
Endothelial Dysfunction Induced by Extracellular Neutrophil Traps Plays Important Role in the Occurrence and Treatment of Extracellular Neutrophil Traps-Related Disease

Abstract
:1. Introduction
2. The Relationship between NETs and ED
2.1. What Is Neutrophil Extracellular Traps
2.2. The Formation of NETs
2.3. Endothelial Dysfunction
2.4. The Linkages of NETs and ED
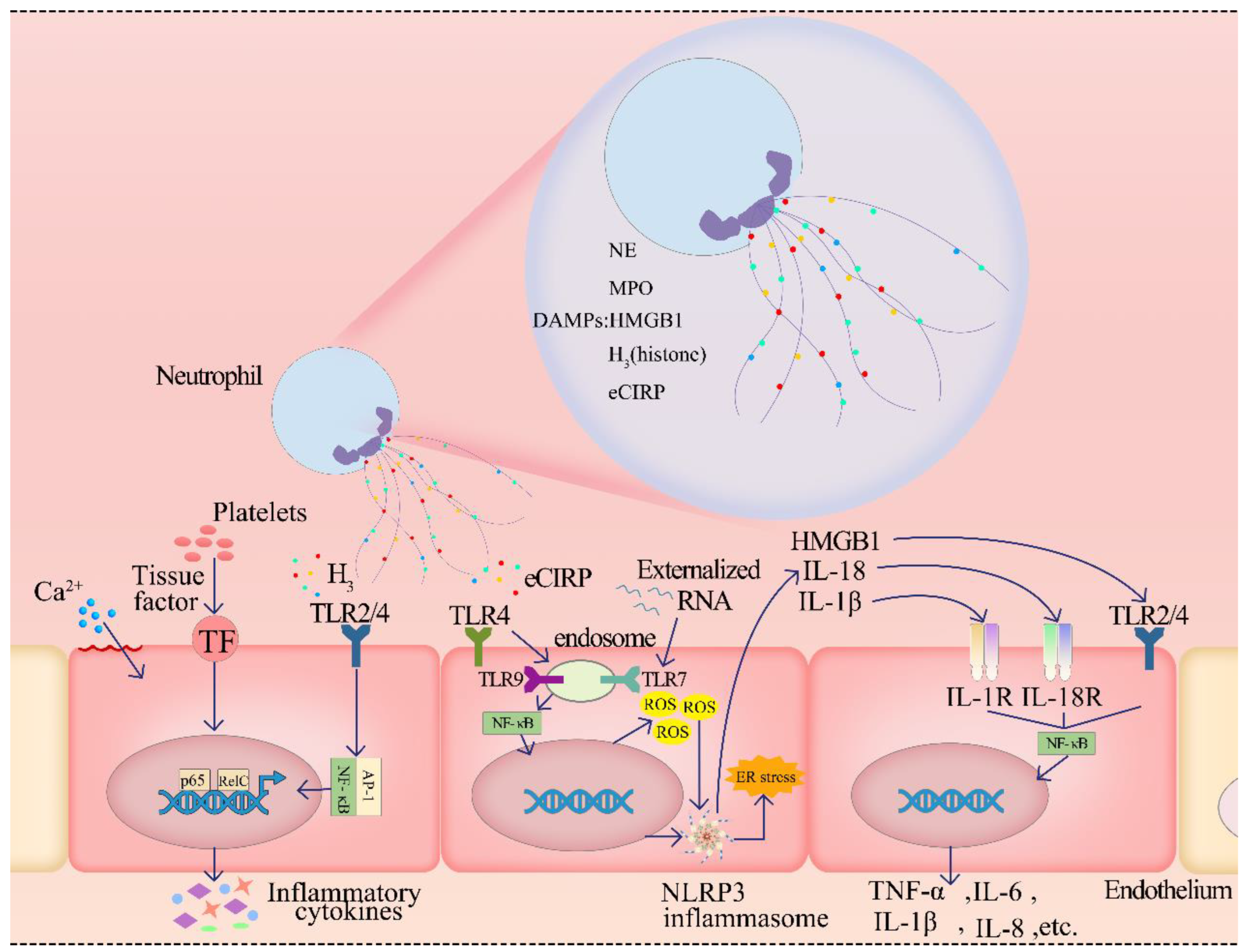
2.4.1. The Components of NETs Cause ED by Damage-Associated Molecular Patterns
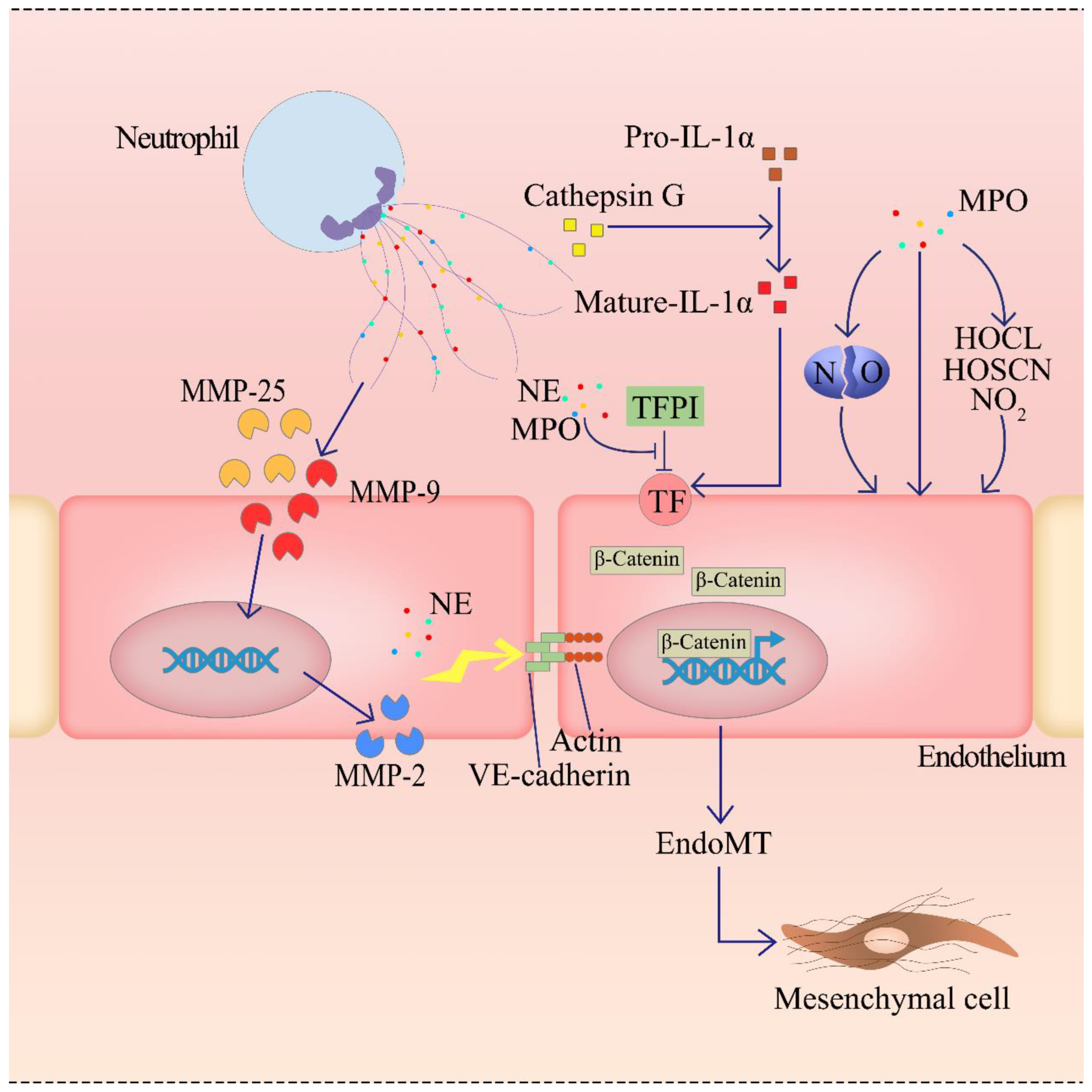
2.4.2. Enzymes of NETs Can Cause the ED
2.4.3. NETs Induce the ED by Activating the Complement System
3. Diseases and Treatment
3.1. Cardiovascular Disease
3.2. Autoimmune Diseases
3.2.1. ANCA-Associated Vasculitis
3.2.2. Systemic Lupus Erythematosus
3.3. Sepsis
3.4. Others
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Köstlin-Gille, N.; Hillmer, S.; Braun, M.; Kranig, S.A.; Dietz, S.; Krause, C.; Rühle, J.; Frommhold, D.; Pöschl, J.; et al. Gut Microbiota-Derived Small Extracellular Vesicles Endorse Memory-like Inflammatory Responses in Murine Neutrophils. Biomedicines 2022, 10, 442. [Google Scholar] [CrossRef]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785. [Google Scholar] [CrossRef]
- Moorlag, S.J.C.F.M.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef]
- Lajqi, T.; Braun, M.; Kranig, S.A.; Frommhold, D.; Pöschl, J.; Hudalla, H. LPS Induces Opposing Memory-like Inflammatory Responses in Mouse Bone Marrow Neutrophils. Int. J. Mol. Sci. 2021, 22, 9803. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.-H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef]
- Damgaard, D.; Bjørn, M.E.; Steffensen, M.A.; Pruijn, G.J.M.; Nielsen, C.H. Reduced glutathione as a physiological co-activator in the activation of peptidylarginine deiminase. Arthritis Res. 2016, 18, 102. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Radstake, T.R.D.; van der Heijden, A.; van Mansum, M.A.M.; Dieteren, C.; de Rooij, D.J.; Barrera, P.; Zendman, A.J.W.; van Venrooij, W.J. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bawadekar, M.; Shim, D.; Johnson, C.J.; Warner, T.F.; Rebernick, R.; Damgaard, D.; Nielsen, C.H.; Pruijn, G.J.M.; Nett, J.E.; Shelef, M.A. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J. Autoimmun. 2017, 80, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Kobayashi, S.D.; Quinn, M.T.; Deleo, F.R. A NET Outcome. Front. Immunol. 2012, 3, 365. [Google Scholar] [CrossRef] [Green Version]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Vallance, P. Importance of asymmetrical dimethylarginine in cardiovascular risk. Lancet 2001, 358, 2096–2097. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arter. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Dhananjayan, R.; Koundinya, K.S.S.; Malati, T.; Kutala, V.K. Endothelial Dysfunction in Type 2 Diabetes Mellitus. Indian J. Clin. Biochem. 2016, 31, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: Endothelial activation and dysfunction. Cell. Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Anderson, T.J. Fundamentals of endothelial function for the clinical cardiologist. Circulation 2002, 105, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmashankar, K.; Widlansky, M.E. Vascular endothelial function and hypertension: Insights and directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Land, W. Allograft injury mediated by reactive oxygen species: From conserved proteins of Drosophila to acute and chronic rejection of human transplants. Part II: Role of reactive oxygen species in the induction of the heat shock response as a regulator of innate. Transplant. Rev. 2003, 17, 31–44. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Liu, J.; Lv, B.; Chen, F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb. Res. 2016, 137, 211–218. [Google Scholar] [CrossRef]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence from Vascular Pathology in Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Alhamdi, Y.; Abrams, S.T.; Cheng, Z.; Jing, S.; Su, D.; Liu, Z.; Lane, S.; Welters, I.; Wang, G.; Toh, C.-H. Circulating Histones Are Major Mediators of Cardiac Injury in Patients with Sepsis. Crit. Care Med. 2015, 43, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.A. An endogenous factor mediates shock-induced injury. Nat. Med. 2013, 19, 1368–1369. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Yao, M.; Liu, Y.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin. Sci. 2019, 133, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Khan, M.M.; Yang, W.L.; Brenner, M.; Bolognese, A.C.; Wang, P. Cold-inducible RNA-binding protein (CIRP) causes sepsis-associated acute lung injury via induction of endoplasmic reticulum stress. Sci. Rep. 2017, 7, 41363. [Google Scholar] [CrossRef]
- Yang, W.L.; Sharma, A.; Wang, Z.; Li, Z.; Fan, J.; Wang, P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 2016, 6, 26571. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sun, B. Neutrophil pyroptosis: New perspectives on sepsis. Cell. Mol. Life Sci. 2019, 76, 2031–2042. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.Y.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Van Avondt, K.; Maegdefessel, L.; Soehnlein, O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb. Haemost. 2019, 119, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ward, M.F.; Sama, A.E. Targeting HMGB1 in the treatment of sepsis. Expert Opin. Targets 2014, 18, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, J.; Salcedo, R.; Mivechi, N.F.; Trinchieri, G.; Horuzsko, A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012, 72, 3977–3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, L.P.; Wang, X.; Carlucci, P.M.; Torres-Ruiz, J.J.; Romo-Tena, J.; Sun, H.-W.; Hafner, M.; Kaplan, M.J. RNA Externalized by Neutrophil Extracellular Traps Promotes Inflammatory Pathways in Endothelial Cells. Arthritis Rheumatol. 2021, 73, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arter. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef]
- Maiocchi, S.L.; Ku, J.; Thai, T.; Chan, E.; Rees, M.D.; Thomas, S.R. Myeloperoxidase: A versatile mediator of endothelial dysfunction and therapeutic target during cardiovascular disease. Pharm. Ther. 2021, 221, 107711. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Kremer Hovinga, J.A.; Schatzberg, D.; Wagner, D.D.; Lämmle, B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood 2012, 120, 1157–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production through Interleukin-1α and Cathepsin G. Arter. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. The membrane attack complex as an inflammatory trigger. Immunobiology 2016, 221, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef] [Green Version]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, H.; Qu, M.; Nan, K.; Cao, H.; Cata, J.P.; Chen, W.; Miao, C. Review: The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front. Cell. Infect. Microbiol. 2021, 11, 653228. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular.r Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front. Immunol. 2017, 8, 928. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.A.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Hermida, N.; Balligand, J.-L. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: The role of statins. Antioxid. Redox Signal. 2014, 20, 1216–1237. [Google Scholar] [CrossRef]
- Verma, S.; Buchanan, M.R.; Anderson, T.J. Endothelial function testing as a biomarker of vascular disease. Circulation 2003, 108, 2054–2059. [Google Scholar] [CrossRef]
- Patiño-Trives, A.M.; Pérez-Sánchez, C.; Pérez-Sánchez, L.; Luque-Tévar, M.; Ábalos-Aguilera, M.C.; Alcaide-Ruggiero, L.; Arias-de la Rosa, I.; Román-Rodríguez, C.; Seguí, P.; Espinosa, M.; et al. Anti-dsDNA Antibodies Increase the Cardiovascular Risk in Systemic Lupus Erythematosus Promoting a Distinctive Immune and Vascular Activation. Arter. Thromb. Vasc. Biol. 2021, 41, 2417–2430. [Google Scholar] [CrossRef]
- Pérez-Sánchez, C.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gómez, Y.; Arias-de la Rosa, I.; Ábalos-Aguilera, M.C.; Rodriguez-Ariza, A.; Castro-Villegas, M.C.; Ortega-Castro, R.; Segui, P.; et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in Rheumatoid Arthritis patients. J. Autoimmun. 2017, 82, 31–40. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arter. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arter. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Martínez-Cuesta, M.Á.; Álvarez, Á. The Neutrophil Secretome as a Crucial Link between Inflammation and Thrombosis. Int. J. Mol. Sci. 2021, 22, 4170. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Kranzhöfer, R.; Clark, S.; Libby, P. Macrophage foam cells from experimental atheroma constitutively produce matrix-degrading proteinases. Proc. Natl. Acad. Sci. USA 1995, 92, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasterkamp, G.; den Ruijter, H.M.; Libby, P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat. Rev. Cardiol. 2017, 14, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Van Lammeren, G.W.; den Ruijter, H.M.; Vrijenhoek, J.E.P.; van der Laan, S.W.; Velema, E.; de Vries, J.-P.P.M.; de Kleijn, D.P.V.; Vink, A.; de Borst, G.J.; Moll, F.L.; et al. Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation 2014, 129, 2269–2276. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Totani, L.; Amore, C.; Di Santo, A.; Dell’Elba, G.; Piccoli, A.; Martelli, N.; Tenor, H.; Beume, R.; Evangelista, V. Roflumilast inhibits leukocyte-platelet interactions and prevents the prothrombotic functions of polymorphonuclear leukocytes and monocytes. J. Thromb. Haemost. 2016, 14, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharm. Exp. 2013, 345, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Hashiguchi, N.; Nagaoka, I.; Tabe, Y.; Kadota, K.; Sato, K. Heparins attenuated histone-mediated cytotoxicity in vitro and improved the survival in a rat model of histone-induced organ dysfunction. Intensive Care Med. Exp. 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J. Low Molecular Weight Heparins—A new tool to disetangle from the NETs. Pharm. Res. 2017, 123, 157. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Rovere-Querini, P.; D’Angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharm. Res. 2017, 123, 146–156. [Google Scholar] [CrossRef]
