
Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells


 and
and 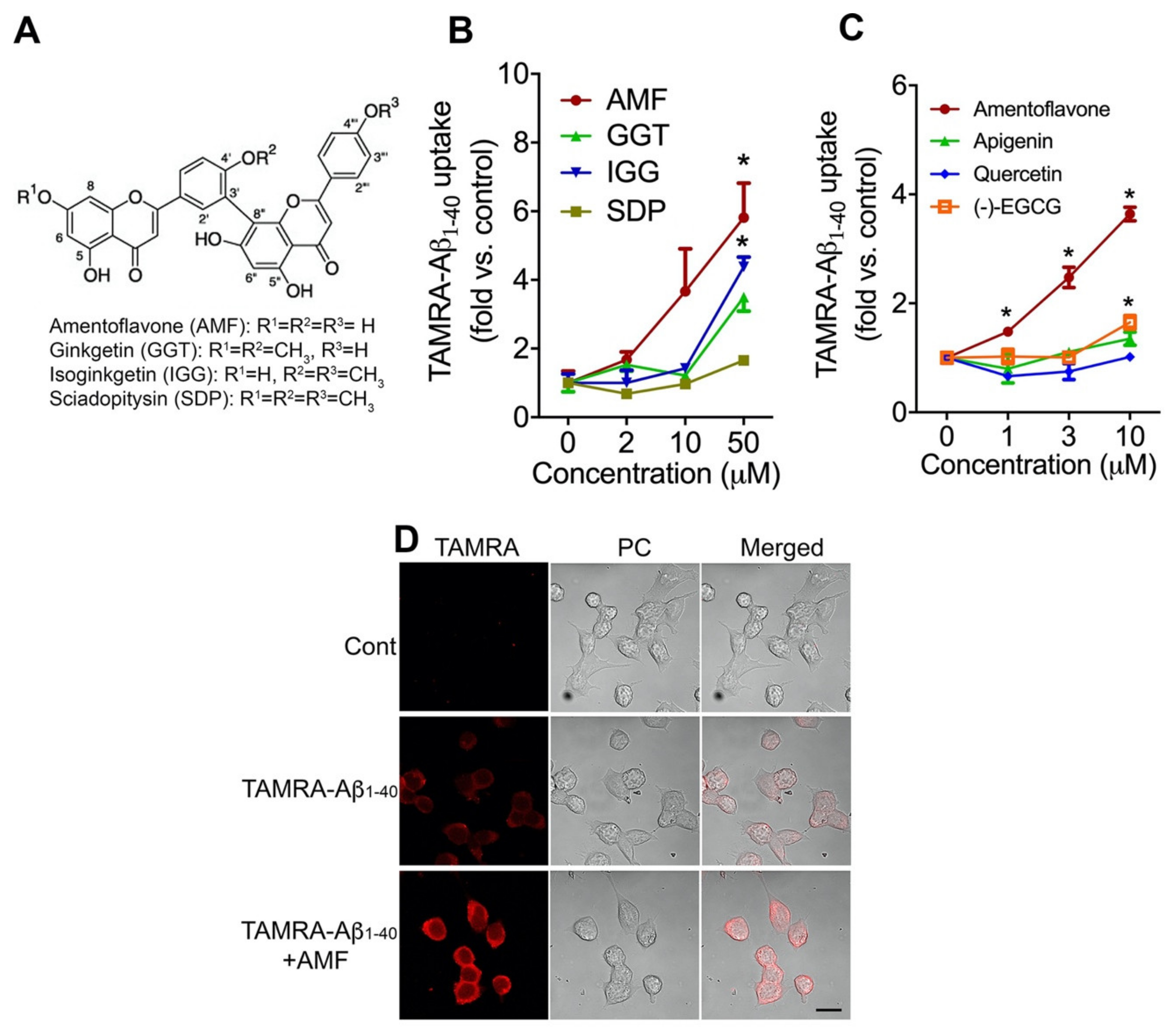{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Amentoflavone Most Potently Increases Cellular Uptake of Aβ1-40 in N2a Neurons
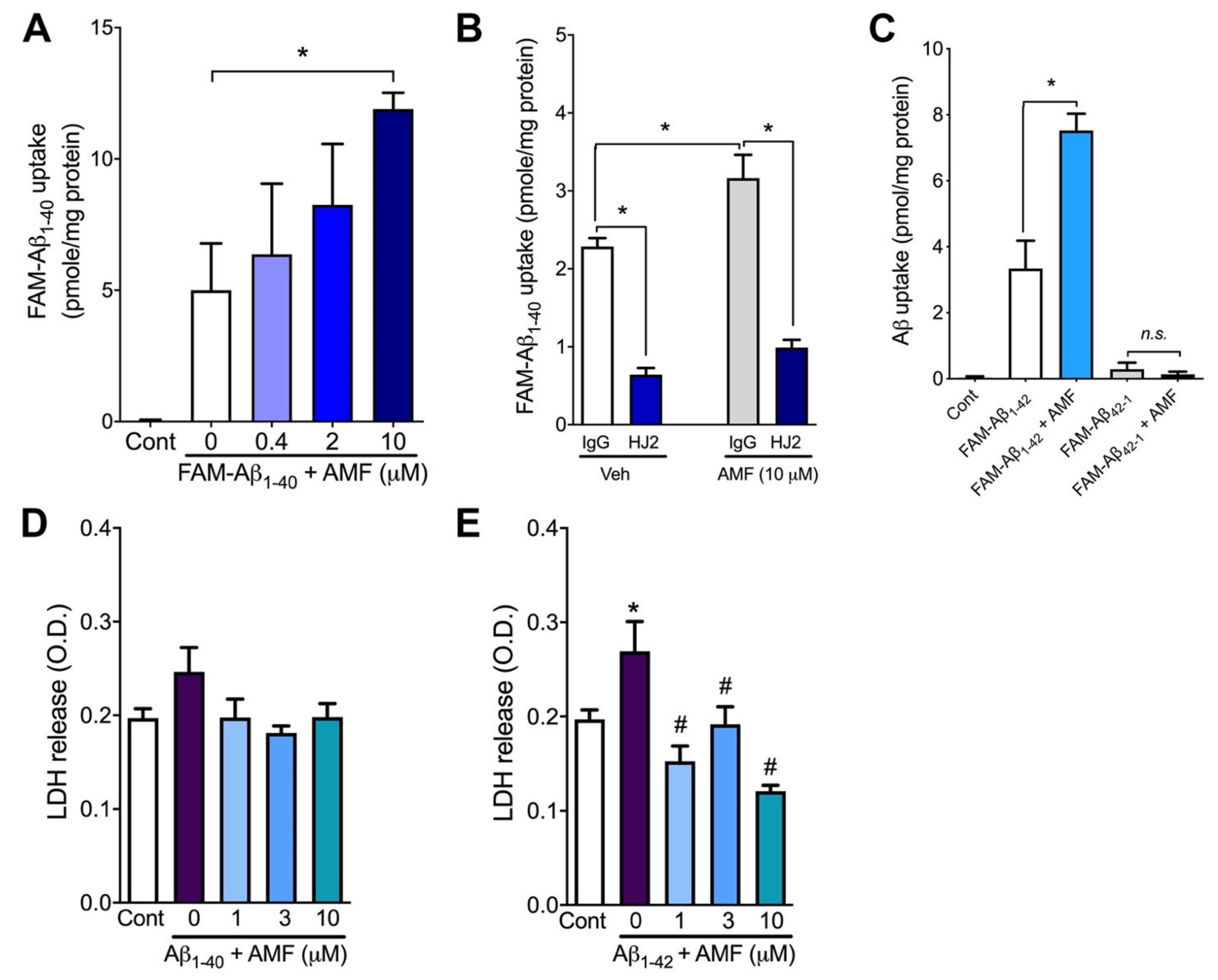
2.2. Amentoflavone Increases Cellular Uptake of Both Aβ1-40 and Aβ1-42 without Causing Cytotoxicity
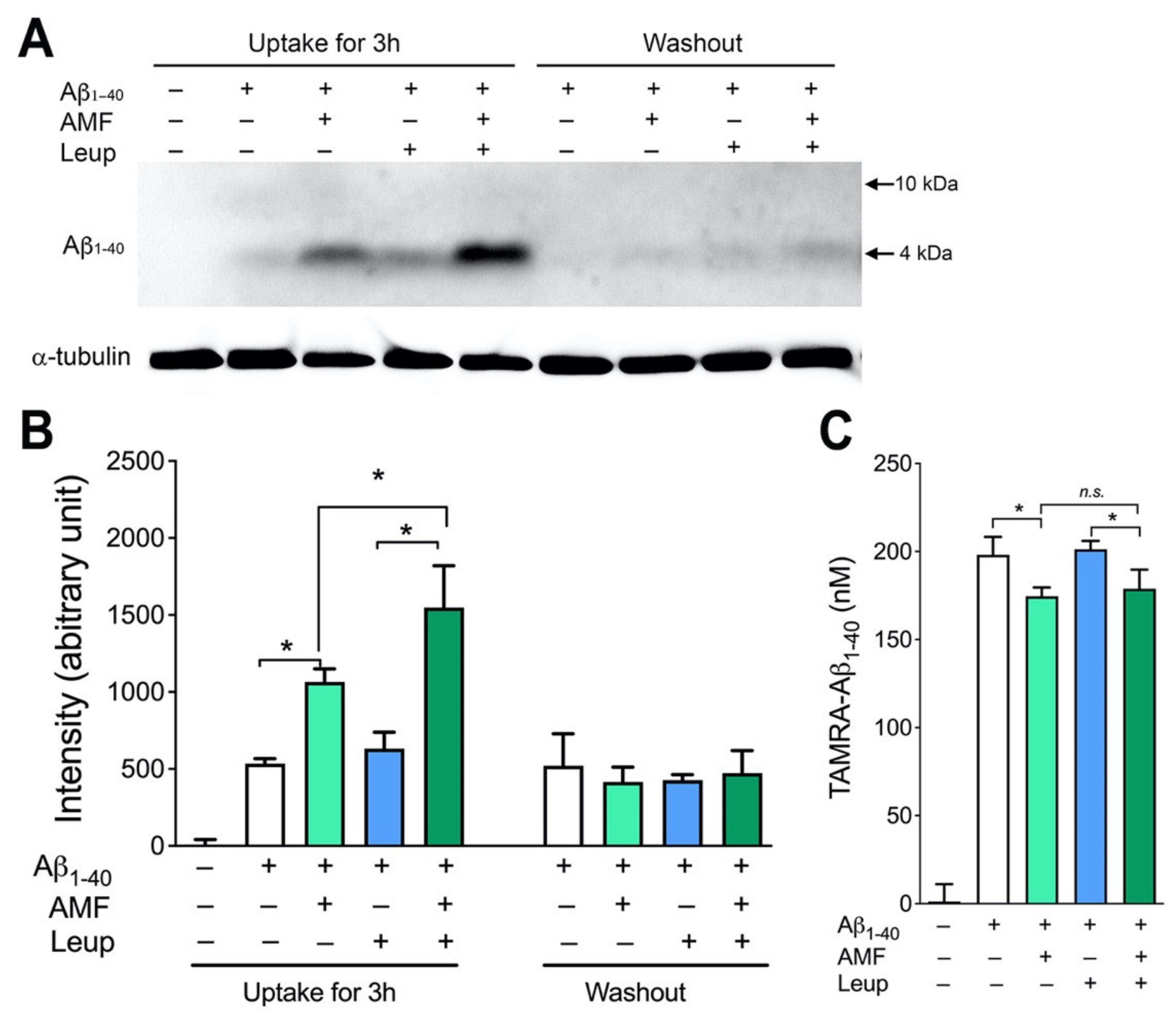
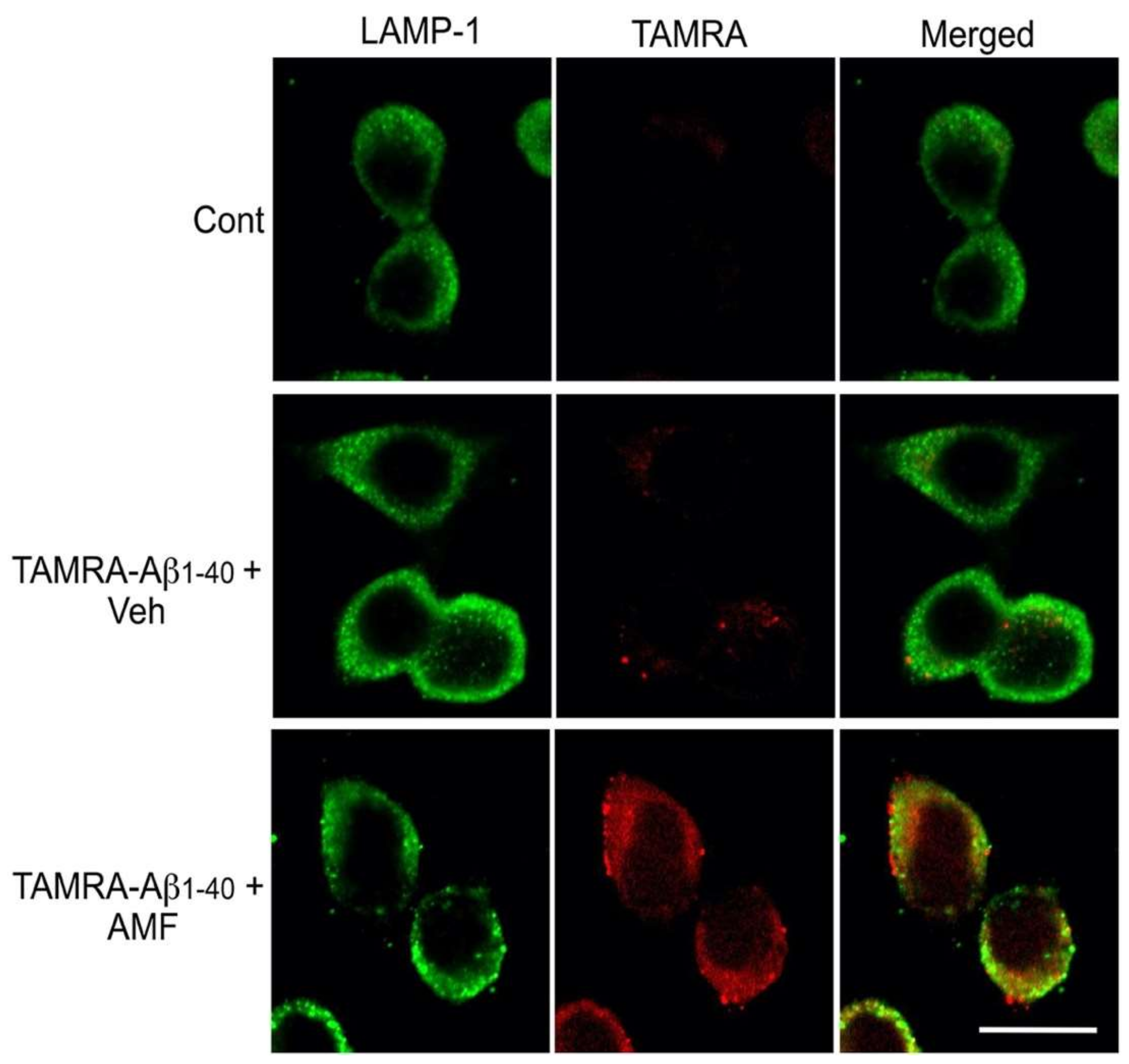
2.3. Internalized Aβ1-40 Peptide Undergoes Lysosomal Degradation in N2a Cells
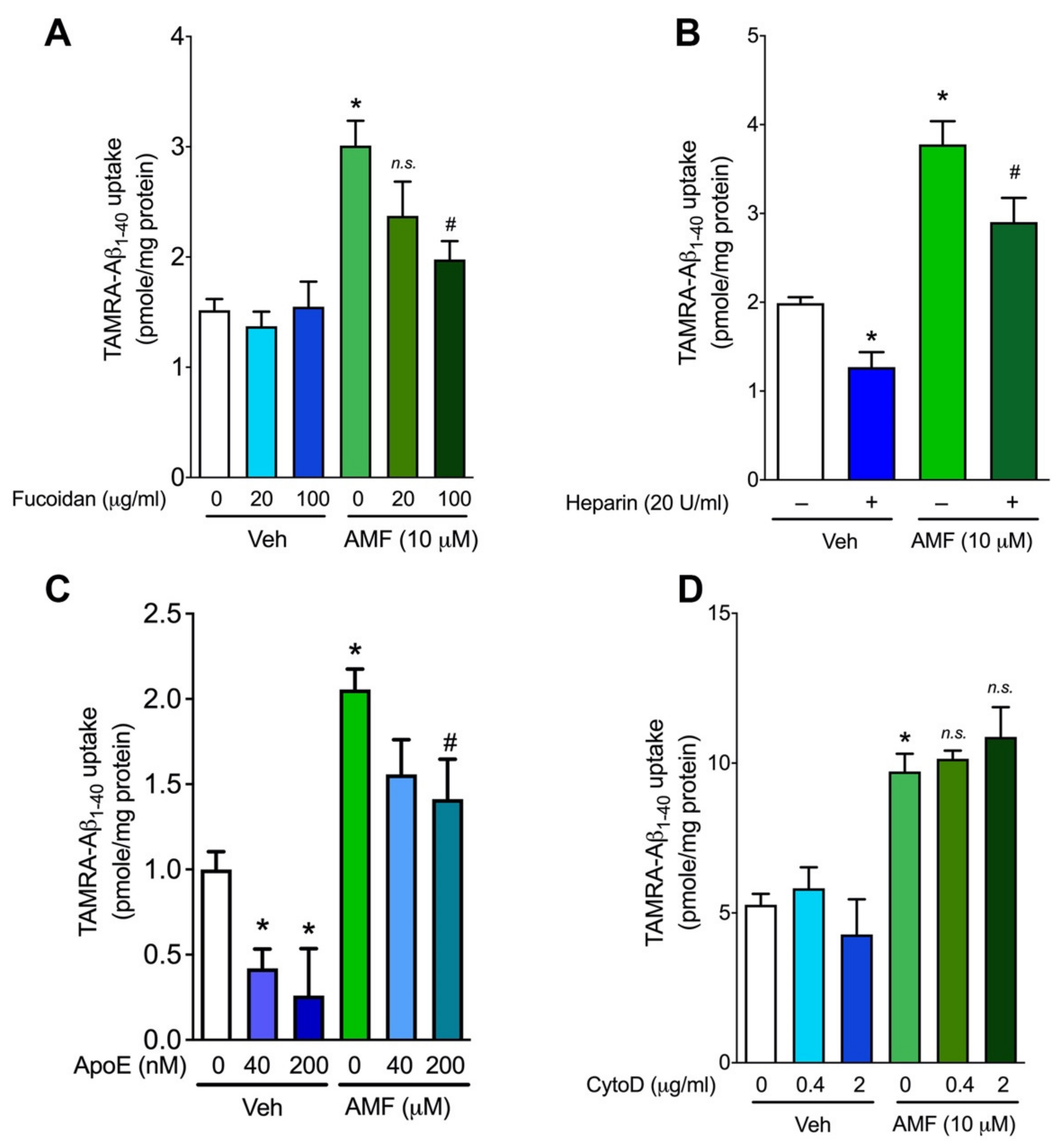
2.4. Evidence That AMF Promotes Aβ1-40 Uptake via the Receptor-Mediated Endocytosis
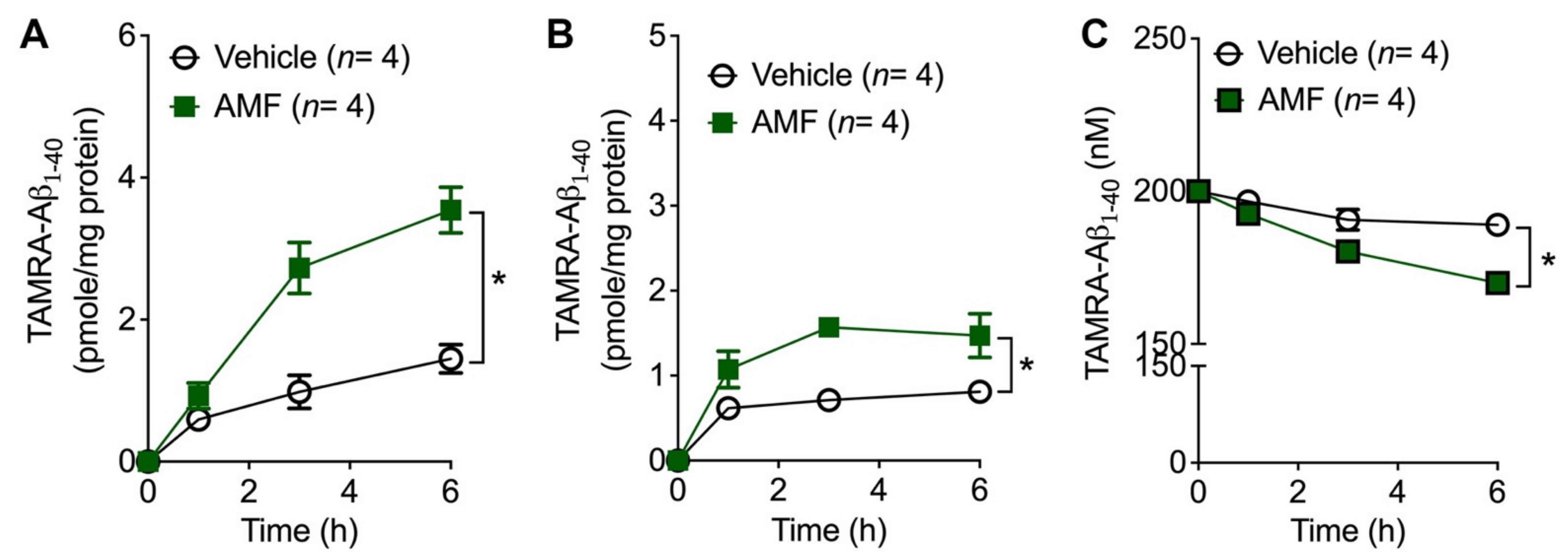
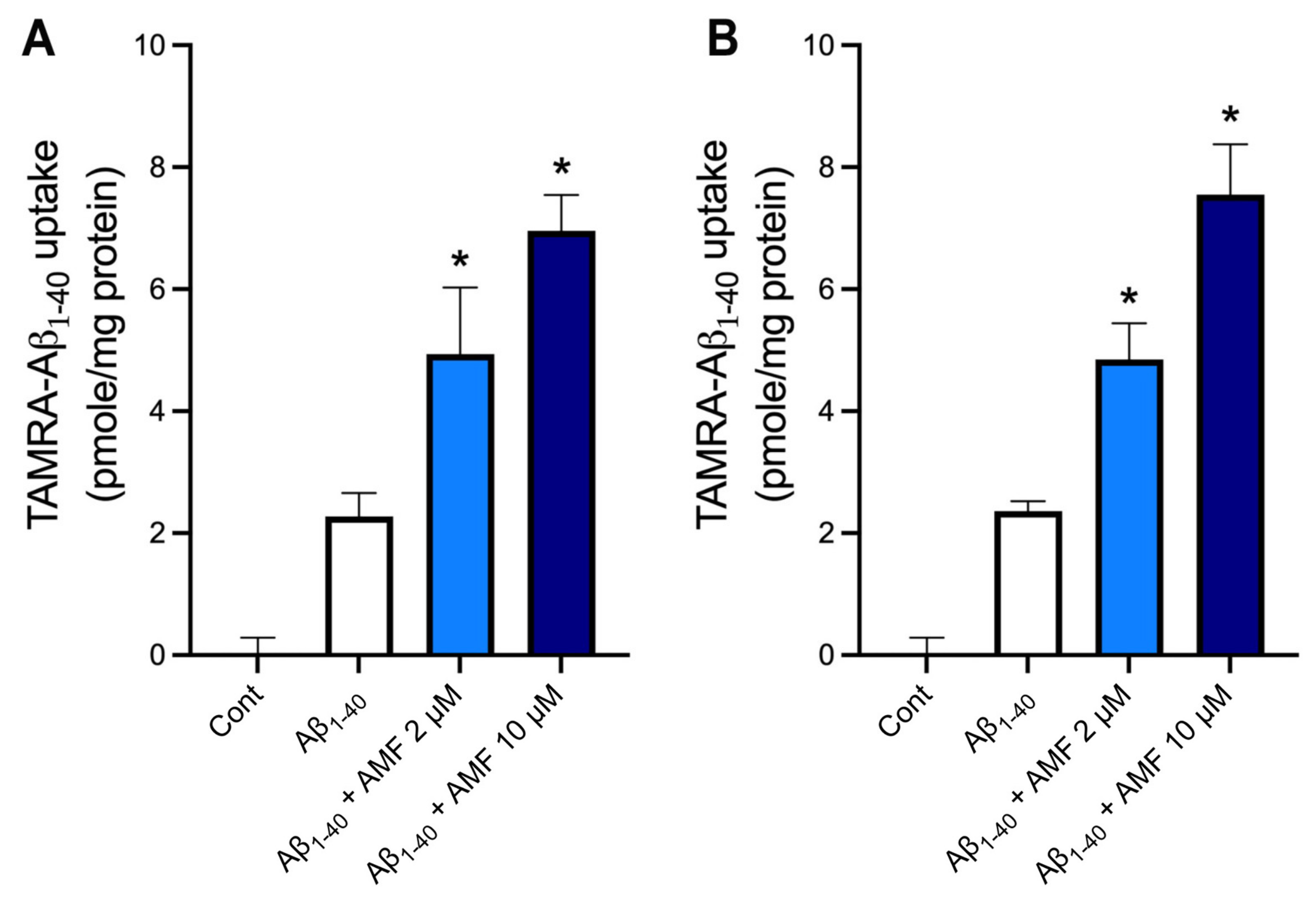
2.5. AMF Promotes Aβ1-40 Uptake in Microglia and Astrocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Aβ Uptake Assay
4.4. Lactate Dehydrogenase (LDH) Activity Assay
4.5. SDS-PAGE Electrophoresis and Immunoblotting
4.6. Immunofluorescent Labeling and Confocal Microscopy
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ubhi, K.; Masliah, E. Alzheimer’s disease: Recent advances and future perspectives. J. Alzheimers Dis. 2013, 33, S185–S194. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.F. Perspective on race and ethnicity in Alzheimer’s disease research. Alzheimers Dement. 