
Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome

 , , ,
, , ,  , ,
, ,  ,
, 
Abstract
:1. Introduction
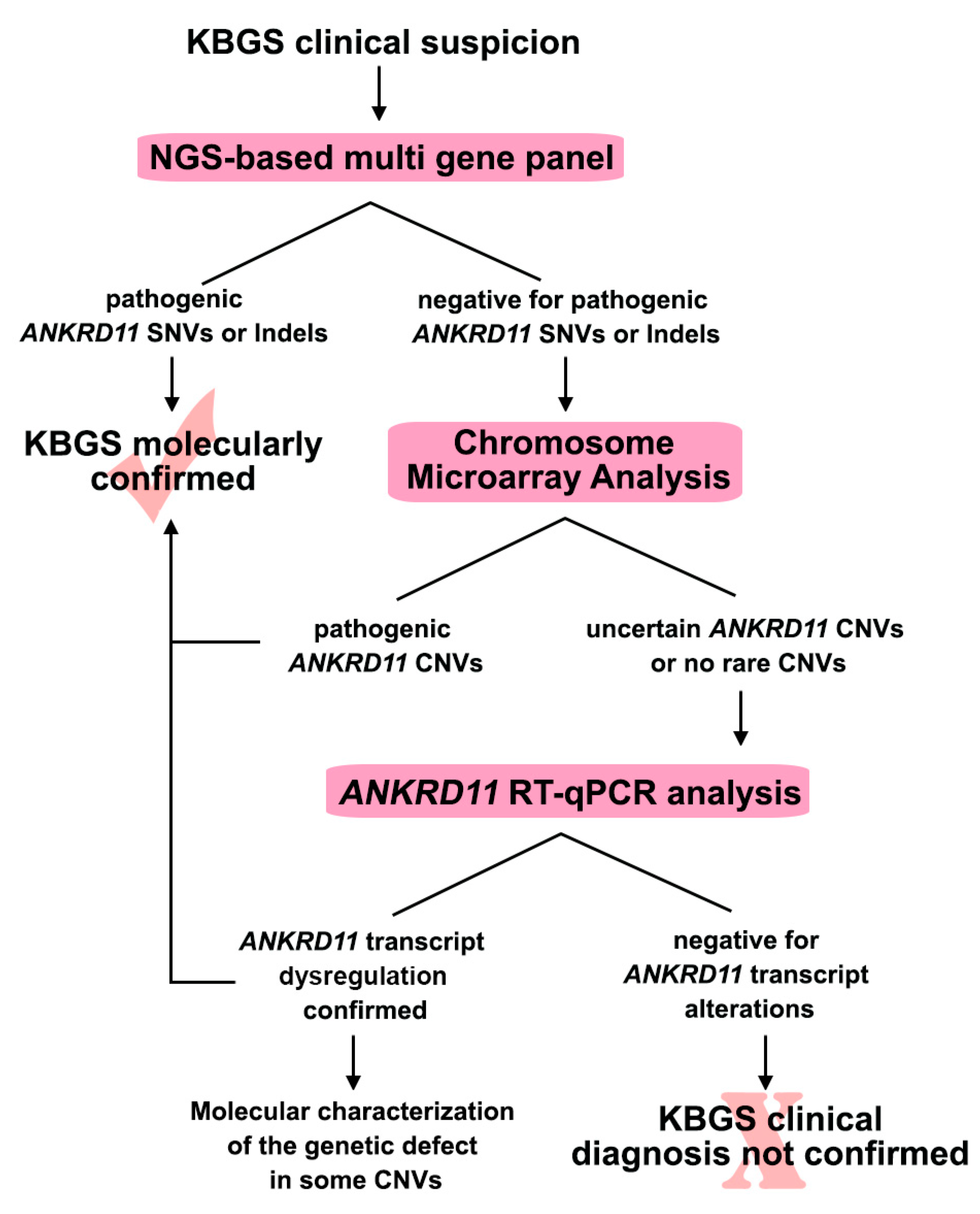
2. Results
2.1. Sequence Analysis Discovered New ANKRD11 Pathogenic Variants
2.2. High-Resolution CMA Evidenced Never Reported ANKRD11 Rearrangements
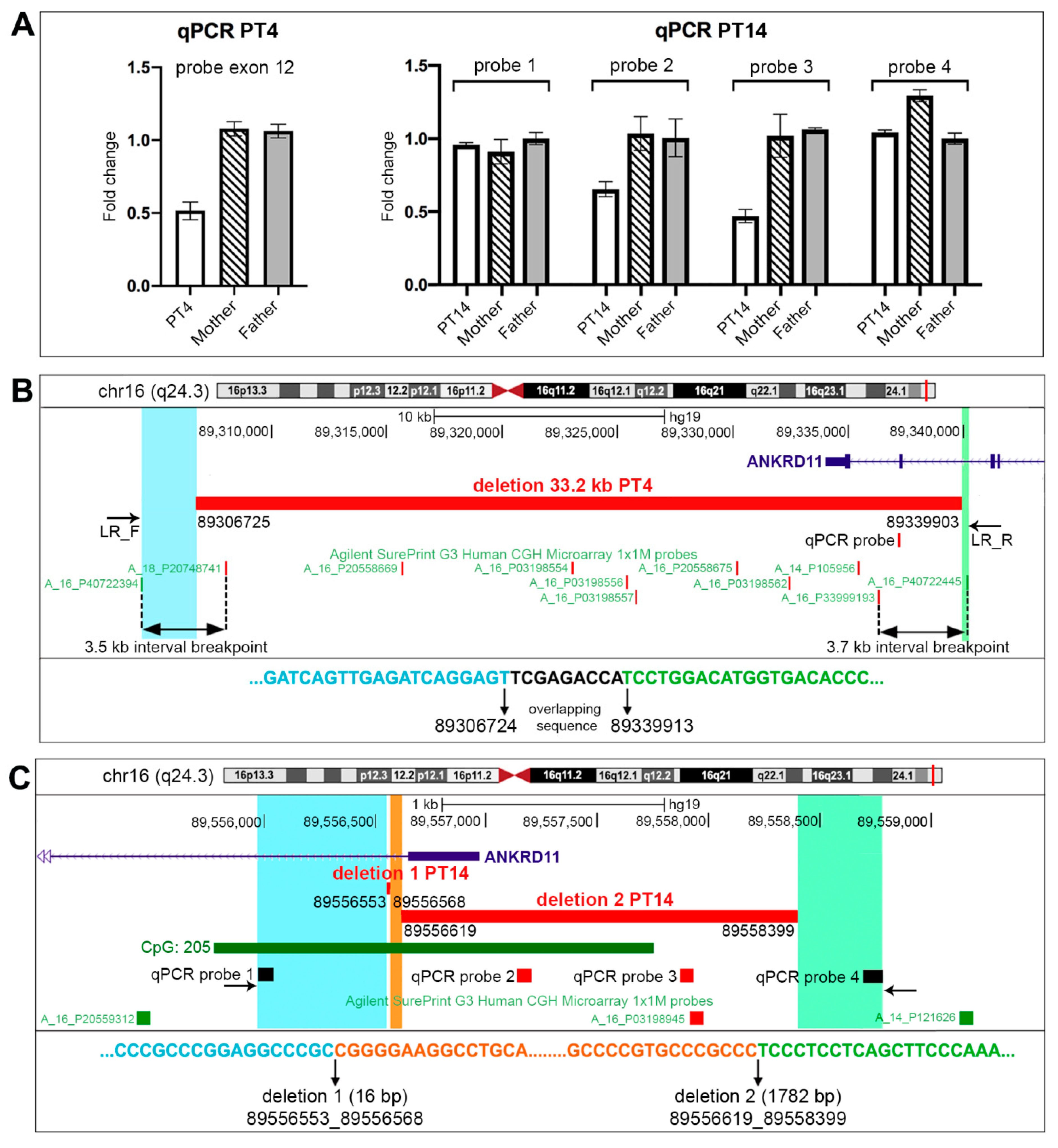
2.3. RT-qPCR Analysis: A Valuable Diagnostic Approach
2.4. Molecular Characterization of PT4 and PT14 Patients’ CNVs
2.5. Statistical Analysis
3. Discussion
4. Materials and Methods
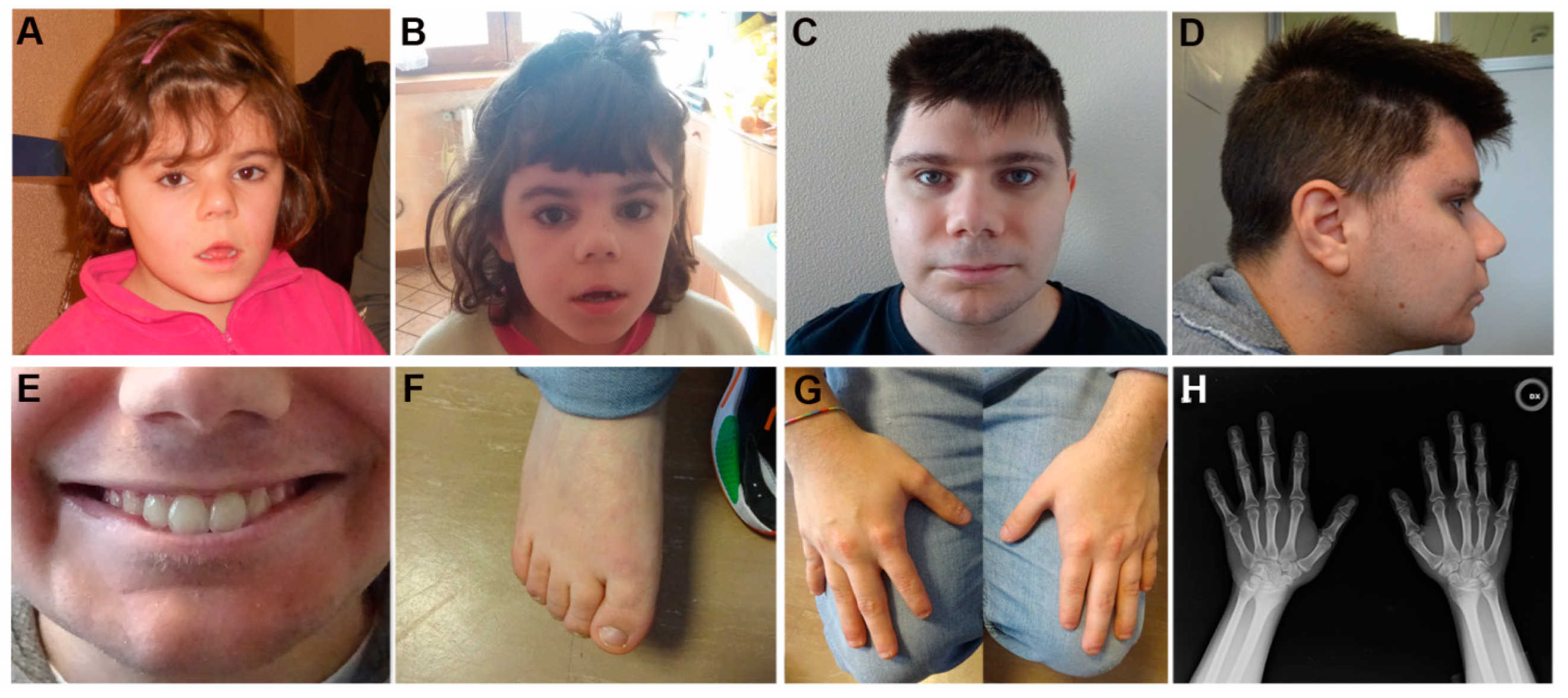
4.1. Patients
4.2. ANKRD11 Sequencing
4.3. High-Resolution Chromosome Microarray Analysis
4.4. Evaluation of ANKRD11 RELATIVE Expression
4.5. CNVs Molecular Characterization
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, J.; Pallister, P.D.; Tiddy, W.; Opitz, J.M. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects. Orig. Artic. Ser. 1975, 11, 7–18. [Google Scholar] [PubMed]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crippa, M.; Rusconi, D.; Castronovo, C.; Bestetti, I.; Russo, S.; Cereda, A.; Selicorni, A.; Larizza, L.; Finelli, P. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet. 2015, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, I.; Mallozzi, M.B.; Hüning, I.; Gervasini, C.; Kuechler, A.; Agolini, E.; Albrecht, B.; Baquero-Montoya, C.; Bohring, A.; Bramswig, N.C.; et al. ANKRD11 variants: KBG syndrome and beyond. Clin. Genet. 2021, 100, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Low, K.; Ashraf, T.; Canham, N.; Clayton-Smith, J.; Deshpande, C.; Donaldson, A.; Fisher, R.; Flinter, F.; Foulds, N.; Fryer, A.; et al. Clinical and genetic aspects of KBG syndrome. Am. J. Med. Genet. A 2016, 170, 2835–2846. [Google Scholar] [CrossRef] [Green Version]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Hanly, C.; Shah, H.; Au, P.Y.B.; Murias, K. Description of neurodevelopmental phenotypes associated with 10 genetic neurodevelopmental disorders: A scoping review. Clin. Genet. 2021, 99, 335–346. [Google Scholar] [CrossRef]