- Qiao, Y.; Jiang, J.; Zhang, Z.; Ma, X. Heparin reduces endothelial cell damage induced by neutrophil extracellular traps. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2017, 29, 342–346. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Andrassy, M.; Volz, H.C.; Igwe, J.C.; Funke, B.; Eichberger, S.N.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.T.; Lukic, I.K.; et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar] [CrossRef] [Green Version]
- Tadie, J.-M.; Bae, H.-B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.-F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zhang, Q.; Hou, W.; Yan, Z.; Chen, R.; Bonaroti, J.; Bansal, P.; Billiar, T.R.; Tsung, A.; Wang, Q.; et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014, 146, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handono, K.; Sidarta, Y.O.; Pradana, B.A.; Nugroho, R.A.; Hartono, I.A.; Kalim, H.; Endharti, A.T. Vitamin D prevents endothelial damage induced by increased neutrophil extracellular traps formation in patients with systemic lupus erythematosus. Acta Med. Indones. 2014, 46, 189–198. [Google Scholar]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Fetz, A.E.; Wallace, S.E.; Bowlin, G.L. Electrospun Polydioxanone Loaded with Chloroquine Modulates Template-Induced NET Release and Inflammatory Responses From Human Neutrophils. Front. Bioeng. Biotechnol. 2021, 9, 652055. [Google Scholar] [CrossRef] [PubMed]
- Lesiak, A.; Narbutt, J.; Sysa-Jedrzejowska, A.; Lukamowicz, J.; McCauliffe, D.P.; Wózniacka, A. Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus 2010, 19, 683–688. [Google Scholar] [CrossRef]
- Wozniacka, A.; Lesiak, A.; Narbutt, J.; McCauliffe, D.P.; Sysa-Jedrzejowska, A. Chloroquine treatment influences proinflammatory cytokine levels in systemic lupus erythematosus patients. Lupus 2006, 15, 268–275. [Google Scholar] [CrossRef]
- Grayson, P.C.; Kaplan, M.J. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Stelton, C.R.; Connors, D.B.; Walia, S.S.; Walia, H.S. Hydrochloroquine retinopathy: Characteristic presentation with review of screening. Clin. Rheumatol. 2013, 32, 895–898. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Gallo, A.; Ceolotto, G.; Pinton, P.; Iori, E.; Murphy, E.; Rutter, G.A.; Rizzuto, R.; Semplicini, A.; Avogaro, A. Metformin prevents glucose-induced protein kinase C-beta2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes 2005, 54, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uozumi, R.; Iguchi, R.; Masuda, S.; Nishibata, Y.; Nakazawa, D.; Tomaru, U.; Ishizu, A. Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Mod. Rheumatol. 2020, 30, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kronbichler, A.; Park, D.D.-Y.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.D.; Puy, C.; Fernández, J.A.; Mitrugno, A.; Keshari, R.S.; Taku, N.A.; Chu, T.T.; Xu, X.; Gruber, A.; Lupu, F.; et al. Activated protein C inhibits neutrophil extracellular trap formation and activation. J. Biol. Chem. 2017, 292, 8616–8629. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.H.; Fernández, J.A.; Gale, A.J.; Mosnier, L.O. Activated protein C. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 73–80. [Google Scholar] [CrossRef]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated protein C: Biased for translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, I.; Tamura, H.; Reich, J. Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model. Int. J. Mol. Sci. 2020, 21, 5973. [Google Scholar] [CrossRef]
- Czaikoski, P.G.; Mota, J.M.S.C.; Nascimento, D.C.; Sônego, F.; Castanheira, F.V.E.S.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef] [Green Version]
- Boufenzer, A.; Carrasco, K.; Jolly, L.; Brustolin, B.; Di-Pillo, E.; Derive, M.; Gibot, S. Potentiation of NETs release is novel characteristic of TREM-1 activation and the pharmacological inhibition of TREM-1 could prevent from the deleterious consequences of NETs release in sepsis. Cell. Mol. Immunol. 2021, 18, 452–460. [Google Scholar] [CrossRef]
- Meara, C.H.O.; Coupland, L.A.; Kordbacheh, F.; Quah, B.J.C.; Chang, C.-W.; Simon Davis, D.A.; Bezos, A.; Browne, A.M.; Freeman, C.; Hammill, D.J.; et al. Neutralizing the pathological effects of extracellular histones with small polyanions. Nat. Commun. 2020, 11, 6408. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Gandhi, A.A.; Smith, S.A.; Wang, Q.; Chiang, D.; Yalavarthi, S.; Ali, R.A.; Liu, C.; Sule, G.; Tsou, P.-S.; et al. Endothelium-protective, histone-neutralizing properties of the polyanionic agent defibrotide. medRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Welsh, N.; Farrah, T.E.; Gallacher, P.J.; Dhaun, N. ANCA associated vasculitis. BMJ 2020, 369, m1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, F.A.; Lanyon, P.C.; Grainge, M.J.; Shaunak, R.; Mahr, A.; Hubbard, R.B.; Watts, R.A. Incidence of ANCA-associated vasculitis in a UK mixed ethnicity population. Rheumatology 2016, 55, 1656–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef] [Green Version]
- Charles, L.A.; Caldas, M.L.; Falk, R.J.; Terrell, R.S.; Jennette, J.C. Antibodies against granule proteins activate neutrophils in vitro. J. Leukoc. Biol. 1991, 50, 539–546. [Google Scholar] [CrossRef]
- Kumar, S.V.R.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [Green Version]
- Binda, V.; Moroni, G.; Messa, P. ANCA-associated vasculitis with renal involvement. J. Nephrol. 2018, 31, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Sacoto, G.; Boukhlal, S.; Specks, U.; Flores-Suárez, L.F.; Cornec, D. Lung involvement in ANCA-associated vasculitis. Presse Med. 2020, 49, 104039. [Google Scholar] [CrossRef] [PubMed]
- Geetha, D.; Jefferson, J.A. ANCA-Associated Vasculitis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C.; Falk, R.J. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat. Rev. Rheumatol. 2014, 10, 463–473. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Lara, A.R.; Schwarz, M.I. Diffuse alveolar hemorrhage. Chest 2010, 137, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [Google Scholar] [CrossRef]
- Sherer, Y.; Gorstein, A.; Fritzler, M.J.; Shoenfeld, Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Semin. Arthritis Rheum. 2004, 34, 501–537. [Google Scholar] [CrossRef]
