
Surface Chemical and Morphological Analysis of Chitosan/1,3-β-d-Glucan Polysaccharide Films Cross-Linked at 90 °C

 , ,
, ,  , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results
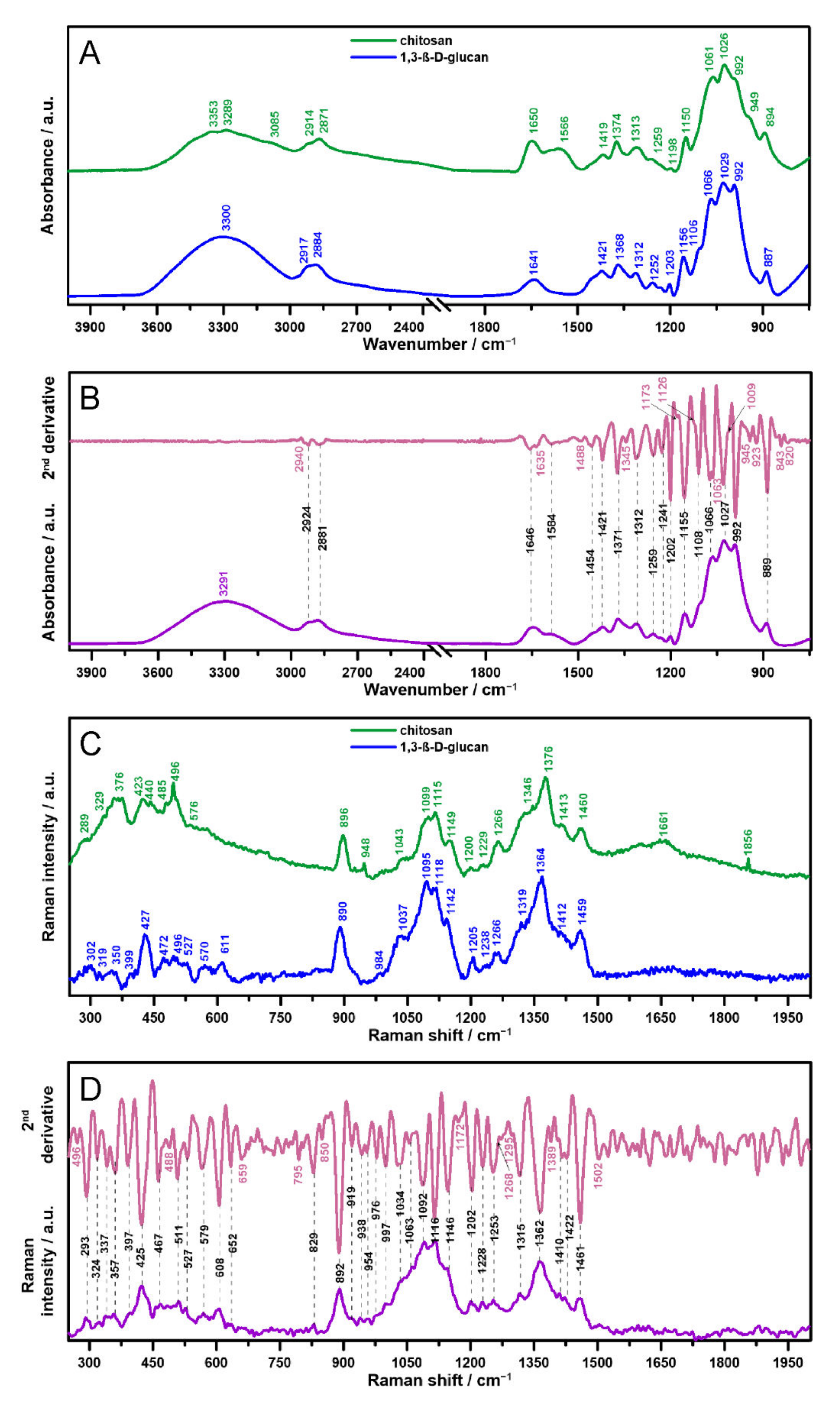
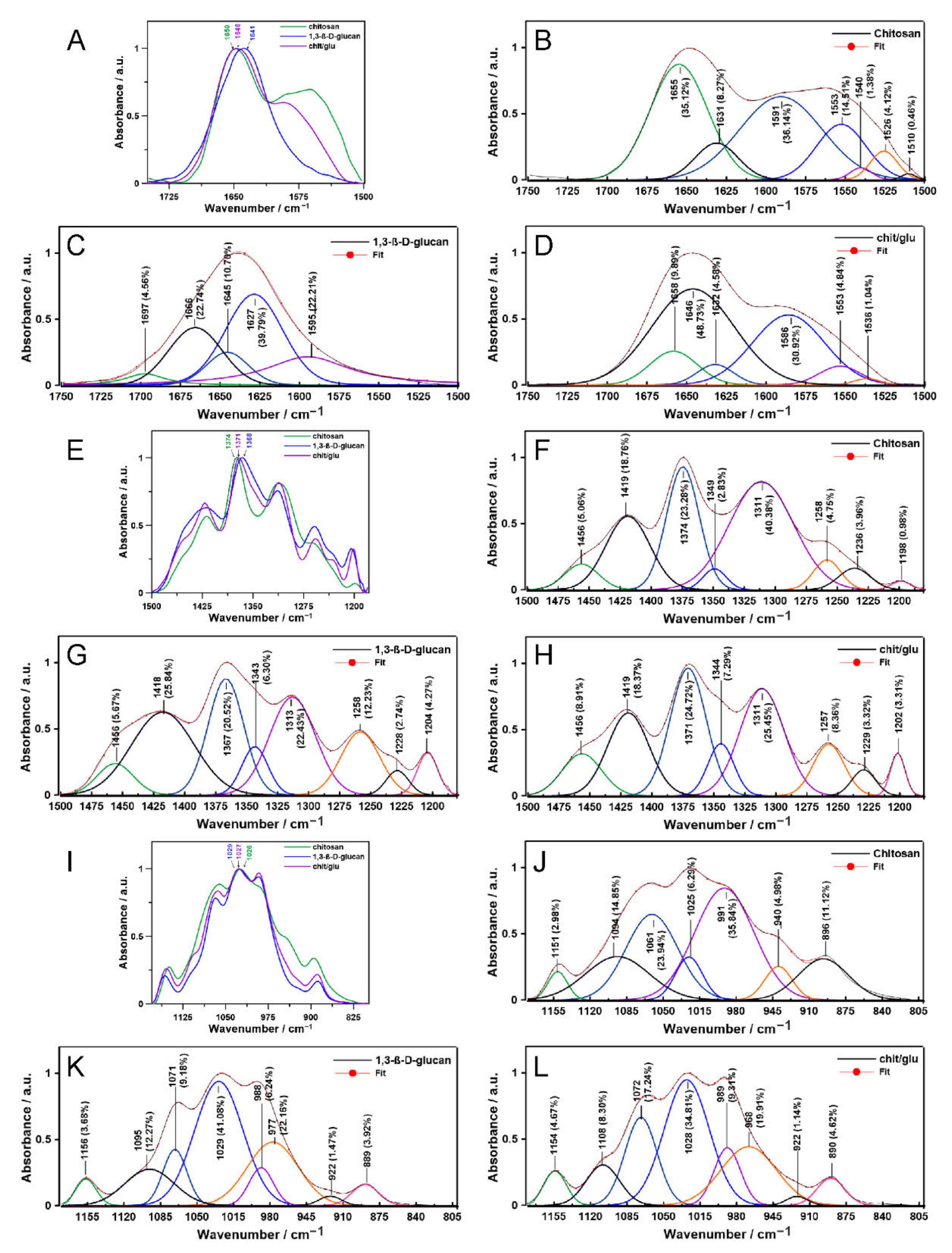
2.1. Vibrational Spectroscopy
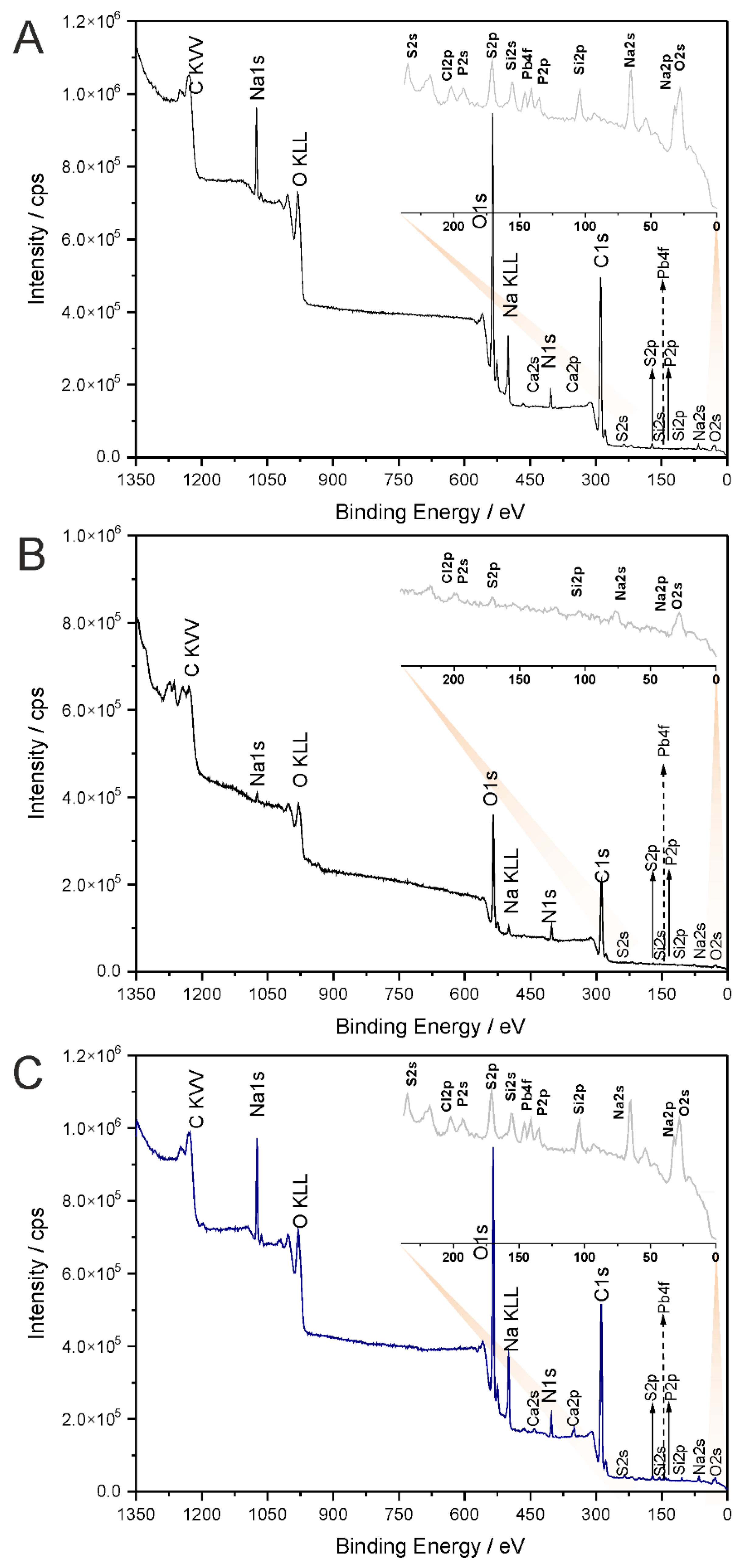
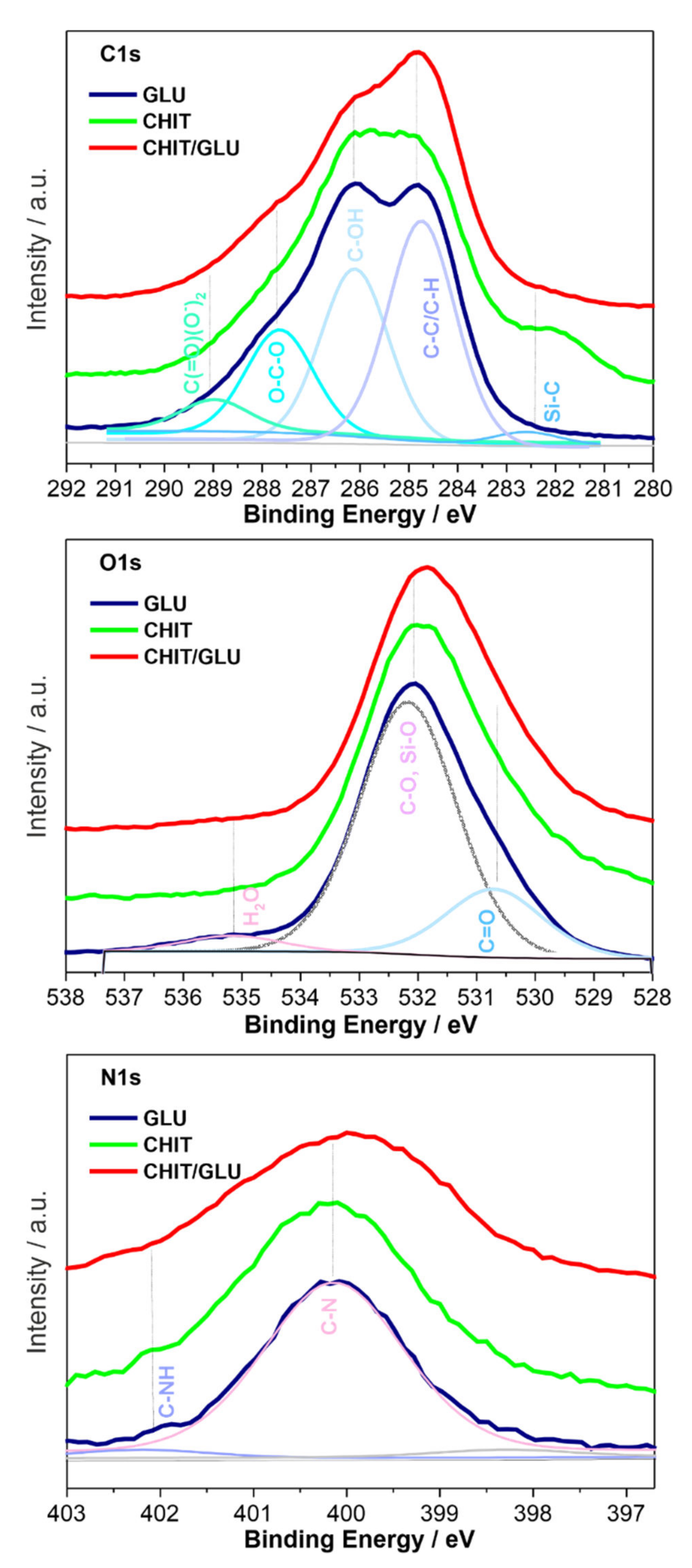
2.2. XPS Spectroscopy
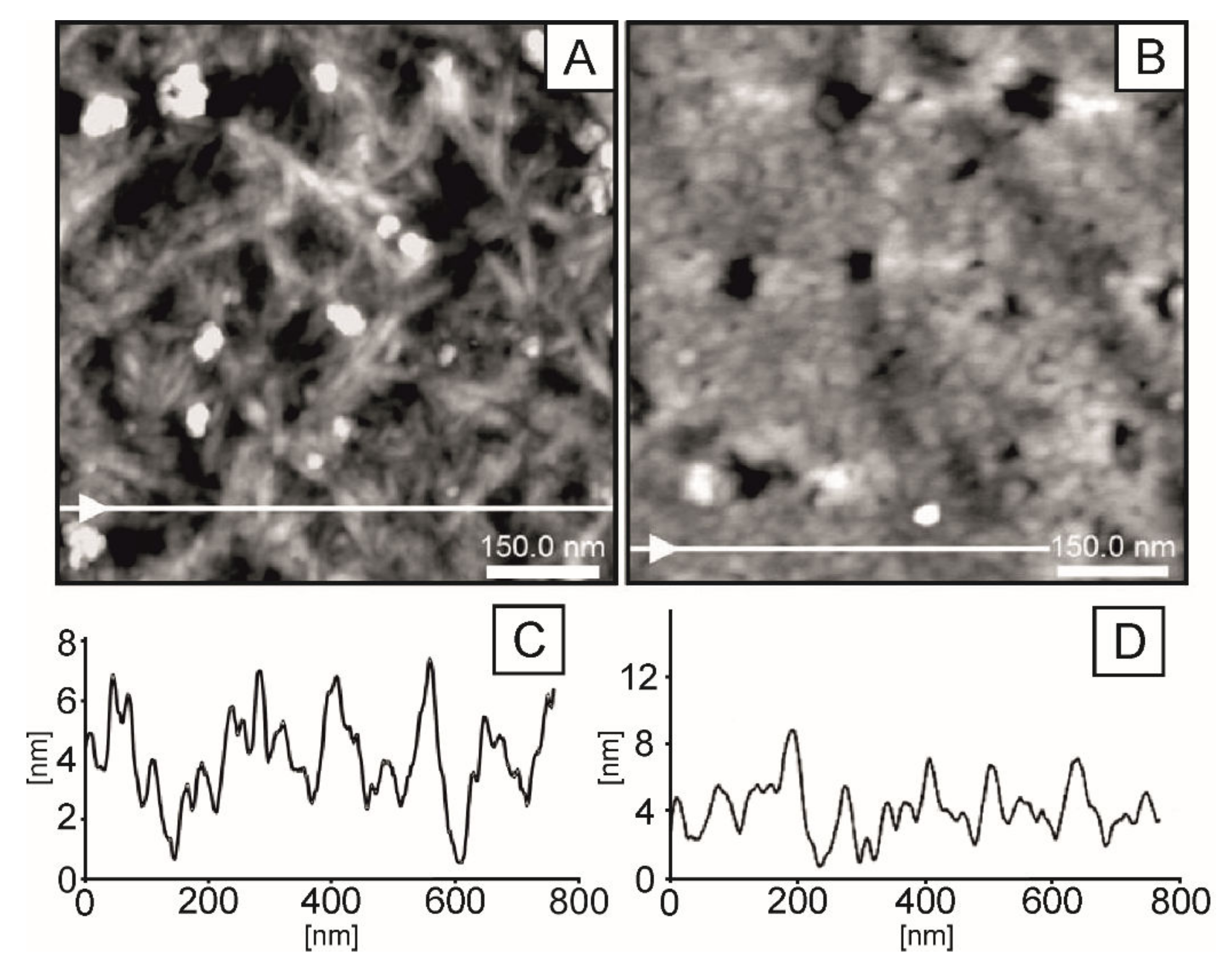
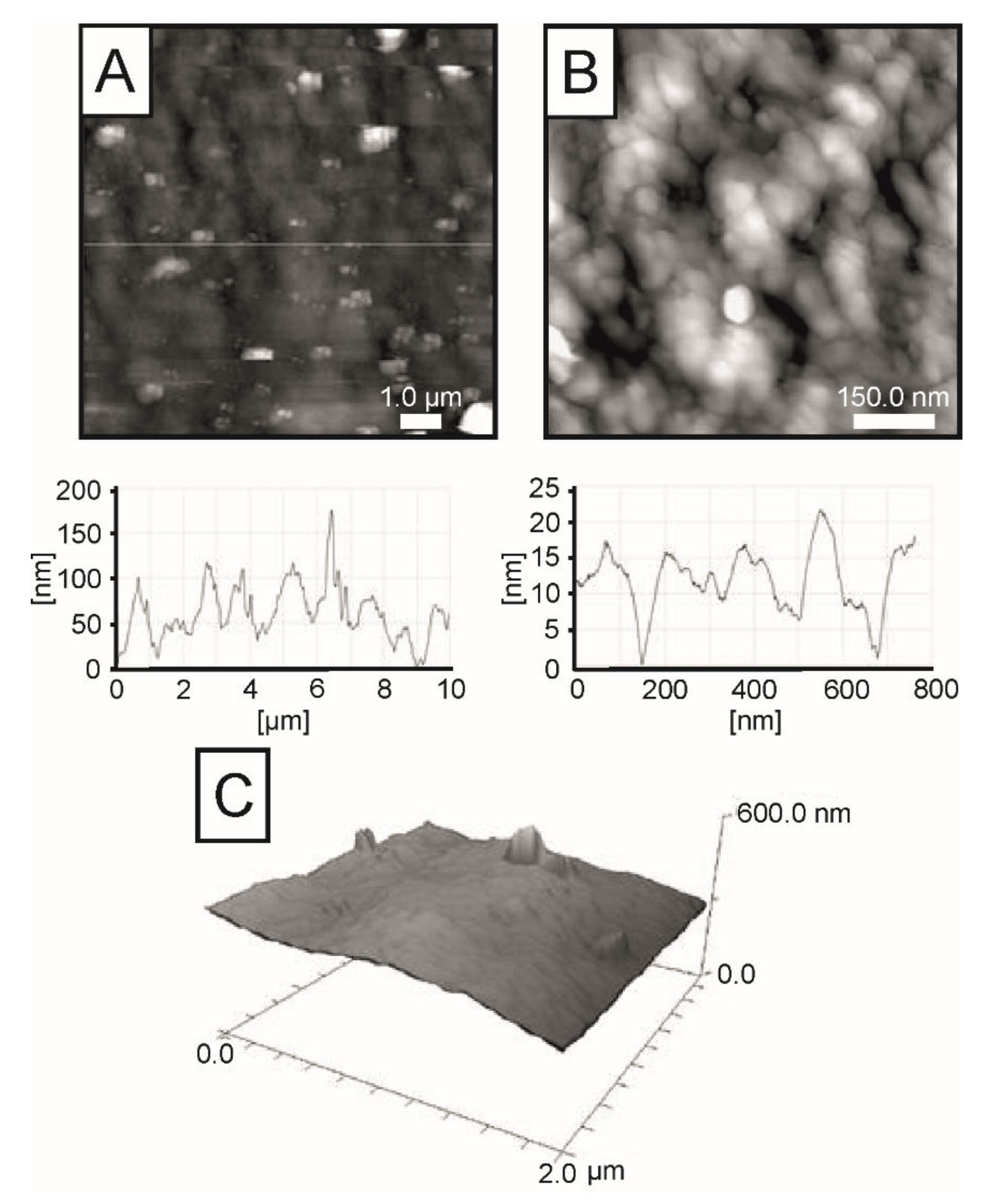
2.3. AFM Imaging
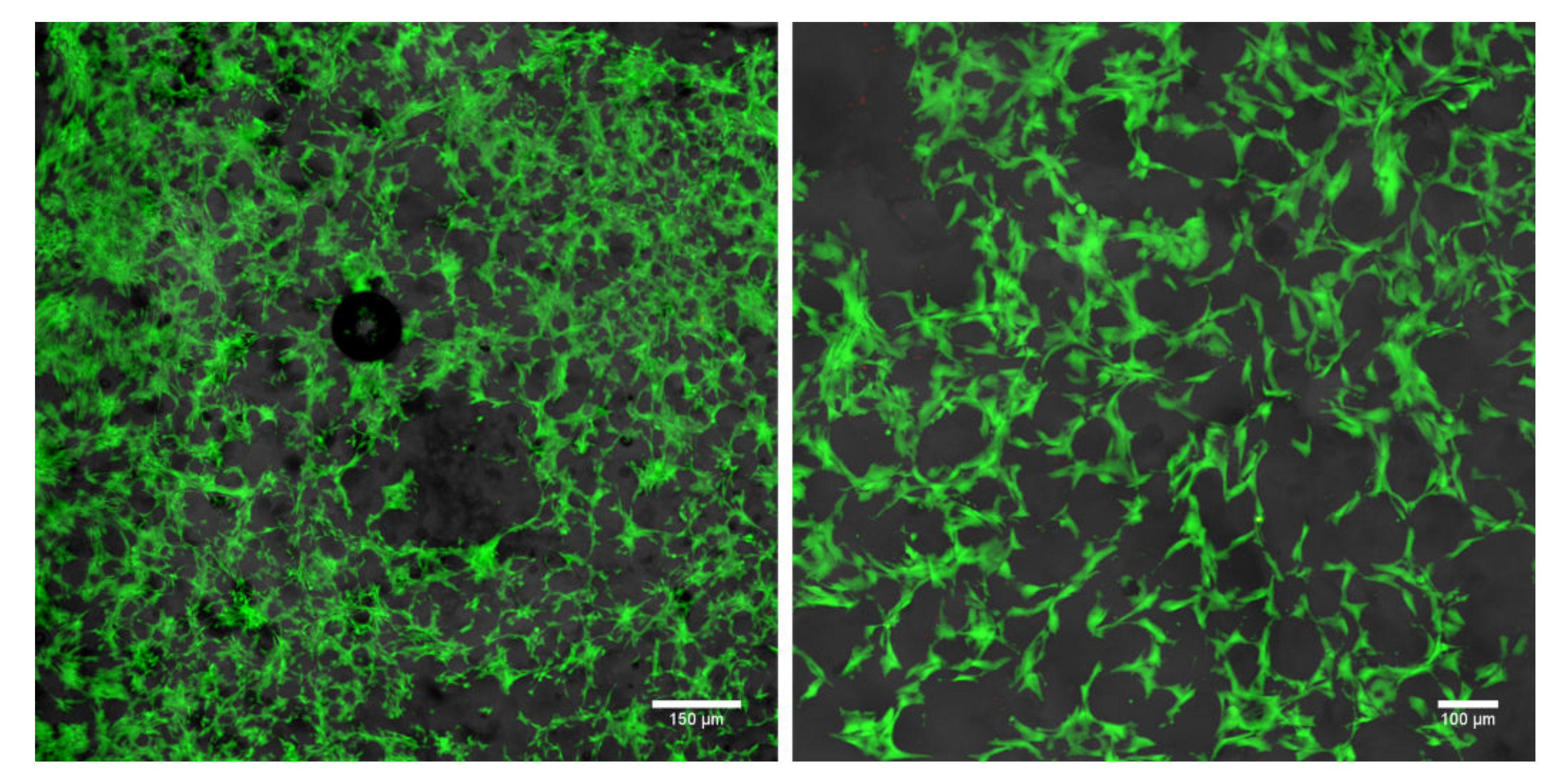
2.4. Cytotoxicity Evaluation
3. Discussion
4. Materials and Methods
4.1. Formation of Hybrid Chitosan/1,3-β-d-Glucan Matrices
4.2. Attenuated Total Reflection Fourier Transform Infrared (ATR FT-IR) Spectroscopy
4.3. Raman Spectroscopy
4.4. Second-Order Derivative Determination, Spectral Deconvolution and Ratios Calculations
4.5. X-ray Photoelectron Spectroscopy (XPS)
4.6. Atomic Force Microscopy (AFM)
4.7. Cytotoxicity Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nwe, N.; Stevens, W.F.; Tokura, S.; Tamura, H. Characterization of chitosan and chitosan–glucan complex extracted from the cell wall of fungus Gongronella butleri USDB 0201 by enzymatic method. Enzym. Microb. Technol. 2008, 42, 242–251. [Google Scholar] [CrossRef]
