
The Transcription Factor EB (TFEB) Sensitizes the Heart to Chronic Pressure Overload

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
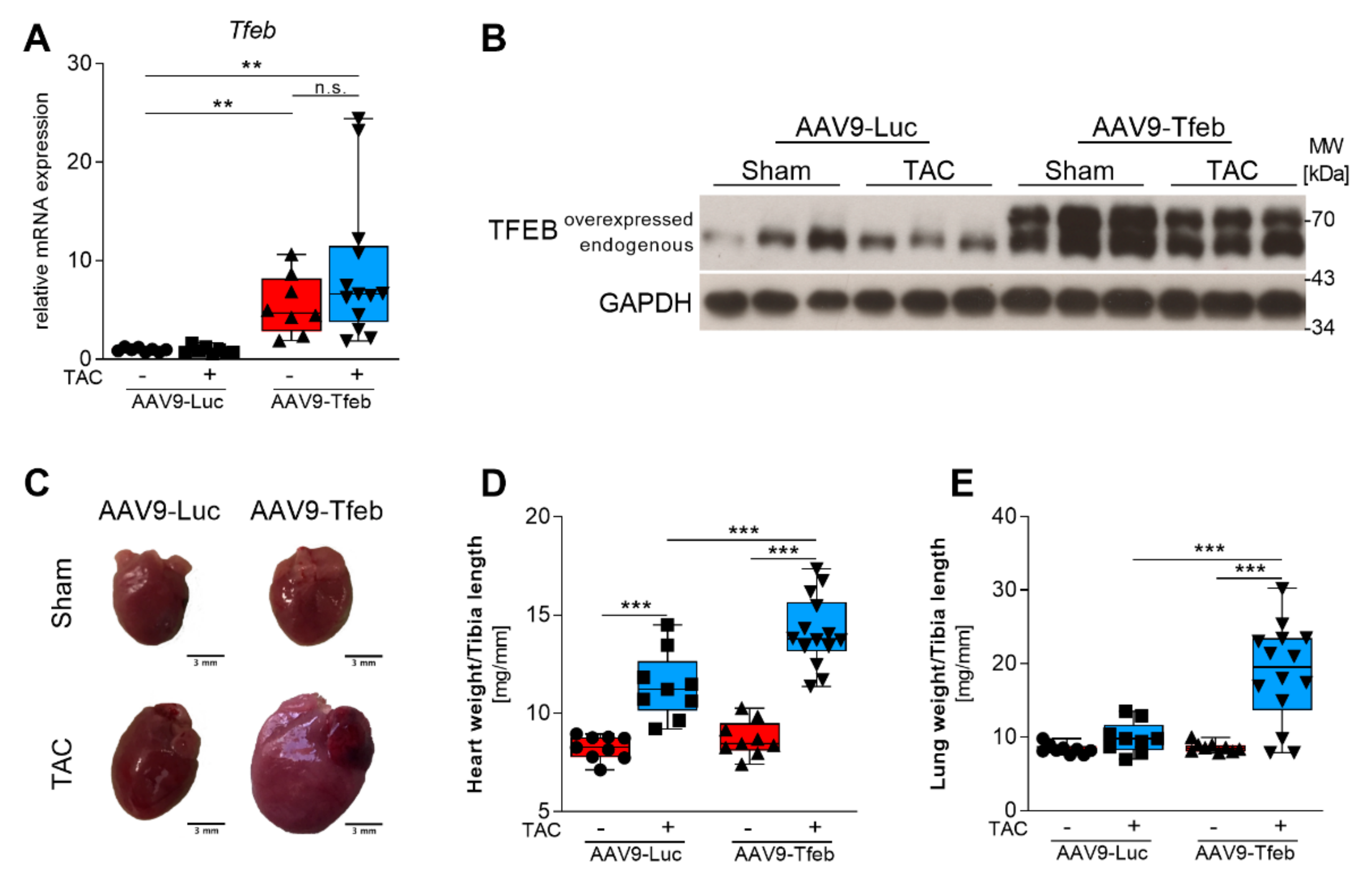
2.1. TFEB Overexpression Leads to Heart Failure in Response to Pressure Overload
2.2. TFEB Overexpression Aggravates Pressure-Overload Induced Cardiac Stress Response
2.3. TAC-Induced Interstitial Fibrosis Is Augmented by TFEB Overexpression
2.4. TFEB Increases ALP Gene Expression during Chronic Pressure Overload
3. Discussion
4. Materials and Methods
4.1. Construction of Adeno-Associated Vectors
4.2. Animal Model
4.3. Transthoracic Echocardiography
4.4. Histological Analyses
4.5. RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR
4.6. RNA-Sequencing
4.7. Protein Extraction
4.8. Statistical Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, D.; Garrison, R.J.; Savage, D.D.; Kannel, W.B.; Castelli, W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990, 322, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.L.; Horwich, T.B.; Fonarow, G.C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Ike, C.; Li, L.; Wang, D.Z.; Glass, D.J.; Patterson, C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ. Res. 2007, 100, 456–459. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodriguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Fielitz, J.; van Rooij, E.; Spencer, J.A.; Shelton, J.M.; Latif, S.; van der Nagel, R.; Bezprozvannaya, S.; de Windt, L.; Richardson, J.A.; Bassel-Duby, R.; et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc. Natl. Acad. Sci. USA 2007, 104, 4377–4382. [Google Scholar] [CrossRef] [Green Version]
- Lodka, D.; Pahuja, A.; Geers-Knorr, C.; Scheibe, R.J.; Nowak, M.; Hamati, J.; Kohncke, C.; Purfurst, B.; Kanashova, T.; Schmidt, S.; et al. Muscle RING-finger 2 and 3 maintain striated-muscle structure and function. J. Cachexia Sarcopenia Muscle 2016, 7, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Tannous, P.; Zhu, H.; Nemchenko, A.; Berry, J.M.; Johnstone, J.L.; Shelton, J.M.; Miller, F.J., Jr.; Rothermel, B.A.; Hill, J.A. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 2008, 117, 3070–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, M.; Abdul-Hussein, S.; Oldfors, A.; Gonzalez-Costello, J.; van der Ven, P.F.; Furst, D.O.; Gonzalez, L.; Moreno, D.; Torrejon-Escribano, B.; Alio, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef] [PubMed]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Undyala, V.V.; Gottlieb, R.A.; Mentzer, R.M., Jr.; Przyklenk, K. Autophagy: Definition, molecular machinery, and potential role in myocardial ischemia-reperfusion injury. J. Cardiovasc. Pharm. Ther. 2010, 15, 220–230. [Google Scholar] [CrossRef]