- Safi, R.; Al-Hage, J.; Abbas, O.; Kibbi, A.-G.; Nassar, D. Investigating the presence of neutrophil extracellular traps in cutaneous lesions of different subtypes of lupus erythematosus. Exp. Derm. 2019, 28, 1348–1352. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sil, P.; Wicklum, H.; Surell, C.; Rada, B. Macrophage-derived IL-1β enhances monosodium urate crystal-triggered NET formation. Inflamm. Res. 2017, 66, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, J.; Zahn, S.; Tüting, T. Pathogenesis of cutaneous lupus erythematosus: Common and different features in distinct subsets. Lupus 2010, 19, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Tripodo, C.; Gong, M.; Sangaletti, S.; Colombo, M.P.; Coffman, R.L.; Barrat, F.J. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med. 2010, 207, 2931–2942. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.D.; Wang, G.; Toh, C.-H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2019, 200, 869–880. [Google Scholar] [CrossRef]
- Liang, Y.; Pan, B.; Alam, H.B.; Deng, Q.; Wang, Y.; Chen, E.; Liu, B.; Tian, Y.; Williams, A.M.; Duan, X.; et al. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur. J. Pharm. 2018, 833, 432–440. [Google Scholar] [CrossRef]
- Maruchi, Y.; Tsuda, M.; Mori, H.; Takenaka, N.; Gocho, T.; Huq, M.A.; Takeyama, N. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit. Care 2018, 22, 176. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Russo, R.M.; Li, Y.; Karmakar, M.; Liu, B.; Puskarich, M.A.; Jones, A.E.; Stringer, K.A.; Standiford, T.J.; Alam, H.B. Serum citrullinated histone H3 concentrations differentiate patients with septic verses non-septic shock and correlate with disease severity. Infection 2021, 49, 83–93. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Liu, B.; Zhao, T.; Chong, W.; Wang, Y.; Alam, H.B. Citrullinated histone H3: A novel target for the treatment of sepsis. Surgery 2014, 156, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhou, Y.; Qu, M.; Yu, Y.; Chen, Z.; Zhu, S.; Guo, K.; Chen, W.; Miao, C. Tissue Factor-Enriched Neutrophil Extracellular Traps Promote Immunothrombosis and Disease Progression in Sepsis-Induced Lung Injury. Front. Cell. Infect. Microbiol. 2021, 11, 677902. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, N.; Sayed, M.A.; Poonsuph, C.J.; Sehgal, R.; Shirke, M.M.; Harky, A. Septic Cardiomyopathy: From Basics to Management Choices. Curr. Probl. Cardiol. 2021, 46, 100767. [Google Scholar] [CrossRef] [PubMed]
- Woźnica, E.A.; Inglot, M.; Woźnica, R.K.; Łysenko, L. Liver dysfunction in sepsis. Adv. Clin. Exp. Med. 2018, 27, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, H.; Li, Y.; Dai, M.; Zhang, L.; Liu, S.; Tan, H.; Deng, P.; Liu, J.; Mao, Z.; et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp. Cell Res. 2019, 382, 111486. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, W.; Wang, W.; Tong, X.; Xia, R.; Fan, J.; Du, J.; Zhang, C.; Shi, X. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit. Care 2020, 24, 380. [Google Scholar] [CrossRef]
