
In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein



Abstract
:1. Introduction
2. Results
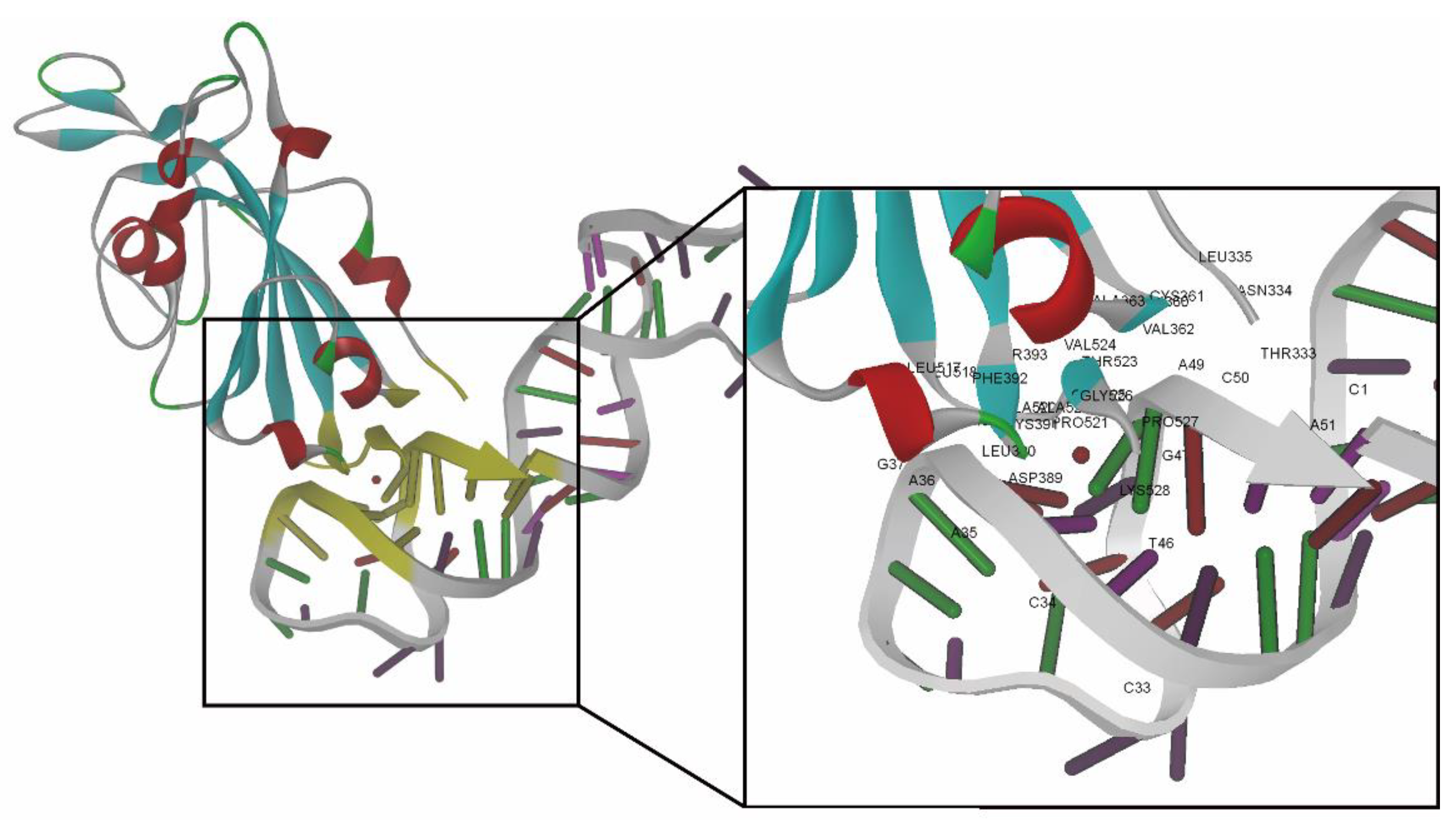
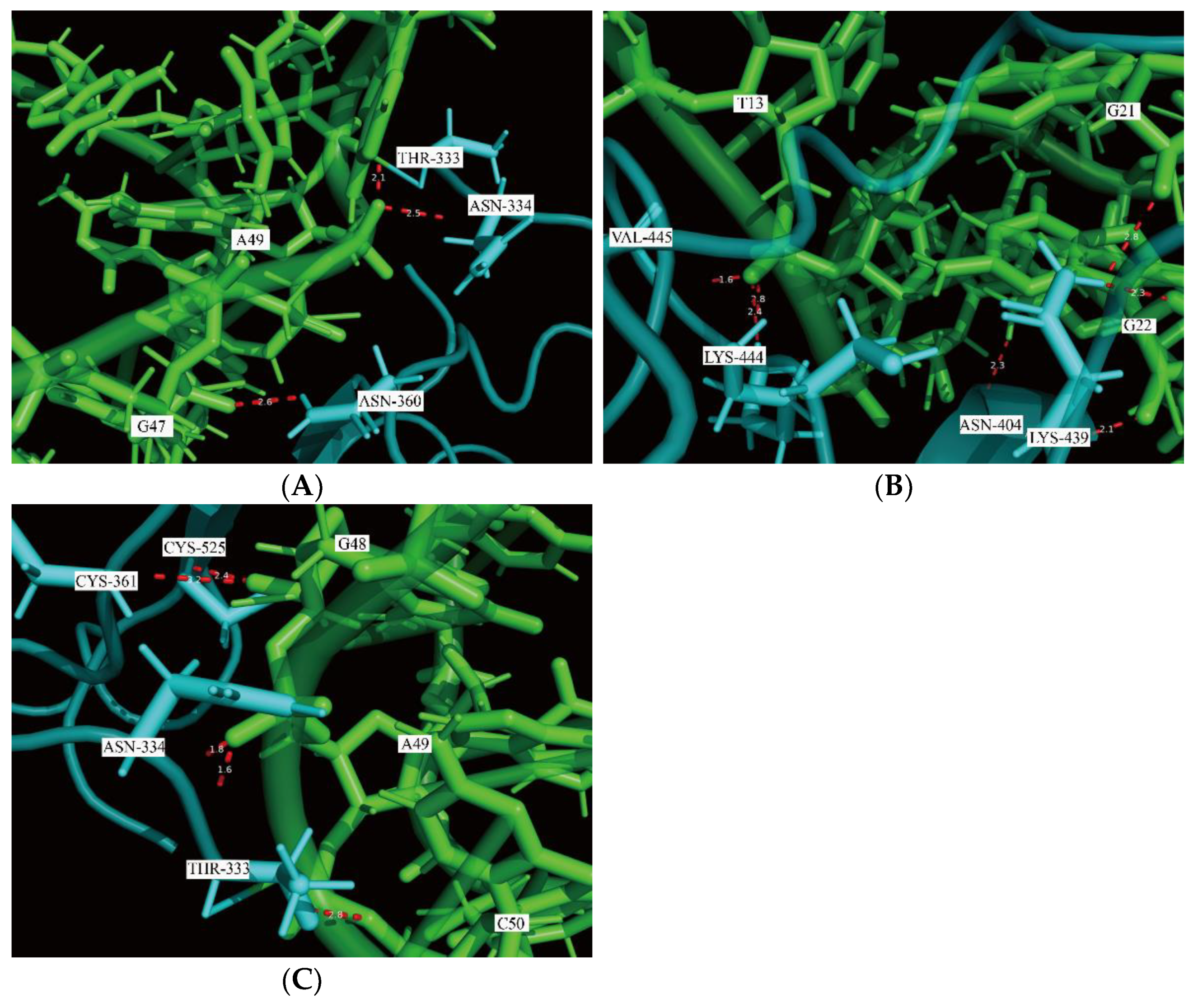
2.1. Docking Simulation of the RBD-1C and Mutated Aptamers
2.2. MD Simulation of Protein–Aptamer Complexes
2.2.1. RMSD
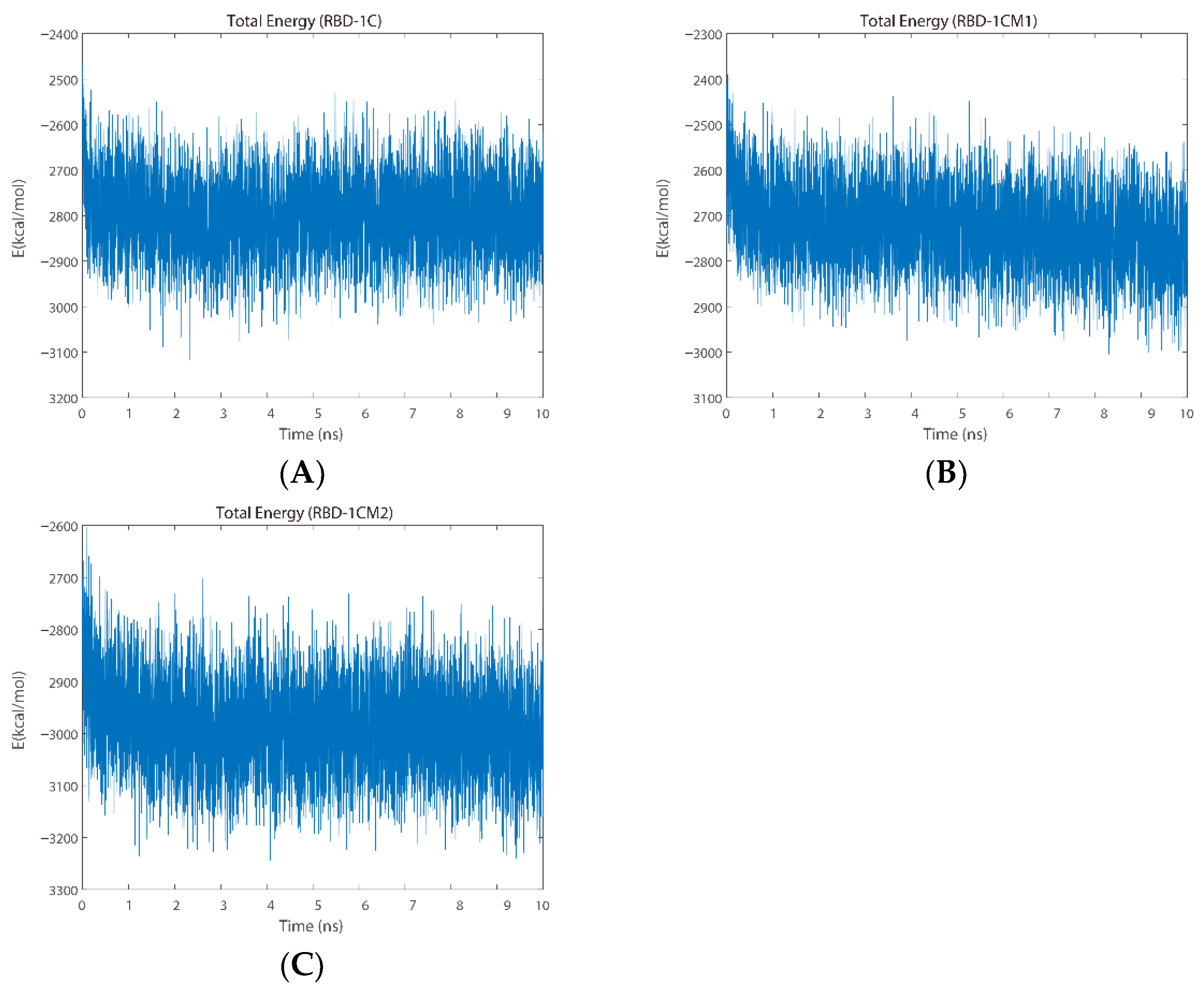
2.2.2. Total Energy Analysis
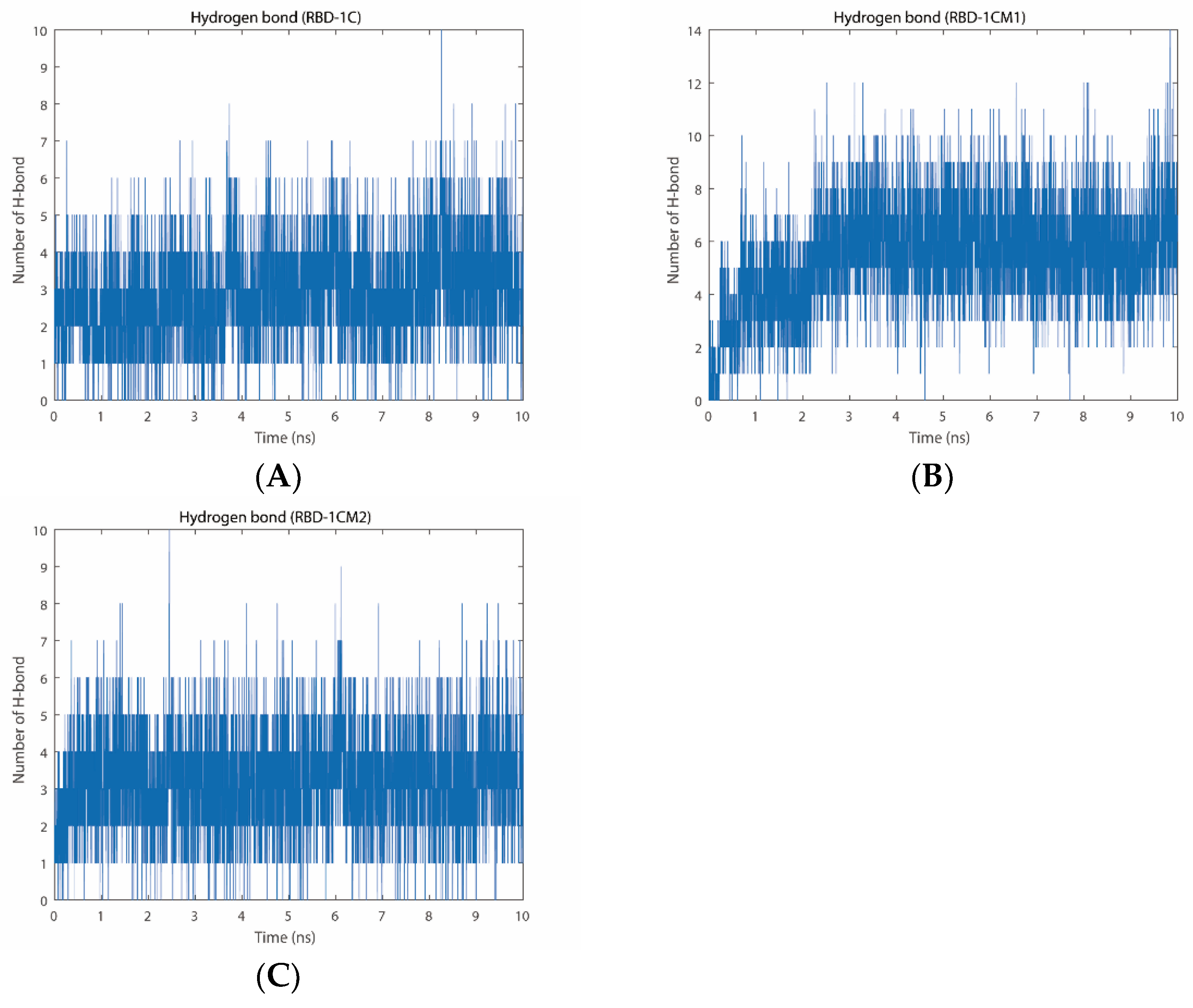
2.2.3. Number of Hydrogen Bonds
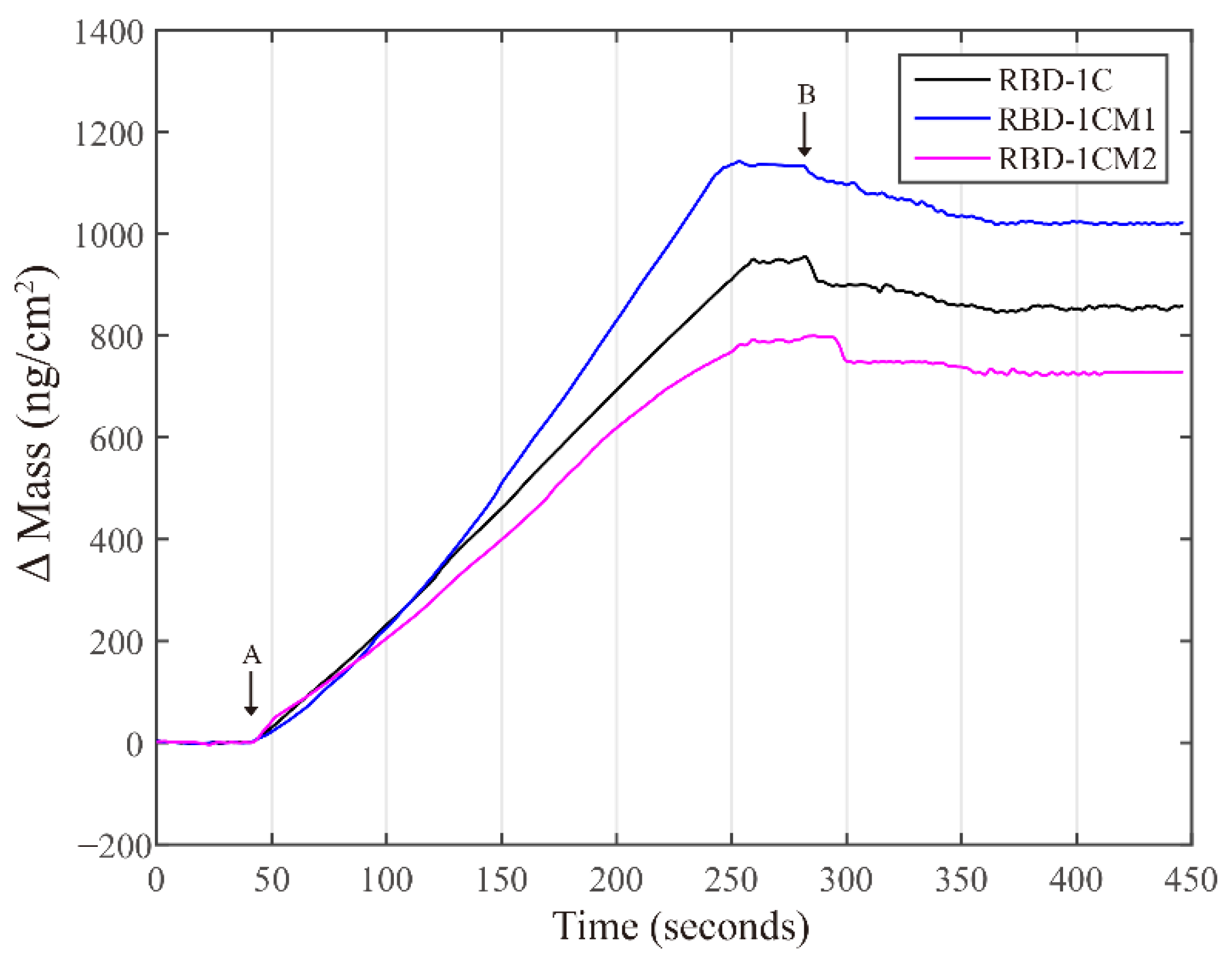
2.3. Quartz Crystal Microbalance (QCM) Experiments
3. Discussion
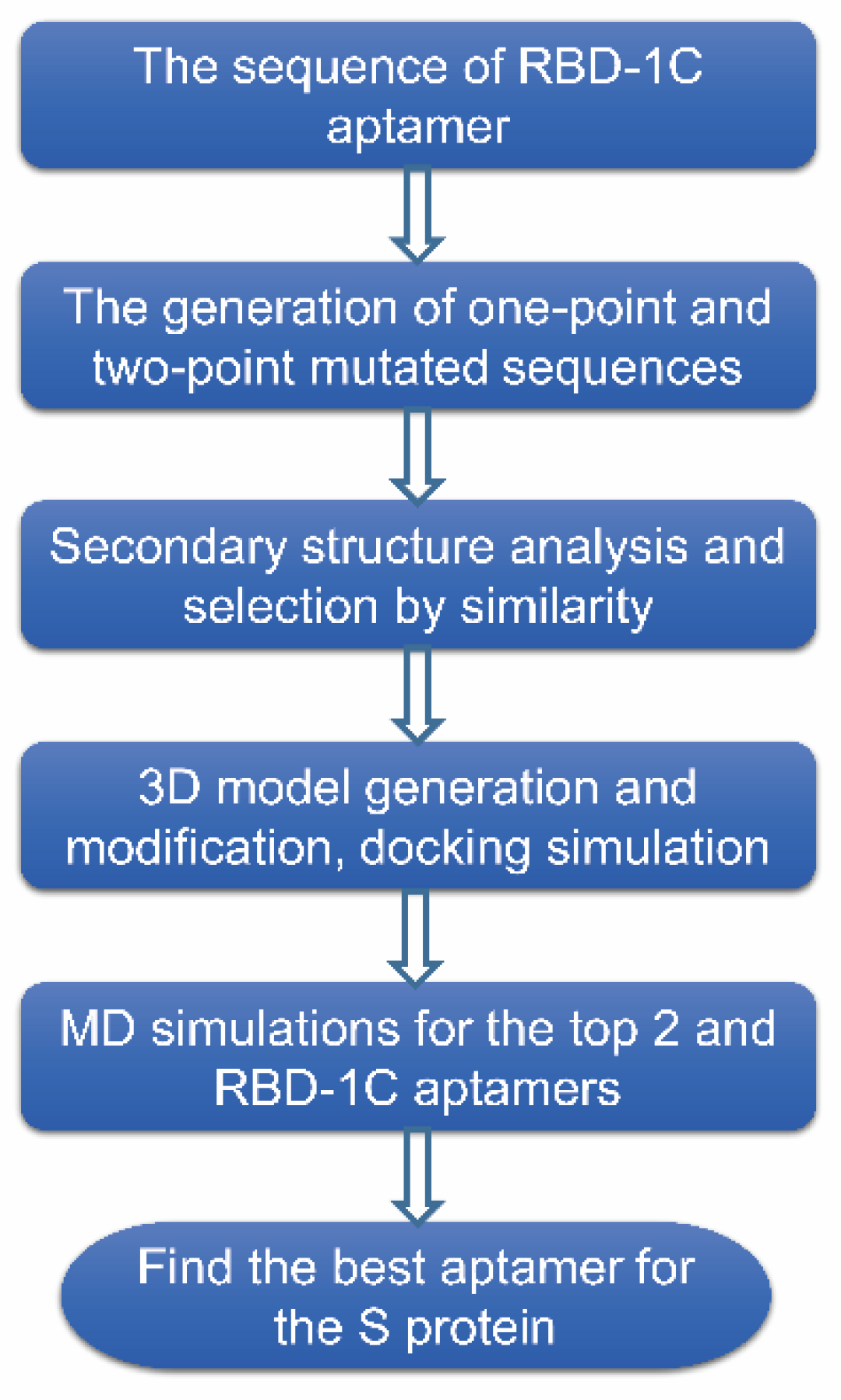
4. Material and Methods
4.1. Sources of Protein and Aptamer Data
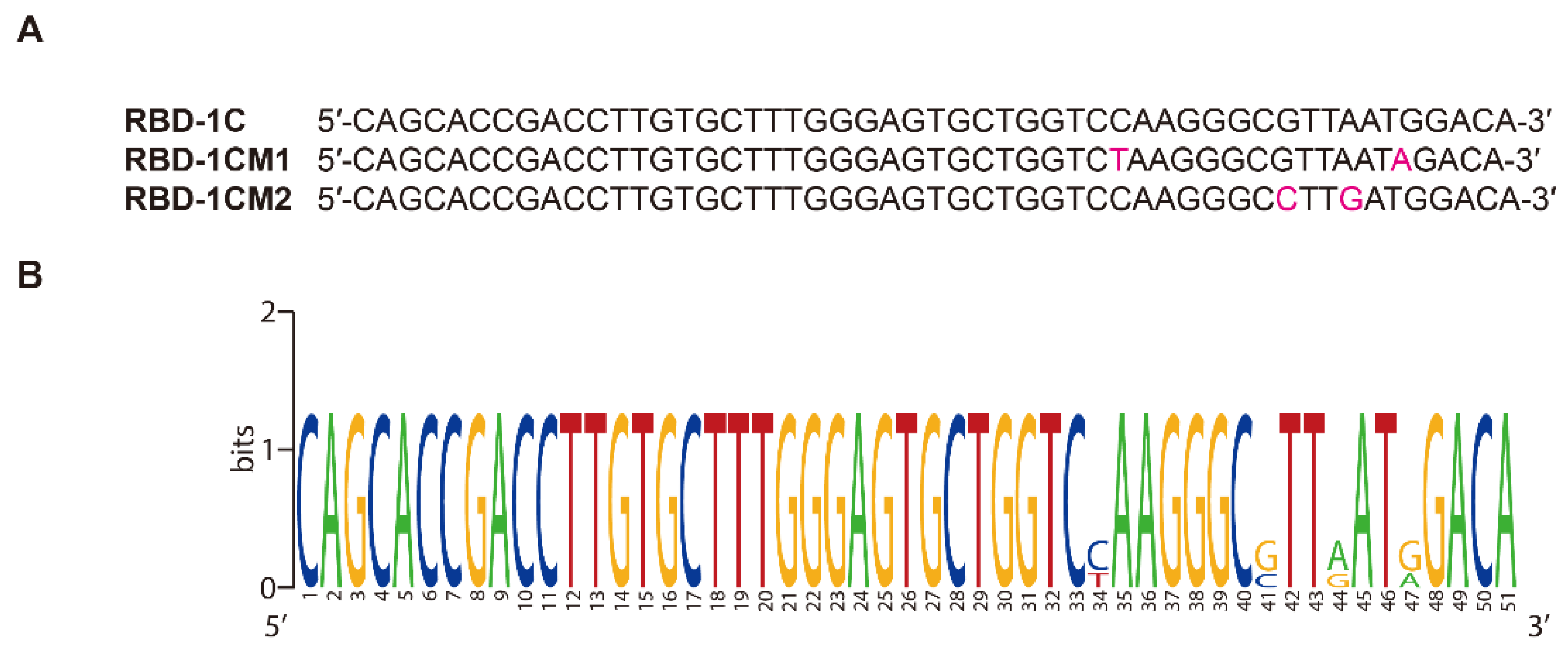
4.2. Generation of Mutated Sequences
4.3. Comparison of Similarity in the Secondary Structure
4.4. Generation of the 3D Aptamer Structure and Molecular Simulations
4.5. Experimental Section
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.P.; Zhou, R.; Chan, K.H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e12. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Igloi, Z.; Velzing, J.; van Beek, J.; van de Vijver, D.; Aron, G.; Ensing, R.; Benschop, K.; Han, W.; Boelsums, T.; Koopmans, M.; et al. Clinical evaluation of Roche SD Biosensor rapid antigen test for SARS-CoV-2 in municipal health service testing site, the Netherlands. Emerg. Infect. Dis. 2021, 27, 1323–1329. [Google Scholar] [CrossRef]