- Mahmoud, M.G.; El Kady, E.M.; Asker, M.S. Chitin, Chitosan and Glucan, Properties and Applications. World J. Agric. Soil Sci. 2019, 3, 1–19. [Google Scholar] [CrossRef]
- Chen, F.-M.; Liu, X. Advancing biomaterials of human origin for tissue engineering. Prog. Polym. Sci. 2016, 53, 86–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-I.; Wang, Y. Cell Responses to Surface and Architecture of Tissue Engineering Scaffolds. In Regenerative Medicine and Tissue Engineering—Cells and Biomaterials; Eberli, D., Ed.; InTech: London, UK, 2011; pp. 569–588. ISBN 9789533076638. [Google Scholar]
- Ratner, B.D. Surface Properties and Surface Characterization of Biomaterials. In Biomaterials Science; Ratner, B.D., Hoffman, A.S., Schoen, F.J., Lemons, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 34–55. ISBN 9780123746269. [Google Scholar]
- Mirabella, F. Internal Reflection Spectroscopy: Theory and Applications; Marcel Dekker Inc.: New York, NY, USA, 1993. [Google Scholar]
- Agrawal, G.; Samal, S.K. Raman Spectroscopy for Advanced Polymeric Biomaterials. ACS Biomater. Sci. Eng. 2018, 4, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.L. Applications of XPS in bioengineering. Surf. Interface Anal. 2006, 38, 1380–1385. [Google Scholar] [CrossRef]
- Jandt, K.D. Atomic force microscopy of biomaterials surfaces and interfaces. Surf. Sci. 2001, 491, 303–332. [Google Scholar] [CrossRef]
- Kumirska, J.; Czerwicka, M.; Kaczyński, Z.; Bychowska, A.; Brzozowski, K.; Thöming, J.; Stepnowski, P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drugs 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [Green Version]
- Gieroba, B.; Sroka-Bartnicka, A.; Kazimierczak, P.; Kalisz, G.; Pieta, I.S.; Nowakowski, R.; Pisarek, M.; Przekora, A. Effect of Gelation Temperature on the Molecular Structure and Physicochemical Properties of the Curdlan Matrix: Spectroscopic and Microscopic Analyses. Int. J. Mol. Sci. 2020, 21, 6154. [Google Scholar] [CrossRef]
- Gieroba, B.; Sroka-Bartnicka, A.; Kazimierczak, P.; Kalisz, G.; Lewalska-Graczyk, A.; Vivcharenko, V.; Nowakowski, R.; Pieta, I.S.; Przekora, A. Spectroscopic studies on the temperature-dependent molecular arrangements in hybrid chitosan/1,3-β-D-glucan polymeric matrices. Int. J. Biol. Macromol. 2020, 159, 911–921. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, H. Recent progress on curdlan provided by functionalization strategies. Food Hydrocoll. 2017, 68, 128–135. [Google Scholar] [CrossRef]
