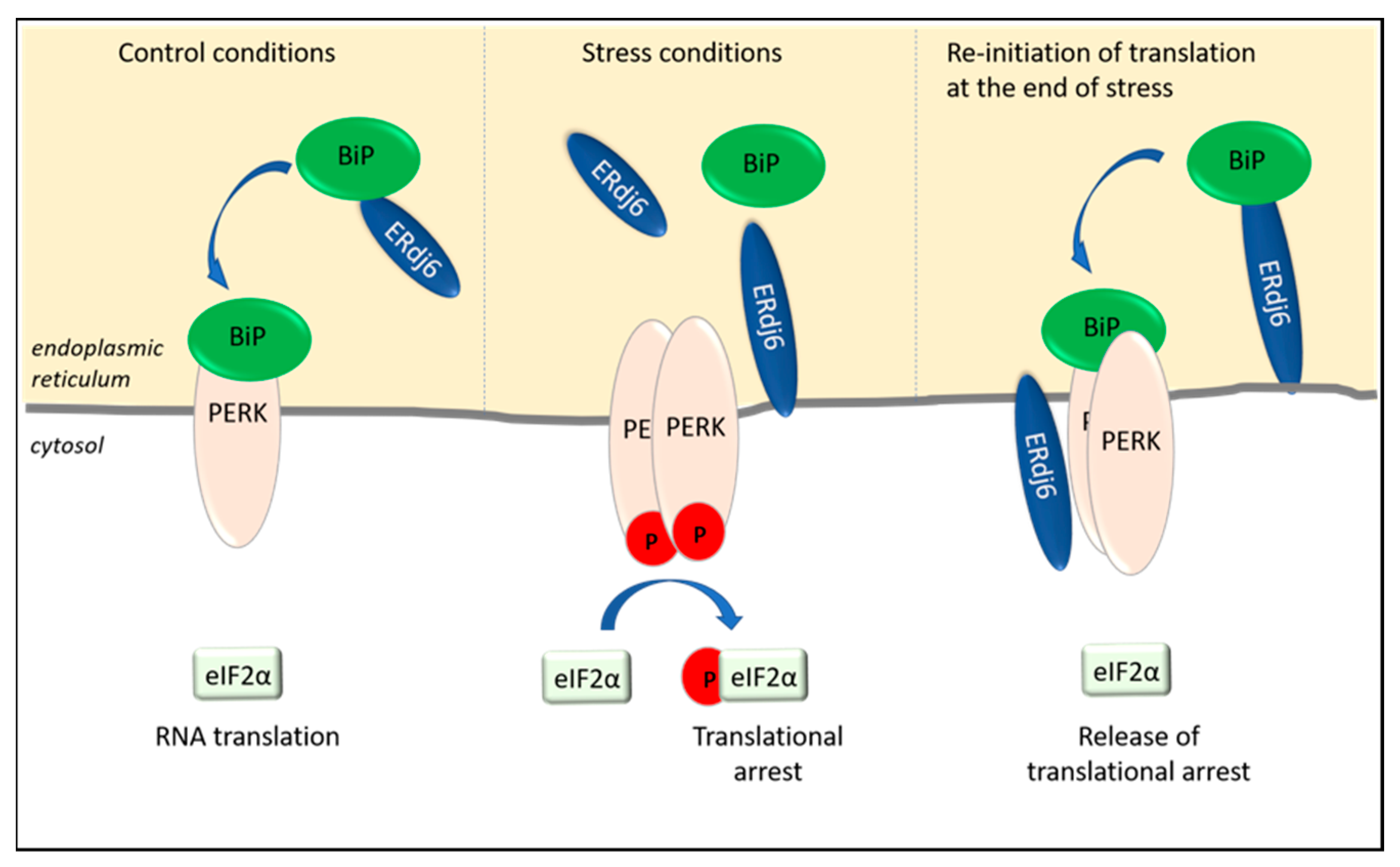
2.1. Translational Arrest Is Regulated by ERdj1 and ERdj2
2.2. ERdj6 Reinitiates Translation at the End of a Stress Period
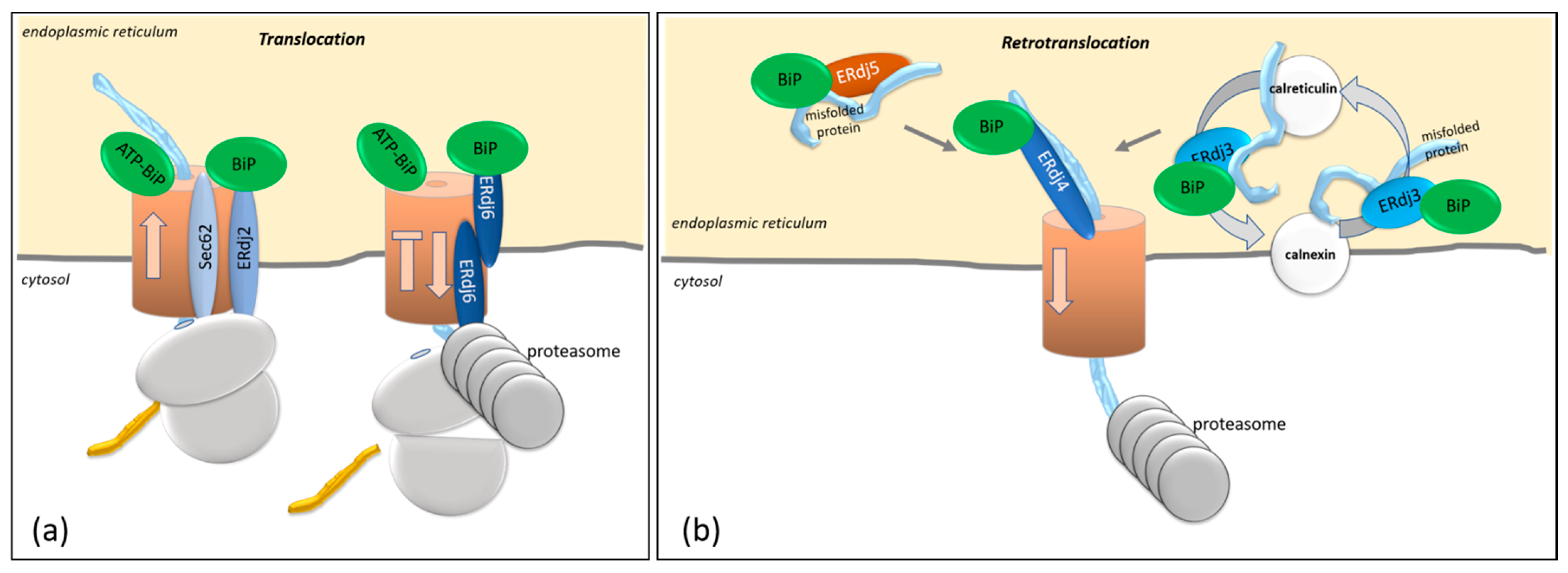
3. Co- and Posttranslational Translocation in Mammalian Cells by ERdj2 and ERdj6
3.1. Features Determining ERdj2-Mediated Cotranslational Translocation of Proteins
3.2. ERdj6-Mediated Cotranslational Degradation
4. Protein Degradation Controlled by ERdj3, ERdj4, and ERdj5
4.1. Processing of Target Proteins: Futile Folding Versus Degradation of Target Proteins
- Alzheimer Disease
- α1-Antitrypsin Deficiency
- Diabetes
- Gaucher’s Disease
- Cystic Fibrosis
- Interstitial Lung Disease
4.2. Features Determining the Fate of Target Proteins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Read, A.; Schroder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef]
- Karagoz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef] [Green Version]
- Radanovic, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Yun, C.; Fisher, E.A.; Kreglinger, N.; Kreibich, G.; Oyadomari, M.; Harding, H.P.; Goodman, A.G.; Harant, H.; Garrison, J.L.; et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 2006, 126, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Miesbauer, M.; Pfeiffer, N.V.; Rambold, A.S.; Muller, V.; Kiachopoulos, S.; Winklhofer, K.F.; Tatzelt, J. alpha-Helical domains promote translocation of intrinsically disordered polypeptides into the endoplasmic reticulum. J. Biol. Chem. 2009, 284, 24384–24393. [Google Scholar] [CrossRef] [Green Version]
- Hamman, B.D.; Hendershot, L.M.; Johnson, A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 1998, 92, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Haigh, N.G.; Johnson, A.E. A new role for BiP: Closing the aqueous translocon pore during protein integration into the ER membrane. J. Cell Biol. 2002, 156, 261–270. [Google Scholar] [CrossRef]
- Alder, N.N.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef]
- Ziska, A.; Tatzelt, J.; Dudek, J.; Paton, A.W.; Paton, J.C.; Zimmermann, R.; Hassdenteufel, S. The signal peptide plus a cluster of positive charges in prion protein dictate chaperone-mediated Sec61 channel gating. Biol. Open 2019, 8, bio040691. [Google Scholar] [CrossRef] [Green Version]
- Hendrick, J.P.; Hartl, F.U. The role of molecular chaperones in protein folding. FASEB J. 1995, 9, 1559–1569. [Google Scholar] [CrossRef]
- Rosam, M.; Krader, D.; Nickels, C.; Hochmair, J.; Back, K.C.; Agam, G.; Barth, A.; Zeymer, C.; Hendrix, J.; Schneider, M.; et al. Bap (Sil1) regulates the molecular chaperone BiP by coupling release of nucleotide and substrate. Nat. Struct. Mol. Biol. 2018, 25, 90–100. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef] [Green Version]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Matlack, K.E.; Misselwitz, B.; Plath, K.; Rapoport, T.A. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 1999, 97, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Tomkiewicz, D.; Nouwen, N.; Driessen, A.J. Pushing, pulling and trapping—Modes of motor protein supported protein translocation. FEBS Lett. 2007, 581, 2820–2828. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J.; Volkmer, J.; Bies, C.; Guth, S.; Muller, A.; Lerner, M.; Feick, P.; Schafer, K.H.; Morgenstern, E.; Hennessy, F.; et al. A novel type of co-chaperone mediates transmembrane recruitment of DnaK-like chaperones to ribosomes. EMBO J. 2002, 21, 2958–2967. [Google Scholar] [CrossRef] [Green Version]