- Van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Du Bois, P.; Pablo Tortola, C.; Lodka, D.; Kny, M.; Schmidt, F.; Song, K.; Schmidt, S.; Bassel-Duby, R.; Olson, E.N.; Fielitz, J. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ. Res. 2015, 117, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Pablo Tortola, C.; Fielitz, B.; Li, Y.; Rudebusch, J.; Luft, F.C.; Fielitz, J. Activation of Tripartite Motif Containing 63 Expression by Transcription Factor EB and Transcription Factor Binding to Immunoglobulin Heavy Chain Enhancer 3 Is Regulated by Protein Kinase D and Class IIa Histone Deacetylases. Front. Physiol. 2020, 11, 550506. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle. Mol. Ther. Methods Clin. Dev. 2016, 3, 16054. [Google Scholar] [CrossRef]
- Gatto, F.; Rossi, B.; Tarallo, A.; Polishchuk, E.; Polishchuk, R.; Carrella, A.; Nusco, E.; Alvino, F.G.; Iacobellis, F.; De Leonibus, E.; et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci. Rep. 2017, 7, 15089. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, H.; Guan, Y.; Wang, Q.; Zhou, F.; Jie, L.; Ju, J.; Pu, L.; Du, H.; Wang, X. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am. J. Transl. Res. 2015, 7, 1574–1587. [Google Scholar] [PubMed]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef] [Green Version]
- Fielitz, J.; Hein, S.; Mitrovic, V.; Pregla, R.; Zurbrugg, H.R.; Warnecke, C.; Schaper, J.; Fleck, E.; Regitz-Zagrosek, V. Activation of the cardiac renin-angiotensin system and increased myocardial collagen expression in human aortic valve disease. J. Am. Coll. Cardiol. 2001, 37, 1443–1449. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; Facchinetti, V.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Schepens, M.; Thijssen, J.; Schoenmakers, E.F.; van Kessel, A.G. Regulation of the MiTF/TFE bHLH-LZ transcription factors through restricted spatial expression and alternative splicing of functional domains. Nucleic Acids Res. 2004, 32, 2315–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ma, B.; Han, X. The role of autophagy in angiotensin II-induced pathological cardiac hypertrophy. J. Mol. Endocrinol. 2016, 57, R143–R152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.V.; Rothermel, B.A.; Hill, J.A. Autophagy in hypertensive heart disease. J. Biol. Chem. 2010, 285, 8509–8514. [Google Scholar] [CrossRef] [Green Version]
- Givvimani, S.; Munjal, C.; Tyagi, N.; Sen, U.; Metreveli, N.; Tyagi, S.C. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE 2012, 7, e32388. [Google Scholar] [CrossRef]
- Dammrich, J.; Pfeifer, U. Cardiac hypertrophy in rats after supravalvular aortic constriction. I. Size and number of cardiomyocytes, endothelial and interstitial cells. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1983, 43, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chi, R.F.; Qin, F.Z.; Guo, X.F. Distinct changes of myocyte autophagy during myocardial hypertrophy and heart failure: Association with oxidative stress. Exp. Physiol. 2016, 101, 1050–1063. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, Y.; Li, C.; Li, Y.; Jiang, S.; Zhang, X.; Ding, Z.; Tu, F.; Wu, J.; Gao, X.; et al. Class III PI3K-mediated prolonged activation of autophagy plays a critical role in the transition of cardiac hypertrophy to heart failure. J. Cell Mol. Med. 2015, 19, 1710–1719. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Ruegg, M.A.; et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 2011, 123, 1073–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drolet, M.C.; Desbiens-Brassard, V.; Roussel, E.; Tu, V.; Couet, J.; Arsenault, M. Blockade of the acute activation of mTOR complex 1 decreases hypertrophy development in rats with severe aortic valve regurgitation. Springerplus 2015, 4, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, B.C.; Kim, M.S.; van Rooij, E.; Plato, C.F.; Papst, P.J.; Vega, R.B.; McAnally, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N.; et al. Regulation of cardiac stress signaling by protein kinase d1. Mol. Cell Biol. 2006, 26, 3875–3888. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.D.; Zhao, J.J. TFEB Participates in the Abeta-Induced Pathogenesis of Alzheimer’s Disease by Regulating the Autophagy-Lysosome Pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef]
- Meng, X.; Luo, Y.; Liang, T.; Wang, M.; Zhao, J.; Sun, G.; Sun, X. Gypenoside XVII Enhances Lysosome Biogenesis and Autophagy Flux and Accelerates Autophagic Clearance of Amyloid-beta through TFEB Activation. J. Alzheimers Dis. 2016, 52, 1135–1150. [Google Scholar] [CrossRef]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef]
- Su, H.; Lu, R.; Kan, Y.W. Adeno-associated viral vector-mediated vascular endothelial growth factor gene transfer induces neovascular formation in ischemic heart. Proc. Natl. Acad. Sci. USA 2000, 97, 13801–13806. [Google Scholar] [CrossRef] [Green Version]
- Hulot, J.S.; Salem, J.E.; Redheuil, A.; Collet, J.P.; Varnous, S.; Jourdain, P.; Logeart, D.; Gandjbakhch, E.; Bernard, C.; Hatem, S.N.; et al. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: Results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail. 2017, 19, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Schlegel, P.; Huditz, R.; Meinhardt, E.; Rapti, K.; Geis, N.; Most, P.; Katus, H.A.; Muller, O.J.; Bekeredjian, R.; Raake, P.W. Locally Targeted Cardiac Gene Delivery by AAV Microbubble Destruction in a Large Animal Model. Hum. Gene Ther. Methods 2016, 27, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Muller, O.J.; Leuchs, B.; Pleger, S.T.; Grimm, D.; Franz, W.M.; Katus, H.A.; Kleinschmidt, J.A. Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc. Res. 2006, 70, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Goehringer, C.; Rutschow, D.; Bauer, R.; Schinkel, S.; Weichenhan, D.; Bekeredjian, R.; Straub, V.; Kleinschmidt, J.A.; Katus, H.A.; Muller, O.J. Prevention of cardiomyopathy in delta-sarcoglycan knockout mice after systemic transfer of targeted adeno-associated viral vectors. Cardiovasc. Res. 2009, 82, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; McTiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Pacak, C.A.; Mah, C.S.; Thattaliyath, B.D.; Conlon, T.J.; Lewis, M.A.; Cloutier, D.E.; Zolotukhin, I.; Tarantal, A.F.; Byrne, B.J. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 2006, 99, e3–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werfel, S.; Jungmann, A.; Lehmann, L.; Ksienzyk, J.; Bekeredjian, R.; Kaya, Z.; Leuchs, B.; Nordheim, A.; Backs, J.; Engelhardt, S.; et al. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV-mediated expression of Cre recombinase. Cardiovasc. Res. 2014, 104, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kny, M.; Csalyi, K.D.; Klaeske, K.; Busch, K.; Meyer, A.M.; Merks, A.M.; Darm, K.; Dworatzek, E.; Fliegner, D.; Baczko, I.; et al. Ninjurin1 regulates striated muscle growth and differentiation. PLoS ONE 2019, 14, e0216987. [Google Scholar] [CrossRef] [Green Version]
- Busch, K.; Kny, M.; Huang, N.; Klassert, T.E.; Stock, M.; Hahn, A.; Graeger, S.; Todiras, M.; Schmidt, S.; Chamling, B.; et al. Inhibition of the NLRP3/IL-1beta axis protects against sepsis-induced cardiomyopathy. J. Cachexia Sarcopenia Muscle 2021, 12, 1653–1668. [Google Scholar] [CrossRef]
- Schmidt, F.; Kny, M.; Zhu, X.; Wollersheim, T.; Persicke, K.; Langhans, C.; Lodka, D.; Kleber, C.; Weber-Carstens, S.; Fielitz, J. The E3 ubiquitin ligase TRIM62 and inflammation-induced skeletal muscle atrophy. Crit. Care 2014, 18, 545. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.; Kny, M.; Pablo-Tortola, C.; Todiras, M.; Willenbrock, M.; Schmidt, S.; Schmoeckel, K.; Jorde, I.; Nowak, M.; Jarosch, E.; et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 2020, 11, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanders, L.; Kny, M.; Hahn, A.; Schmidt, S.; Wundersitz, S.; Todiras, M.; Lahmann, I.; Bandyopadhyay, A.; Wollersheim, T.; Kaderali, L.; et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FastQC. Available online: www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 29 April 2022).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wundersitz, S.; Pablo Tortola, C.; Schmidt, S.; Oliveira Vidal, R.; Kny, M.; Hahn, A.; Zanders, L.; Katus, H.A.; Sauer, S.; Butter, C.; et al. The Transcription Factor EB (TFEB) Sensitizes the Heart to Chronic Pressure Overload. Int. J. Mol. Sci. 2022, 23, 5943. https://doi.org/10.3390/ijms23115943
Wundersitz S, Pablo Tortola C, Schmidt S, Oliveira Vidal R, Kny M, Hahn A, Zanders L, Katus HA, Sauer S, Butter C, et al. The Transcription Factor EB (TFEB) Sensitizes the Heart to Chronic Pressure Overload. International Journal of Molecular Sciences. 2022; 23(11):5943. https://doi.org/10.3390/ijms23115943
Chicago/Turabian StyleWundersitz, Sebastian, Cristina Pablo Tortola, Sibylle Schmidt, Ramon Oliveira Vidal, Melanie Kny, Alexander Hahn, Lukas Zanders, Hugo A. Katus, Sascha Sauer, Christian Butter, and et al. 2022. "The Transcription Factor EB (TFEB) Sensitizes the Heart to Chronic Pressure Overload" International Journal of Molecular Sciences 23, no. 11: 5943. https://doi.org/10.3390/ijms23115943