- Varma, R.; Vasudevan, S. Extraction, Characterization, and Antimicrobial Activity of Chitosan from Horse Mussel Modiolus modiolus. ACS Omega 2020, 5, 20224–20230. [Google Scholar] [CrossRef]
- Ren, X.D.; Liu, Q.S.; Feng, H.; Yin, X.Y. The Characterization of Chitosan Nanoparticles by Raman Spectroscopy. Appl. Mech. Mater. 2014, 665, 367–370. [Google Scholar] [CrossRef]
- Šandula, J.; Kogan, G.; Kačuráková, M.; Machová, E. Microbial (1→3)-β-d-glucans, their preparation, physico-chemical characterization and immunomodulatory activity. Carbohydr. Polym. 1999, 38, 247–253. [Google Scholar] [CrossRef]
- Noothalapati, H.; Sasaki, T.; Kaino, T.; Kawamukai, M.; Ando, M.; Hamaguchi, H.; Yamamoto, T. Label-free Chemical Imaging of Fungal Spore Walls by Raman Microscopy and Multivariate Curve Resolution Analysis. Sci. Rep. 2016, 6, 27789. [Google Scholar] [CrossRef] [Green Version]
- Eddya, M.; Tbib, B.; EL-Hami, K. A comparison of chitosan properties after extraction from shrimp shells by diluted and concentrated acids. Heliyon 2020, 6, e03486. [Google Scholar] [CrossRef] [Green Version]