- Blau, M.; Mullapudi, S.; Becker, T.; Dudek, J.; Zimmermann, R.; Penczek, P.A.; Beckmann, R. ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat. Struct. Mol. Biol. 2005, 12, 1015–1016. [Google Scholar] [CrossRef]
- Dudek, J.; Greiner, M.; Muller, A.; Hendershot, L.M.; Kopsch, K.; Nastainczyk, W.; Zimmermann, R. ERj1p has a basic role in protein biogenesis at the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2005, 12, 1008–1014. [Google Scholar] [CrossRef]
- Benedix, J.; Lajoie, P.; Jaiswal, H.; Burgard, C.; Greiner, M.; Zimmermann, R.; Rospert, S.; Snapp, E.L.; Dudek, J. BiP modulates the affinity of its co-chaperone ERj1 for ribosomes. J. Biol. Chem. 2010, 285, 36427–36433. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.A.; Grau, H.; Kraft, R.; Kostka, S.; Prehn, S.; Kalies, K.U.; Hartmann, E. Mammalian Sec61 is associated with Sec62 and Sec63. J. Biol. Chem. 2000, 275, 14550–14557. [Google Scholar] [CrossRef] [Green Version]
- Daverkausen-Fischer, L.; Prols, F. Dual topology of co-chaperones at the membrane of the endoplasmic reticulum. Cell Death Discov. 2021, 7, 203. [Google Scholar] [CrossRef]
- Mary, C.; Scherrer, A.; Huck, L.; Lakkaraju, A.K.; Thomas, Y.; Johnson, A.E.; Strub, K. Residues in SRP9/14 essential for elongation arrest activity of the signal recognition particle define a positively charged functional domain on one side of the protein. RNA 2010, 16, 969–979. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; de Escauriaza, M.D.; Lajoie, P.; Theis, M.; Jung, M.; Muller, A.; Burgard, C.; Greiner, M.; Snapp, E.L.; Dudek, J.; et al. Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 2010, 21, 691–703. [Google Scholar] [CrossRef] [Green Version]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Jeyaraman, K.; Yusoff, P.; Shenolikar, S. Phosphorylation at tyrosine 262 promotes GADD34 protein turnover. J. Biol. Chem. 2013, 288, 33146–33155. [Google Scholar] [CrossRef] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyedmers, J.; Lerner, M.; Bies, C.; Dudek, J.; Skowronek, M.H.; Haas, I.G.; Heim, N.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7214–7219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampofo, E.; Welker, S.; Jung, M.; Muller, L.; Greiner, M.; Zimmermann, R.; Montenarh, M. CK2 phosphorylation of human Sec63 regulates its interaction with Sec62. Biochim. Biophys. Acta 2013, 1830, 2938–2945. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Pfeffer, S.; Lee, P.H.; Cavalie, A.; Helms, V.; Forster, F.; Zimmermann, R. An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [Green Version]