- Abe-Hatano, C.; Iida, A.; Kosugi, S.; Momozawa, Y.; Terao, C.; Ishikawa, K.; Okubo, M.; Hachiya, Y.; Nishida, H.; Nakamura, K.; et al. Whole genome sequencing of 45 Japanese patients with intellectual disability. Am. J. Med. Genet. A 2021, 185, 1468–1480. [Google Scholar] [CrossRef]
- Spengler, S.; Oehl-Jaschkowitz, B.; Begemann, M.; Hennes, P.; Zerres, K.; Eggermann, T. Haploinsufficiency of ANKRD11 (16q24.3) Is Not Obligatorily Associated with Cognitive Impairment but Shows a Clinical Overlap with Silver-Russell Syndrome. Mol. Syndromol. 2013, 4, 246–249. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, S.; Okamoto, N.; Stark, Z.; Nabetani, M.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Mizuguchi, T.; Ohtake, A.; Saitsu, H.; et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J. Hum. Genet. 2017, 62, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, I.; Teresa-Rodrigo, M.E.; Pozojevic, J.; Ruiz Gil, S.; Bader, I.; Braunholz, D.; Bramswig, N.C.; Gervasini, C.; Larizza, L.; Pfeiffer, L.; et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum. Genet. 2017, 136, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.; Bestetti, I.; Maitz, S.; Weiss, K.; Spano, A.; Masciadri, M.; Smithson, S.; Larizza, L.; Low, K.; Cohen, L.; et al. SETD5 Gene Haploinsufficiency in Three Patients with Suspected KBG Syndrome. Front. Neurol. 2020, 11, 631. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, G.; Gnazzo, M.; Novelli, A.; Grammatico, P. Clinical refinement of the SETD5-associated phenotype in a child displaying novel features and KBG syndrome-like appearance. Am. J. Med. Genet. A 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Larizza, L.; Finelli, P. Developmental disorders with intellectual disability driven by chromatin dysregulation: Clinical overlaps and molecular mechanisms. Clin. Genet. 