2008, 4, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. A summary risk score for the prediction of Alzheimer disease in elderly persons. Arch. Neurol. 2010, 67, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Rensink, A.A.; de Waal, R.M.; Kremer, B.; Verbeek, M.M. Pathogenesis of cerebral amyloid angiopathy. Brain Res. Rev. 2003, 43, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [Green Version]
- Bredesen, D.E. Neurodegeneration in Alzheimer’s disease: Caspases and synaptic element interdependence. Mol. Neurodegener. 2009, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral Abeta in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell. Mol. Life Sci. 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, T.; Liu, C.C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, T.; Cirrito, J.R.; Liu, C.C.; Shinohara, M.; Li, J.; Schuler, D.R.; Shinohara, M.; Holtzman, D.M.; Bu, G. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J. Neurosci. 2013, 33, 19276–19283. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.C.; Zhang, L.; Bu, G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ye, R.D. Microglial Abeta receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef]
- Kang, S.S.; Lee, J.Y.; Choi, Y.K.; Song, S.S.; Kim, J.S.; Jeon, S.J.; Han, Y.N.; Son, K.H.; Han, B.H. Neuroprotective effects of naturally occurring biflavonoids. Bioorg. Med. Chem. Lett. 2005, 15, 3588–3591. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kang, S.S.; Lee, S.K.; Han, B.H. Polyphenolic Biflavonoids Inhibit Amyloid-Beta Fibrillation and Disaggregate Preformed Amyloid-Beta Fibrils. Biomol. Ther. (Seoul) 2020, 28, 145–151. [Google Scholar] [CrossRef]
- Thapa, A.; Woo, E.R.; Chi, E.Y.; Sharoar, M.G.; Jin, H.G.; Shin, S.Y.; Park, I.S. Biflavonoids are superior to monoflavonoids in inhibiting amyloid-beta toxicity and fibrillogenesis via accumulation of nontoxic oligomer-like structures. Biochemistry 2011, 50, 2445–2455. [Google Scholar] [CrossRef] [PubMed]
- Windsor, P.K.; Plassmeyer, S.P.; Mattock, D.S.; Bradfield, J.C.; Choi, E.Y.; Miller, B.R., 3rd; Han, B.H. Biflavonoid-Induced Disruption of Hydrogen Bonds Leads to Amyloid-beta Disaggregation. Int. J. Mol. Sci. 2021, 22, 2888. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Sharma, A.K.; Han, B.H.; Mirica, L.M. Amentoflavone: A Bifunctional Metal Chelator that Controls the Formation of Neurotoxic Soluble Aβ42 Oligomers. ACS Chem. Neurosci. 