- Van derMoeren, N.; Zwart, V.F.; Lodder, E.B.; Van denBijllaardt, W.; VanEsch, H.R.J.M.; Stohr, J.J.J.M.; Pot, J.; Welschen, I.; VanMechelen, P.M.F.; Pas, S.D.; et al. Performance evaluation of a Sars-CoV-2 rapid antigentest: Test performance in the community in the Netherlands. medRxiv 2020. [Google Scholar] [CrossRef]
- Sisay, A.; Tesfaye, A.; Desale, A.; Ataro, I.; Woldesenbet, Z.; Nigusse, B.; Tayachew, A.; Kebede, A.; Desta, A.F. Diagnostic performance of SARS-CoV-2 IgM/IgG rapid test kits for the detection of the novel coronavirus in ethiopia. J. Multidiscip. Healthc. 2021, 14, 171–180. [Google Scholar] [CrossRef]
- Liu, C.; Mao, B.; Martinez, V.; Chen, X.; Li, Y.; He, L.; Chen, S.; Guo, X.; Shen, X.; Bao, X.; et al. A facile assay for rapid detection of COVID-19 antibodies. RSC Adv. 2020, 10, 28041–28048. [Google Scholar] [CrossRef]
- Dunn, M.R.; Jimenez, R.M.; Chaput, J.C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 2017, 1, 76. [Google Scholar] [CrossRef]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Idili, A.; Parolo, C.; Alvarez-Diduk, R.; Merkoçi, A. Rapid and efficient detection of the SARS-CoV-2 spike protein using an electrochemical aptamer-based sensor. ACS Sens. 2021, 6, 3093–3101. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tian, S.; Zhao, W.; Liu, K.; Ma, X.; Guo, J. Aptamer-based lateral flow assay on-site biosensors. Biosens. Bioelectron. 2021, 186, 113279. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef] [PubMed]