2019, 95, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.; Voronova, A.; Zander, M.A.; Cancino, G.I.; Bramall, A.; Krause, M.P.; Abad, C.; Tekin, M.; Neilsen, P.M.; Callen, D.F.; et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev. Cell. 2015, 32, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Ka, M.; Kim, W.Y. ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol. Dis. 2018, 111, 138–152. [Google Scholar] [CrossRef]
- Zhang, A.; Li, C.W.; Chen, J.D. Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem Biophys. Res. Commun. 2007, 358, 1034–1040. [Google Scholar] [CrossRef] [Green Version]
- Walz, K.; Cohen, D.; Neilsen, P.M.; Foster, J., 2nd; Brancati, F.; Demir, K.; Fisher, R.; Moffat, M.; Verbeek, N.E.; Bjørgo, K.; et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum. Genet. 2015, 134, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.; Luk, H.M.; Lo, I.F.M. KBG syndrome in a Chinese population: A case series. Am. J. Med. Genet. A 2022, in press. [Google Scholar] [CrossRef]
- Goldenberg, A.; Riccardi, F.; Tessier, A.; Pfundt, R.; Busa, T.; Cacciagli, P.; Capri, Y.; Coutton, C.; Delahaye-Duriez, A.; Frebourg, T.; et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am. J. Med. Genet. A 2016, 170, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Morel Swols, D.; Tekin, M. KBG Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Kutkowska-Kaźmierczak, A.; Boczar, M.; Kalka, E.; Castañeda, J.; Klapecki, J.; Pietrzyk, A.; Barczyk, A.; Malinowska, O.; Landowska, A.; Gambin, T.; et al. Wide Fontanels, Delayed Speech Development and Hoarse Voice as Useful Signs in the Diagnosis of KBG Syndrome: A Clinical Description of 23 Cases with Pathogenic Variants Involving the ANKRD11 Gene or Submicroscopic Chromosomal Rearrangements of 16q24.3. Genes 2021, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, E.; Ockeloen, C.W.; Kampen, R.A.; Hampstead, J.E.; Dingemans, A.J.M.; Rots, D.; Lütje, L.; Ashraf, T.; Baker, T.; Barat-Houari, M.; et al. Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein. medRxiv 2021. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Sun, C.; Yang, L.; Lu, W.; Luo, F. Comprehensive analysis of clinical spectrum and genotype associations in Chinese and literature reported KBG syndrome. Transl. Pediatr. 2021, 10, 834–842. [Google Scholar] [CrossRef]
- Bucerzan, S.; Miclea, D.; Lazea, C.; Asavoaie, C.; Kulcsar, A.; Grigorescu-Sido, P. 16q24.3 Microduplication in a Patient With Developmental Delay, Intellectual Disability, Short Stature, and Nonspecific Dysmorphic Features: Case Report and Review of the Literature. Front. Pediatr. 2020, 8, 390. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Pezzani, L.; Marchetti, D.; Cereda, A.; Caffi, L.G.; Manara, O.; Mamoli, D.; Pezzoli, L.; Lincesso, A.R.; Perego, L.; Pellicioli, I.; et al. Atypical presentation of pediatric BRAF RASopathy with acute encephalopathy. Am. J. Med. Genet. A 2018, 176, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Crippa, M.; Bonati, M.T.; Calzari, L.; Picinelli, C.; Gervasini, C.; Sironi, A.; Bestetti, I.; Guzzetti, S.; Bellone, S.; Selicorni, A.; et al. Molecular Etiology Disclosed by Array CGH in Patients with Silver-Russell Syndrome or Similar Phenotypes. Front. Genet. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Sequencing Method | Chromosomal Position (hg19) | Exon | HGVS Nomenclature * | Variant Type | Inheritance | ACMG Classification # | rs Number (dbSNP) | Allele Frequency (GnomAD) | Novelty | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stopgain and Indel variants | ||||||||||||
| PT1 | M | Sanger Sequencing | chr16:89350427 | 9 | c.2523G > A, (p.Trp841Ter) | Stopgain | De novo | P | unreported | unreported | This cohort | - |