2020, 11, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, B.; Cheng, G.; Yang, X.; Zhao, N.; Shi, R. Amentoflavone Ameliorates Abeta1-42-Induced Memory Deficits and Oxidative Stress in Cellular and Rat Model. Neurochem. Res. 2018, 43, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, C.; Zheng, M.; Liu, S.; Shi, R. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci. 2019, 239, 117043. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon, R.; Fuenzalida, C.; Santibanez, M.; von Bernhardi, R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J. Biol. Chem. 2005, 280, 30406–30415. [Google Scholar]
- Brandenburg, L.O.; Konrad, M.; Wruck, C.J.; Koch, T.; Lucius, R.; Pufe, T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1-42-induced signal transduction in glial cells. J. Neurochem. 2010, 113, 749–760. [Google Scholar] [CrossRef]
- Thelen, T.; Hao, Y.; Medeiros, A.I.; Curtis, J.L.; Serezani, C.H.; Kobzik, L.; Harris, L.H.; Aronoff, D.M. The class A scavenger receptor, macrophage receptor with collagenous structure, is the major phagocytic receptor for Clostridium sordellii expressed by human decidual macrophages. J. Immunol. 2010, 185, 4328–4335. [Google Scholar] [CrossRef] [Green Version]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef] [Green Version]