- Cleri, F.; Lensink, M.F. Blossey, R. DNA aptamers block the receptor binding domain at the spike protein of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 778. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, Q.; Chen, J.; Ni, X.; Dai, J. A DNA aptamer based method for detection of SARS-CoV-2 nucleocapsid protein. Virol. Sin. 2020, 35, 351–354. [Google Scholar] [CrossRef]
- Devi, A.; Chaitanya, N.S.N. Designing of peptide aptamer targeting the receptor-binding domain of spike protein of SARS-CoV-2: An in silico study. Mol. Divers. 2021, 26, 157–169. [Google Scholar] [CrossRef]
- Behbahani, M.; Mohabatkar, H.; Hosseini, B. In silico design of quadruplex aptamers against the spike protein of SARS-CoV-2. Inform. Med. Unlocked 2021, 26, 100757. [Google Scholar] [CrossRef]
- Zloh, M.; Kirton, S.B. The benefits of in silico modeling to identify possible small-molecule drugs and their off-target interactions. Future Med. Chem. 2018, 10, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Agamah, F.E.; Mazandu, G.K.; Hassan, R.; Bope, C.D.; Thomford, N.E.; Ghansah, A.; Chimusa, E.R. Computational/in silico methods in drug target and lead prediction. Brief. Bioinform. 2020, 21, 1663–1675. [Google Scholar] [CrossRef]