| PT2 | F | Sanger Sequencing | chr16:89346392 | 9 | c.6552_6558dupTGAGGAG, (p.Pro2187Ter) | Insertion | De novo | P | unreported | unreported | This cohort | - |
| PT5 | M | NGS gene panel | chr16:89349628 | 9 | c.3319_3322delAAAG, (p.Lys1107AlafsTer210) | Deletion | Unknown | P | unreported | unreported | This cohort | - |
| PT6 | F | NGS gene panel | chr16:89348689 | 9 | c.4261G > T, (p.Glu1421Ter) | Stopgain | De novo | P | unreported | unreported | This cohort | - |
| PT9 | F | NGS gene panel | chr16:89346113 | 9 | c.6836_6837delTG, (p.Val2279GlyfsTer16) | Deletion | De novo | P | rs1555525296 | unreported | Reported $ | - |
| PT15 | F | NGS gene panel | chr16:89351563 | 9 | c.1388_1389delAA, (p.Lys463ArgfsTer29) | Deletion | De novo | P | unreported | unreported | This cohort | - |
| PT17 | M | NGS gene panel | chr16:89351043 | 9 | c.1903_1907delAAACA, (p.Lys635GlnfsTer26) | Deletion | De novo | P | rs886041125 | unreported | Reported | 25413698 |
| PT18 | M | NGS gene panel | chr16:89350538 | 9 | c.2408_2412delAAAAA, (p.Lys803ArgfsTer5) | Deletion | Unknown | P | rs886039902 | unreported | Reported | 27667800 |
| PT19 | F | NGS gene panel | chr16:89350753 | 9 | c.2197C > T, (p.Arg733Ter) | Stopgain | Unknown | P | rs886041791 | unreported | Reported | 31191201 |
| PT20 | M | NGS gene panel | chr16:89351491 | 9 | c.1459G > T, (p.Glu487Ter) | Stopgain | De novo | P | unreported | unreported | This cohort | - |
| PT22 ‡ | F | NGS gene panel | chr16:89351566 | 9 | c.1381_1384delGAAA, (p.Glu461GlnfsTer48) | Deletion | De novo | P | unreported | unreported | Reported | 27605097 |
| PT23 ‡ | F | Sanger Sequencing | chr16:89351566 | 9 | c.1381_1384delGAAA, (p.Glu461GlnfsTer48) | Deletion | De novo | P | unreported | unreported | Reported | 27605097 |
| PT24 | M | NGS gene panel | chr16:89349181 | 9 | c.3768_3769delCA, (p.His1256GlnfsTer26) | Deletion | De novo | P | unreported | unreported | Reported $ | - |