- Lelubre, C.; Vincent, J.-L. Mechanisms and treatment of organ failure in sepsis. Nat. Rev. Nephrol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Schattner, M. Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J. Leukoc. Biol. 2019, 105, 873–880. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.-S.; Chen, Y.; Wilson, Z.; Fallon, E.A.; Reichner, J.S.; Ayala, A. PAD4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J. Immunol. 2018, 200, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Yang, H.; Hu, X.; Zhang, Z.; Ge, S.; Xu, Z.; Gao, J.; Liu, J.; White, G.C.; Ma, Y.-Q. Kindlin-3 in platelets and myeloid cells differentially regulates deep vein thrombosis in mice. Aging 2019, 11, 6951–6959. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, C.; Zhang, X.; Wang, S.; Zhu, R.; Zhou, A.; Chen, S.; Feng, J. Heparin improves alveolarization and vascular development in hyperoxia-induced bronchopulmonary dysplasia by inhibiting neutrophil extracellular traps. Biochem. Biophys. Res. Commun. 2020, 522, 33–39. [Google Scholar] [CrossRef]
- Snoderly, H.T.; Boone, B.A.; Bennewitz, M.F. Neutrophil extracellular traps in breast cancer and beyond: Current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019, 21, 145. [Google Scholar] [CrossRef] [Green Version]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- DuPre, S.A.; Hunter, K.W. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar] [CrossRef]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef] [PubMed]
- Van Enter, B.J.; von Hauff, E. Challenges and perspectives in continuous glucose monitoring. Chem. Commun. 2018, 54, 5032–5045. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Prim. 2016, 2, 16012. [Google Scholar] [CrossRef]
- Klaassen, I.; van Noorden, C.J.F.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561. [Google Scholar] [CrossRef]
- Tibrewal, S.; Ivanir, Y.; Sarkar, J.; Nayeb-Hashemi, N.; Bouchard, C.S.; Kim, E.; Jain, S. Hyperosmolar stress induces neutrophil extracellular trap formation: Implications for dry eye disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7961–7969. [Google Scholar] [CrossRef] [Green Version]
- Barliya, T.; Dardik, R.; Nisgav, Y.; Dachbash, M.; Gaton, D.; Kenet, G.; Ehrlich, R.; Weinberger, D.; Livnat, T. Possible involvement of NETosis in inflammatory processes in the eye: Evidence from a small cohort of patients. Mol. Vis. 2017, 23, 922–932. [Google Scholar]
- Ushio-Fukai, M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Drugs | Strategies | Outcome | Reference |
|---|---|---|---|---|
| Cardiovascular Disease | DNase 1 | DNA degradation | Digest the DNA constitution of NETs, therefore destruct the NETs, protected murine IVC stenosis model from DVT | [99,101] |
| CI-amidine | PAD4 inhibitor | Block the histone citrullination in NETosis to reduce NETosis and eliminate inflammation in DIO mice | [98,101] | |
| Roflumilast | Phosphodiesterase 4 inhibitor | Eliminate the interaction between NETosis and activated ECs and platelets in order to prevent platelet aggregation | [100,102] | |
| Heparin | Anti-histone | Block the histone-induced NF-κB pathway, thus protect the ECs from inflammation of NETs, therefore avoid mice from organ damage | [103,104,105,106] | |
| Anti-high-mobility group box 1 (HMGB1) pAb | Anti-HMGB1 | Reduce the histone 3 and free DNA in the BAL fluid of LPS-treated mice, thus decrease the inflammation and neutrophil chemotaxis to mitigate NETosis | [107,108,109,110,111] | |