- de Meyer, F.J.-M.; Rodgers, J.M.; Willems, T.F.; Smit, B. Molecular simulation of the effect of cholesterol on lipid-mediated protein–protein interactions. Biophys. J. 2010, 99, 3629–3638. [Google Scholar] [CrossRef] [Green Version]
- Elcock, A.H.; Sept, D.; McCammon, J.A. Computer simulation of protein−protein interactions. J. Phys. Chem. B 2001, 105, 1504–1518. [Google Scholar] [CrossRef]
- Hu, W.P.; Kumar, J.V.; Huang, C.J.; Chen, W.Y. Computational selection of RNA aptamer against angiopoietin-2 and experimental evaluation. BioMed Res. Int. 2015, 2015, 658712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P.C.; Lin, H.T.; Chen, W.Y.; Tsai, J.J.P.; Hu, W.P. The combination of computational and biosensing technologies for selecting aptamer against prostate specific antigen. BioMed Res. Int. 2017, 2017, 5041683. [Google Scholar] [CrossRef]
- Hu, B.; Zhou, R.; Li, Z.; Ouyang, S.; Li, Z.; Hu, W.; Wang, L.; Jiao, B. Study of the binding mechanism of aptamer to palytoxin by docking and molecular simulation. Sci. Rep. 2019, 9, 15494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabri, M.Z.; Abdul Hamid, A.A.; Sayed Hitam, S.M.; Abdul, M.Z.; In, M.Z. In Silico screening of aptamers configuration against hepatitis B surface antigen. Adv. Bioinform. 2019, 2019, 6912914. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.V.; Chen, W.-Y.; Tsai, J.J.P.; Hu, W.-P. Molecular simulation methods for selecting thrombin-binding aptamers. Lect. Notes Electr. Eng. 2013, 253, 977–983. [Google Scholar]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and applications of in silico aptamer design and modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef]