- Rieppo, L.; Saarakkala, S.; Närhi, T.; Helminen, H.J.; Jurvelin, J.S.; Rieppo, J. Application of second derivative spectroscopy for increasing molecular specificity of fourier transform infrared spectroscopic imaging of articular cartilage. Osteoarthr. Cartil. 2012, 20, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Udrea, A.-M.; Dinache, A.; Pagès, J.-M.; Pirvulescu, R.A. Quinazoline Derivatives Designed as Efflux Pump Inhibitors: Molecular Modeling and Spectroscopic Studies. Molecules 2021, 26, 2374. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta-Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, K.A.M.; Suarez, P.A.Z.; Rubim, J.C. Photo-degradation of synthetic and natural polyisoprenes at specific UV radiations. Polym. Degrad. Stab. 2005, 90, 34–43. [Google Scholar] [CrossRef]
- Przekora, A.; Benko, A.; Blazewicz, M.; Ginalska, G. Hybrid chitosan/β-1,3-glucan matrix of bone scaffold enhances osteoblast adhesion, spreading and proliferation via promotion of serum protein adsorption. Biomed. Mater. 2016, 11, 045001. [Google Scholar] [CrossRef]
- Bakels, S.; Porskamp, S.B.A.; Rijs, A.M. Formation of Neutral Peptide Aggregates as Studied by Mass-Selective IR Action Spectroscopy. Angew. Chem. 2019, 58, 10537–10541. [Google Scholar] [CrossRef]
- Vodnar, D.C.; Pop, O.L.; Socaciu, C. Monitoring Lactic Acid Fermentation in Media Containing Dandelion (Taraxacum officinale) by FTIR Spectroscopy. Not. Bot. Horti Agrobot. Cluj-Napoca 2012, 40, 65. [Google Scholar] [CrossRef] [Green Version]
- Brauchle, E.; Knopf, A.; Bauer, H.; Shen, N.; Linder, S.; Monaghan, M.G.; Ellwanger, K.; Layland, S.L.; Brucker, S.Y.; Nsair, A.; et al. Non-invasive Chamber-Specific Identification of Cardiomyocytes in Differentiating Pluripotent Stem Cells. Stem Cell 2016, 6, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Shi, H.; Feng, S.; Chen, W.; Yu, Y.; Lin, D.; Chen, R. Confocal Raman spectroscopic analysis of the cytotoxic response to cisplatin in nasopharyngeal carcinoma cells. Anal. Methods 2013, 5, 260–266. [Google Scholar] [CrossRef]
- Lopes, R.P.; Marques, M.P.M.; Valero, R.; Tomkinson, J.; de Carvalho, L.A.E.B. Guanine: A Combined Study Using Vibrational Spectroscopy and Theoretical Methods. Spectrosc. Int. J. 2012, 27, 273–292. [Google Scholar] [CrossRef]