| PT26 | F | NGS gene panel | chr16:89350549 | 9 | c.2398_2401delGAAA, (p.Glu800AsnfsTer62) | Deletion | Unknown | P | rs797045027 | unreported | Reported | 25464108 |
| PT28 | M | NGS gene panel | chr16:89351578 | 9 | c.1372C > T, (p.Arg458Ter) | Stopgain | Unknown | P | rs900492387 | ƒ = 7.1 × 10−6 | Reported | 30202406 |
| PT29 | F | NGS gene panel | chr16:89350582 | 9 | c.2368G > T, (p.Glu790Ter) | Stopgain | Unknown | P | unreported | unreported | This cohort | - |
| PT30 | M | NGS gene panel | chr16:89351718 | 9 | c.1232C > A, (p.Ser411Ter) | Stopgain | Unknown | P | unreported | unreported | Reported | 32056211 |
| Missense variants | ||||||||||||
| PT3 | M | Sanger Sequencing | chr16:89349963 | 9 | c.2987G > T, (p.Gly996Val) | Missense | Maternal | LB | rs1205687342 | ƒ = 3.99 × 10−6 | This cohort | - |
| PT8 | F | NGS gene panel | chr16:89348536 | 9 | c.4414G > A, (p.Glu1472Lys) | Missense | Paternal | LB | rs1597451653 | unreported | This cohort | - |
| PT10 | F | NGS gene panel | chr16:89347549 | 9 | c.5401G > A, (p.Glu1801Lys) | Missense | Maternal | LB | rs938676909 | unreported | This cohort | - |
| PT21 | M | NGS gene panel | chr16:89345919 chr16:89346516 | 99 | c.7031T > C, (p.Leu2344Pro) c.6434C > T, (p.Thr2145Ile) | Missense Missense | Maternal Maternal | US B | unreportedrs 761862402 | unreported ƒ = 3.7 × 10−5 | This cohort This cohort | - |
| PT25 | M | NGS gene panel | chr16:89347752 | 9 | c.5198C > T, (p.Ala1733Val) | Missense | Maternal | B | rs148243995 | ƒ = 4.4 × 10−4 | Reported $ | - |
| PT27 | F | NGS gene panel | chr16: 89341329 | 11 | c.7606C > T, (p.Arg2536Trp) | Missense | De novo | LP | unreported | unreported | Reported † | - |
| Patient ID | Gain/Loss | CNV Description According to the ISCN Nomenclature * | Size | Classification # | Platform | Origin | Involved OMIM Genes |
|---|---|---|---|---|---|---|---|