- Van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, D.T.; Kang, S.W.; Goodman, A.G.; Garrison, J.L.; Taunton, J.; Katze, M.G.; Kaufman, R.J.; Hegde, R.S. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Petrova, K.; Oyadomari, S.; Hendershot, L.M.; Ron, D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008, 27, 2862–2872. [Google Scholar] [CrossRef] [Green Version]
- Schorr, S.; Klein, M.C.; Gamayun, I.; Melnyk, A.; Jung, M.; Schauble, N.; Wang, Q.; Hemmis, B.; Bochen, F.; Greiner, M.; et al. Co-chaperone Specificity in Gating of the Polypeptide Conducting Channel in the Membrane of the Human Endoplasmic Reticulum. J. Biol. Chem. 2015, 290, 18621–18635. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Sun, S.; Appathurai, S.; Sundaram, A.; Plumb, R.; Mariappan, M. A Molecular Mechanism for Turning Off IRE1alpha Signaling during Endoplasmic Reticulum Stress. Cell Rep. 2020, 33, 108563. [Google Scholar] [CrossRef]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 2017, 171, 1625–1637.e1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.; Powis, K.; High, S. Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2403–2409. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A.; Li, L.; Park, E. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 2017, 33, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.J.; Devaraneni, P.K.; Yang, Z.; David, L.L.; Skach, W.R. Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 2015, 58, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schauble, N.; Jalal, C.; Greiner, M.; Hassdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar]
- Johnson, N.; Hassdenteufel, S.; Theis, M.; Paton, A.W.; Paton, J.C.; Zimmermann, R.; High, S. The signal sequence influences post-translational ER translocation at distinct stages. PLoS ONE 2013, 8, e75394. [Google Scholar]
- Brambillasca, S.; Yabal, M.; Soffientini, P.; Stefanovic, S.; Makarow, M.; Hegde, R.S.; Borgese, N. Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J. 2005, 24, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Abell, B.M.; Jung, M.; Oliver, J.D.; Knight, B.C.; Tyedmers, J.; Zimmermann, R.; High, S. Tail-anchored and signal-anchored proteins utilize overlapping pathways during membrane insertion. J. Biol. Chem. 2003, 278, 5669–5678. [Google Scholar] [CrossRef] [Green Version]
- Gemmer, M.; Forster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340. [Google Scholar] [CrossRef]
- Schorr, S.; Nguyen, D.; Hassdenteufel, S.; Nagaraj, N.; Cavalie, A.; Greiner, M.; Weissgerber, P.; Loi, M.; Paton, A.W.; Paton, J.C.; et al. Identification of signal peptide features for substrate specificity in human Sec62/Sec63-dependent ER protein import. FEBS J. 2020, 287, 4612–4640. [Google Scholar] [CrossRef] [Green Version]