- Katilius, E.; Flores, C.; Woodbury, N.W. Exploring the sequence space of a DNA aptamer using microarrays. Nucleic Acids Res. 2007, 35, 7626–7635. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K.; Qwik, M.D. Integrative Molecular Dynamics Toolkit for Novices and Experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef] [Green Version]
- Song, K.M.; Lee, S.; Ban, C. Aptamers and their biological applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandola, C.; Neerathilingam, M. Aptamers for targeted delivery: Current challenges and future opportunities. In Role of Novel Drug Delivery Vehicles in Nanobiomedicine, 1st ed.; Tyagi, R.K., Garg, N., Shukla, R., Eds.; IntechOpen: London, UK, 2019; pp. 1–22. [Google Scholar]
- Pierce, L.C.; Salomon-Ferrer, R.; Augusto F de Oliveira, C.; McCammon, J.A.; Walker, R.C. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theor. Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, D.M. Equilibrium sampling in biomolecular simulations. Annu. Rev. Biophys. 2011, 40, 41–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of conformational change: Comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Amaro, R.E.; Cheng, X.; Ivanov, I.; Xu, D.; McCammon, J.A. Characterizing loop dynamics and ligand recognition in human- and avian-type influenza neuraminidases via generalized born molecular dynamics and end-point free energy calculations. J. Am. Chem. Soc. 2009, 131, 4702–4709. [Google Scholar] [CrossRef] [Green Version]
- Zagrovic, B.; Snow, C.D.; Shirts, M.R.; Pande, V.S. Simulation of folding of a small alpha-helical protein in atomistic detail using worldwide-distributed computing. J. Mol. Biol. 2002, 323, 927–937. [Google Scholar] [CrossRef]
- Zagrovic, B.; Pande, V. Solvent viscosity dependence of the folding rate of a small protein: Distributed computing study. J. Comput. Chem. 2003, 24, 1432–1436. [Google Scholar] [CrossRef]
- Chocholoušová, J.; Feig, M. Implicit solvent simulations of DNA and DNA−protein complexes: Agreement with explicit solvent vs experiment. J. Phys. Chem. B 2006, 110, 17240–17251. [Google Scholar] [CrossRef]
- Wong, S.E.; Bernacki, K.; Jacobson, M. Competition between intramolecular hydrogen bonds and solvation in phosphorylated peptides: Simulations with explicit and implicit solvent. J. Phys. Chem. B 2005, 109, 5249–5258. [Google Scholar] [CrossRef]
- Mousivand, M.; Anfossi, L.; Bagherzadeh, K.; Barbero, N.; Mirzadi-Gohari, A.; Javan-Nikkhah, M. In silico maturation of affinity and selectivity of DNA aptamers against aflatoxin B1 for biosensor development. Anal. Chim. Acta 2020, 1105, 178–186. [Google Scholar] [CrossRef]