- Oliveira, R.N.; Mancini, M.C.; de Oliveira, F.C.S.; Passos, T.M.; Quilty, B.; da Silva Moreira, R.M.; McGuinness, G.B. FTIR analysis and quantification of phenols and flavonoids of five commercially available plants extracts used in wound healing. Matéria 2016, 21, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Abadri, M.H.S.; Delbari, A.; Fakoor, Z.; Baedi, J. Effects of Annealing Temperature on Infrared Spectra of SiO2 Extracted From Rice Husk. J. Ceram. Sci. Technol. 2015, 6, 41–45. [Google Scholar] [CrossRef]
- Grunert, T.; Monahan, A.; Lassnig, C.; Vogl, C.; Müller, M.; Ehling-Schulz, M. Deciphering Host Genotype-Specific Impacts on the Metabolic Fingerprint of Listeria monocytogenes by FTIR Spectroscopy. PLoS ONE 2014, 9, e115959. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.V.; Maréchal, A.; Rich, P.R. Effects of the Hydration State on the Mid-Infrared Spectra of Urea and Creatinine in Relation to Urine Analyses. Appl. Spectrosc. 2016, 70, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shen, X.; Sheng, D.; Chen, X.; Liu, X. FTIR spectroscopic comparison of serum from lung cancer patients and healthy persons. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 122, 193–197. [Google Scholar] [CrossRef]