- Hassdenteufel, S.; Johnson, N.; Paton, A.W.; Paton, J.C.; High, S.; Zimmermann, R. Chaperone-Mediated Sec61 Channel Gating during ER Import of Small Precursor Proteins Overcomes Sec61 Inhibitor-Reinforced Energy Barrier. Cell Rep. 2018, 23, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.M.; Kim, J.; Menasche, B.L.; Sheppard, J.; Liu, X.; Tan, A.C.; Shen, J. Comparative Haploid Genetic Screens Reveal Divergent Pathways in the Biogenesis and Trafficking of Glycophosphatidylinositol-Anchored Proteins. Cell Rep. 2015, 11, 1727–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauwels, E.; Provinciael, B.; Camps, A.; Hartmann, E.; Vermeire, K. Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide. Int. J. Mol. Sci. 2022, 23, 584. [Google Scholar] [CrossRef]
- Shen, Y.; Hendershot, L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 2005, 16, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129812. [Google Scholar] [CrossRef]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Carvalho, P. Order through destruction: How ER-associated protein degradation contributes to organelle homeostasis. EMBO J. 2022, 41, e109845. [Google Scholar] [CrossRef]
- Kaiser, M.L.; Romisch, K. Proteasome 19S RP binding to the Sec61 channel plays a key role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef]
- Romisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, S.; Burress, H.; Taylor, M.; Nemec, K.N.; Ray, S.; Haslam, D.B.; Teter, K. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect. Immun. 2011, 79, 4739–4747. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.; Van Goor, K.E.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2011, 2, e00101. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Inoue, T.; Chen, G.; Tsai, B. The nucleotide exchange factors Grp170 and Sil1 induce cholera toxin release from BiP to enable retrotranslocation. Mol. Biol. Cell 2015, 26, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Dosey, A.; Herbstman, J.F.; Ravindran, M.S.; Skiniotis, G.; Tsai, B. ERdj5 Reductase Cooperates with Protein Disulfide Isomerase To Promote Simian Virus 40 Endoplasmic Reticulum Membrane Translocation. J. Virol. 2015, 89, 8897–8908. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Bridges, J.P.; Apsley, K.; Xu, Y.; Weaver, T.E. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 2008, 19, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.L.; Genereux, J.C.; Pankow, S.; Aerts, J.M.; Yates, J.R., 3rd; Kelly, J.W. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chem. Biol. 2014, 21, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, T.; Nakaya, T.; Araki, W.; Suzuki, K.; Suzuki, T.; Mizushima, T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem. J. 2007, 402, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Khodayari, N.; Marek, G.; Lu, Y.; Krotova, K.; Wang, R.L.; Brantly, M. Erdj3 Has an Essential Role for Z Variant Alpha-1-Antitrypsin Degradation. J. Cell. Biochem. 2017, 118, 3090–3101. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Cao, D.; Gong, J. The endoplasmic reticulum co-chaperone ERdj3/DNAJB11 promotes hepatocellular carcinoma progression through suppressing AATZ degradation. Future Oncol. 2018, 14, 3001–3013. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Dong, M.; Apsley, K.S.; Martin, E.P.; Na, C.L.; Sitaraman, S.; Weaver, T.E. Deficiency of the BiP co-chaperone ERdj4 causes constitutive endoplasmic reticulum stress and metabolic defects. Mol. Biol. Cell 2014, 25, 431–440. [Google Scholar] [CrossRef]