- Savory, N.; Abe, K.; Yoshida, W.; Ikebukuro, K. In silico maturation: Processing sequences to improve biopolymer functions based on genetic algorithms. In Applications of Metaheuristics in Process Engineering, 1st ed.; Valadi, J., Siarry, P., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 271–288. [Google Scholar]
- Valero, J.; Civit, L.; Dupont, D.M.; Selnihhin, D.; Reinert, L.S.; Idorn, M.; Israels, B.A.; Bednarz, A.M.; Bus, C.; Asbach, B.; et al. A serum-stable RNA aptamer specific for SARS-CoV-2 neutralizes viral entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2112942118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, J.; Jiao, S.; Gu, C.; Xu, W.; Chen, B.; Wang, R.; Chen, H.; Xie, Y.; Wang, A.; et al. Characterization of MW06, a human monoclonal antibody with cross-neutralization activity against both SARS-CoV-2 and SARS-CoV. mAbs 2021, 13, 1953683. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.A.; Zulkifli, R.M.; Hussin, H.; Nadri, M.H. In silico approach for post-SELEX DNA aptamers: A mini-review. J. Mol. Graph. 2021, 105, 107872. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, A.; Nagao, C.; Mizuguchi, K. Prediction of the secondary structure of short DNA aptamers. Biophys. Physicobiol. 2019, 16, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins 2007, 67, 1078–1086. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Decherchi, S.; Spitaleri, A.; Stone, J.; Rocchia, W. NanoShaper-VMD interface: Computing and visualizing surfaces, pockets and channels in molecular systems. Bioinformatics 2019, 35, 1241–1243. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | ZDOCK Score | ZRANK Score | The Number of Hydrogen Bonds |
|---|---|---|---|
| RBD-1C | 44.4 | −88.392 | 1 |
| RBD-1CM1 | 51.5 | −98.551 | 1 |
| RBD-1CM2 | 44.7 | −97.133 | 2 |
| ID | Biggest Δmass (ng/cm2) | Final Δmass (ng/cm2) | Ka (×103 M−1s−1) | Kd (×10−3 s−1) | KA (×105 M−1) |
|---|---|---|---|---|---|
| RBD-1C | 948 ± 23.7 | 847.6 ± 42.2 | 0.118 | 1.193 | 1.0 |
| RBD-1CM1 | 1065 ± 38.6 | 964.2 ± 51.4 | 0.124 | 1.029 | 1.2 |
| RBD-1CM2 | 812.8 ± 34.3 | 727.8 ± 46.5 | 0.125 | 1.020 | 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-C.; Chen, W.-Y.; Hwu, E.-T.; Hu, W.-P. In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein. Int. J. Mol. Sci. 2022, 23, 5810. https://doi.org/10.3390/ijms23105810
Lin Y-C, Chen W-Y, Hwu E-T, Hu W-P. In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein. International Journal of Molecular Sciences. 2022; 23(10):5810. https://doi.org/10.3390/ijms23105810
Chicago/Turabian StyleLin, Yu-Chao, Wen-Yih Chen, En-Te Hwu, and Wen-Pin Hu. 2022. "In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein" International Journal of Molecular Sciences 23, no. 10: 5810. https://doi.org/10.3390/ijms23105810