| Autoimmune Diseases | DNase | DNA degradation | Digest the DNA constitution of NETs, therefore avoid glomerular endothelial injury in murine AAV disease models | [101,112] |
| Vitamin D | Inhibiting NETs activity | Decrease the NETs activity to reduce the damage to ECs, and reduce the early cellular apoptosis in SLE patients | [113,114] | |
| Chloroquine/ Hydroxychloroquine (HDQ) | MMPs-TIMPs modulation | Modulate NETs through the regulation of MMP and TIMP to maintain the extracellular homeostasis in SLE patients; also it can prevent platelet aggregation, resulting in endothelium protection | [115,116,117,118,119] | |
| Metformin | Regulating mtDNA-pDC-IFNα pathway | Inhibit ROS production, and repress NETosis with a reduction in elastase, proteinase-3, histones, and cfDNA with in chronic autoimmune disease of the elderly | [120,121,122] | |
| Intravenous Immunoglobulin (IVIG) | Inhibiting ANCA production | Relieve antigen antibody responses, and inflammation, therefore NET amounts in the peritoneum are significantly decreased | [123,124] | |
| Sepsis | Drotrecogin | Recombinant human activated protein C | Inhibit the formation of coagulation factors Va and VIlla and destroy extracellular histones, preventing activated platelets from inducing NETosis | [125,126,127] |
| LL-37 | Enhancing NETs | Improve sterilization capacity and increase the survival rate of CLP mice | [128] | |
| DNase I | DNA degradation | Combine with antibiotics to improve the outcome | [129] | |
| Anti–TREM-1 | Reducing NETosis | Eliminate associated ED and organic damage in mice LPS models | [130] | |
| Small Polyanions (SPAs) | Histone inhibitor (NET-bound and free) | Improve the outcome in the LPS, TNF and CLP mice models | [131] | |
| Defibrotide | Neutralization of histones (cationic proteins) with polyanionic compounds | In vitro, defibrotide counteracted EC activation and pyroptosis-mediated cell death induced by NETs. In vivo, defibrotide stabilized the endothelium and protected against histone-accelerated inferior vena cava thrombosis in mice. The development of MODS was relieved in the later stage of sepsis | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Liu, J.; Yan, N. Endothelial Dysfunction Induced by Extracellular Neutrophil Traps Plays Important Role in the Occurrence and Treatment of Extracellular Neutrophil Traps-Related Disease. Int. J. Mol. Sci. 2022, 23, 5626. https://doi.org/10.3390/ijms23105626
Yu S, Liu J, Yan N. Endothelial Dysfunction Induced by Extracellular Neutrophil Traps Plays Important Role in the Occurrence and Treatment of Extracellular Neutrophil Traps-Related Disease. International Journal of Molecular Sciences. 2022; 23(10):5626. https://doi.org/10.3390/ijms23105626
Chicago/Turabian StyleYu, Shuyang, Jingyu Liu, and Nianlong Yan. 2022. "Endothelial Dysfunction Induced by Extracellular Neutrophil Traps Plays Important Role in the Occurrence and Treatment of Extracellular Neutrophil Traps-Related Disease" International Journal of Molecular Sciences 23, no. 10: 5626. https://doi.org/10.3390/ijms23105626
APA StyleYu, S., Liu, J., & Yan, N. (2022). Endothelial Dysfunction Induced by Extracellular Neutrophil Traps Plays Important Role in the Occurrence and Treatment of Extracellular Neutrophil Traps-Related Disease. International Journal of Molecular Sciences, 23(10), 5626. https://doi.org/10.3390/ijms23105626