| PT4 | Loss | arr[GRCh37] 16q24.3(89307972-89335487) × 1 dn | 27.5 kb | P | Agilent | De novo | ANKRD11 |
| PT12 | Loss | arr[GRCh37] 16q24.3(89555339-89556020) × 1 dn | 682 bp | LP | OGT | De novo | ANKRD11 |
| PT13 | Gain | arr[GRCh37] 16q24.3(89220725-89420725) × 3 | 200 kb | LP | Agilent | Unknown | ACSF3, CDH15, ANKRD11 |
| PT33 | Loss | arr[GRCh37] 16q24.3(88365786-89584412) × 1 | 1.2 Mb | P | Agilent | Unknown | ZNF469, CYBA, MVD, CTU2, PIEZO1, CDT1, APRT, GALNS, TRAPPC2L, ACSF3, CDH15, ANKRD11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bestetti, I.; Crippa, M.; Sironi, A.; Tumiatti, F.; Masciadri, M.; Smeland, M.F.; Naik, S.; Murch, O.; Bonati, M.T.; Spano, A.; et al. Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome. Int. J. Mol. Sci. 2022, 23, 5912. https://doi.org/10.3390/ijms23115912
Bestetti I, Crippa M, Sironi A, Tumiatti F, Masciadri M, Smeland MF, Naik S, Murch O, Bonati MT, Spano A, et al. Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome. International Journal of Molecular Sciences. 2022; 23(11):5912. https://doi.org/10.3390/ijms23115912
Chicago/Turabian StyleBestetti, Ilaria, Milena Crippa, Alessandra Sironi, Francesca Tumiatti, Maura Masciadri, Marie Falkenberg Smeland, Swati Naik, Oliver Murch, Maria Teresa Bonati, Alice Spano, and et al. 2022. "Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome" International Journal of Molecular Sciences 23, no. 11: 5912. https://doi.org/10.3390/ijms23115912
APA StyleBestetti, I., Crippa, M., Sironi, A., Tumiatti, F., Masciadri, M., Smeland, M. F., Naik, S., Murch, O., Bonati, M. T., Spano, A., Cattaneo, E., Mariani, M., Gotta, F., Crosti, F., Cavalli, P., Pantaleoni, C., Natacci, F., Bedeschi, M. F., Milani, D., ... Finelli, P. (2022). Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome. International Journal of Molecular Sciences, 23(11), 5912. https://doi.org/10.3390/ijms23115912