- Omtri, R.S.; Davidson, M.W.; Arumugam, B.; Poduslo, J.F.; Kandimalla, K.K. Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: Ramifications for the Alzheimer’s drug discovery. Mol. Pharm. 2012, 9, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 718188. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisson, J.; McAlpine, J.B.; Friesen, J.B.; Chen, S.N.; Graham, J.; Pauli, G.F. Can Invalid Bioactives Undermine Natural Product-Based Drug Discovery? J. Med. Chem. 2016, 59, 1671–1690. [Google Scholar] [CrossRef]
- Shin, D.H.; Bae, Y.C.; Kim-Han, J.S.; Lee, J.H.; Choi, I.Y.; Son, K.H.; Kang, S.S.; Kim, W.K.; Han, B.H. Polyphenol amentoflavone affords neuroprotection against neonatal hypoxic-ischemic brain damage via multiple mechanisms. J. Neurochem. 2006, 96, 561–572. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional compounds for controlling metal-mediated aggregation of the abeta42 peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Tang, N.; Lai, X.; Zhang, J.; Wen, W.; Li, X.; Li, A.; Wu, Y.; Liu, Z. Insights Into Amentoflavone: A Natural Multifunctional Biflavonoid. Front. Pharmacol. 2021, 12, 768708. [Google Scholar] [CrossRef]