- Lai, C.W.; Otero, J.H.; Hendershot, L.M.; Snapp, E. ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J. Biol. Chem. 2012, 287, 7969–7978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Arora, K.; Mun, K.S.; Yang, F.; Moon, C.; Yarlagadda, S.; Jegga, A.; Weaver, T.; Naren, A.P. Targeting DNAJB9, a novel ER luminal co-chaperone, to rescue DeltaF508-CFTR. Sci. Rep. 2019, 9, 9808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, T.M.; Kolb, A.R.; Boyd, C.R.; Kleyman, T.R.; Brodsky, J.L. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 2010, 21, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Karatas, E.; Bouchecareilh, M. Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int. J. Mol. Sci. 2020, 21, 1493. [Google Scholar] [CrossRef] [Green Version]
- Khodayari, N.; Oshins, R.; Alli, A.A.; Tuna, K.M.; Holliday, L.S.; Krotova, K.; Brantly, M. Modulation of calreticulin expression reveals a novel exosome-mediated mechanism of Z variant alpha1-antitrypsin disposal. J. Biol. Chem. 2019, 294, 6240–6252. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Zuber, C. Quality control of glycoprotein folding and ERAD: The role of N-glycan handling, EDEM1 and OS-9. Histochem. Cell Biol. 2017, 147, 269–284. [Google Scholar] [CrossRef]
- Brennan, L.C.; O’Sullivan, A.; MacLoughlin, R. Cellular Therapy for the Treatment of Paediatric Respiratory Disease. Int. J. Mol. Sci. 2021, 22, 8906. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef] [PubMed]
- Bies, C.; Blum, R.; Dudek, J.; Nastainczyk, W.; Oberhauser, S.; Jung, M.; Zimmermann, R. Characterization of pancreatic ERj3p, a homolog of yeast DnaJ-like protein Scj1p. Biol. Chem. 2004, 385, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Suno, R.; Iemura, S.I.; Natsume, T.; Wada, I.; Hosokawa, N. Endoplasmic reticulum proteins SDF2 and SDF2L1 act as components of the BiP chaperone cycle to prevent protein aggregation. Genes Cells Devoted Mol. Cell. Mech. 2017, 22, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, K.; Wada, I.; Hosokawa, N. SDF2-like protein 1 (SDF2L1) regulates the endoplasmic reticulum localization and chaperone activity of ERdj3 protein. J. Biol. Chem. 2019, 294, 19335–19348. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E., 3rd; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef]
- Behnke, J.; Mann, M.J.; Scruggs, F.L.; Feige, M.J.; Hendershot, L.M. Members of the Hsp70 Family Recognize Distinct Types of Sequences to Execute ER Quality Control. Mol. Cell 2016, 63, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Cormier, J.H.; Tamura, T.; Sunryd, J.C.; Hebert, D.N. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 2009, 34, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Meunier, L.; Hendershot, L.M. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 2002, 277, 15947–15956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, M.; Maegawa, K.; Suzuki, M.; Ushioda, R.; Araki, K.; Matsumoto, Y.; Hoseki, J.; Nagata, K.; Inaba, K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 2011, 41, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Cunnea, P.M.; Miranda-Vizuete, A.; Bertoli, G.; Simmen, T.; Damdimopoulos, A.E.; Hermann, S.; Leinonen, S.; Huikko, M.P.; Gustafsson, J.A.; Sitia, R.; et al. ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J. Biol. Chem. 2003, 278, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, A.; Tokuda, M.; Akai, R.; Kohno, K.; Iwawaki, T. Positive contribution of ERdj5/JPDI to endoplasmic reticulum protein quality control in the salivary gland. Biochem. J. 2009, 425, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Cormier, J.H.; Hebert, D.N. Characterization of early EDEM1 protein maturation events and their functional implications. J. Biol. Chem. 2011, 286, 24906–24915. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Inoue, T.; Banks, L.; Tsai, B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol. Biol. Cell 2013, 24, 785–795. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Anton, V. Mitochondrial Surveillance by Cdc48/p97: MAD vs. Membrane Fusion. Int. J. Mol. Sci. 2020, 21, 6841. [Google Scholar] [CrossRef]
- Hosoda, A.; Kimata, Y.; Tsuru, A.; Kohno, K. JPDI, a novel endoplasmic reticulum-resident protein containing both a BiP-interacting J-domain and thioredoxin-like motifs. J. Biol. Chem. 2003, 278, 2669–2676. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Disease | Mutated Protein | Co-Chaperone | Function | Reference |
|---|---|---|---|---|
| Alzheimer’s disease | Amyloid β-protein | ERdj3 ERdj4 | Chaperoning Chaperoning | [69] [69] |
| α1-Antitrypsin disease (AATD) | α1-Antitrypsin | ERdj3 | Chaperoning | [70,71,72,73] |
| Diabetes | Insulin | ERdj4 ERdj4 ERdj5 | Chaperoning ERAD ERAD | [74] [67,75] [67] |
| Gaucher’s disease | β-Glucocerebroside | ERdj3 | ERAD | [68] |
| Cystic fibrosis | Transmembrane conductive regulator protein | |||
| Chloride channel | ERdj3 ERdj4 | ? ERAD | [76] [76] | |
| ENaC channel | ERdj3 ERdj4 | ERAD ERAD | [77] [77] | |
| Interstitial lung disease (ILD) | Surfactant protein-C | ERdj4 ERdj5 | ERAD ERAD | [67] [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daverkausen-Fischer, L.; Draga, M.; Pröls, F. Regulation of Translation, Translocation, and Degradation of Proteins at the Membrane of the Endoplasmic Reticulum. Int. J. Mol. Sci. 2022, 23, 5576. https://doi.org/10.3390/ijms23105576
Daverkausen-Fischer L, Draga M, Pröls F. Regulation of Translation, Translocation, and Degradation of Proteins at the Membrane of the Endoplasmic Reticulum. International Journal of Molecular Sciences. 2022; 23(10):5576. https://doi.org/10.3390/ijms23105576
Chicago/Turabian StyleDaverkausen-Fischer, Lea, Margarethe Draga, and Felicitas Pröls. 2022. "Regulation of Translation, Translocation, and Degradation of Proteins at the Membrane of the Endoplasmic Reticulum" International Journal of Molecular Sciences 23, no. 10: 5576. https://doi.org/10.3390/ijms23105576