- Yu, S.; Yan, H.; Zhang, L.; Shan, M.; Chen, P.; Ding, A.; Li, S.F. A Review on the Phytochemistry, Pharmacology, and Pharmacokinetics of Amentoflavone, a Naturally-Occurring Biflavonoid. Molecules 2017, 22, 299. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Bu, G. Receptor-associated protein interacts with amyloid-beta peptide and promotes its cellular uptake. J. Biol. Chem. 2009, 284, 33352–33359. [Google Scholar] [CrossRef] [Green Version]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in amyloid-beta production and metabolism. Ann. N. Y. Acad. Sci. 2006, 1086, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Otte-Holler, I.; David, G.; Maat-Schieman, M.L.; van den Heuvel, L.P.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol. (Berlin) 2001, 102, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Kleinnijenhuis, J.; Maass, C.N.; Rensink, A.A.; Otte-Holler, I.; David, G.; van den Heuvel, L.P.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol. Aging 2002, 23, 537–545. [Google Scholar] [CrossRef]
- Cotman, S.L.; Halfter, W.; Cole, G.J. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 2000, 15, 183–198. [Google Scholar] [CrossRef]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef]
- Schulz, J.G.; Megow, D.; Reszka, R.; Villringer, A.; Einhaupl, K.M.; Dirnagl, U. Evidence that glypican is a receptor mediating beta-amyloid neurotoxicity in PC12 cells. Eur. J. Neurosci. 1998, 10, 2085–2093. [Google Scholar] [CrossRef]
- Caruso, G.; Torrisi, S.A.; Mogavero, M.P.; Currenti, W.; Castellano, S.; Godos, J.; Ferri, R.; Galvano, F.; Leggio, G.M.; Grosso, G.; et al. Polyphenols and neuroprotection: Therapeutic implications for cognitive decline. Pharmacol. Ther. 2022, 232, 108013. [Google Scholar] [CrossRef]
- Wicinski, M.; Domanowska, A.; Wodkiewicz, E.; Malinowski, B. Neuroprotective Properties of Resveratrol and Its Derivatives-Influence on Potential Mechanisms Leading to the Development of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2749. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s Disease and in the Gut-Brain Axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.H.; Akter, R.; Bhattacharya, T.; Abdel-Daim, M.M.; Alkahtani, S.; Arafah, M.W.; Al-Johani, N.S.; Alhoshani, N.M.; Alkeraishan, N.; Alhenaky, A.; et al. Resveratrol and Neuroprotection: Impact and Its Therapeutic Potential in Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 619024. [Google Scholar] [CrossRef]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, M.; Fryer, J.D.; Sullivan, P.M.; Christopher, E.A.; Wahrle, S.E.; DeMattos, R.B.; O’Dell, M.A.; Fagan, A.M.; Lashuel, H.A.; Walz, T.; et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 2005, 19, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.H.; Zhou, M.L.; Abousaleh, F.; Brendza, R.P.; Dietrich, H.H.; Koenigsknecht-Talboo, J.; Cirrito, J.R.; Milner, E.; Holtzman, D.M.; Zipfel, G.J. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J. Neurosci. 2008, 28, 13542–13550. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, B.H.; Cofell, B.; Everhart, E.; Humpal, C.; Kang, S.-S.; Lee, S.K.; Kim-Han, J.S. Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells. Int. J. Mol. Sci. 2022, 23, 5885. https://doi.org/10.3390/ijms23115885
Han BH, Cofell B, Everhart E, Humpal C, Kang S-S, Lee SK, Kim-Han JS. Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells. International Journal of Molecular Sciences. 2022; 23(11):5885. https://doi.org/10.3390/ijms23115885
Chicago/Turabian StyleHan, Byung Hee, Brooke Cofell, Emily Everhart, Courtney Humpal, Sam-Sik Kang, Sang Kook Lee, and Jeong Sook Kim-Han. 2022. "Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells" International Journal of Molecular Sciences 23, no. 11: 5885. https://doi.org/10.3390/ijms23115885
APA StyleHan, B. H., Cofell, B., Everhart, E., Humpal, C., Kang, S.-S., Lee, S. K., & Kim-Han, J. S. (2022). Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells. International Journal of Molecular Sciences, 23(11), 5885. https://doi.org/10.3390/ijms23115885