- Traoré, M.; Kaal, J.; Martínez Cortizas, A. Differentiation between pine woods according to species and growing location using FTIR-ATR. Wood Sci. Technol. 2018, 52, 487–504. [Google Scholar] [CrossRef] [Green Version]
- Tognana, S.; Salgueiro, W.; Valcarce, M.B. A micro-Raman study of Cu-particulate-filled epoxy matrix composites. Express Polym. Lett. 2014, 8, 312–321. [Google Scholar] [CrossRef]
- Feng, S.; Huang, S.; Lin, D.; Chen, G.; Xu, Y.; Li, Y.; Huang, Z.; Pan, J.; Chen, R.; Zeng, H. Surface-enhanced Raman spectroscopy of saliva proteins for the noninvasive differentiation of benign and malignant breast tumors. Int. J. Nanomed. 2015, 10, 537. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.P.; Griffiths, P.R. Comparison of the Amide I/II Intensity Ratio of Solution and Solid-State Proteins Sampled by Transmission, Attenuated Total Reflectance, and Diffuse Reflectance Spectrometry. Appl. Spectrosc. 1993, 47, 584–589. [Google Scholar] [CrossRef]
- Kebukawa, Y.; Kobayashi, H.; Urayama, N.; Baden, N.; Kondo, M.; Zolensky, M.E.; Kobayashi, K. Nanoscale infrared imaging analysis of carbonaceous chondrites to understand organic-mineral interactions during aqueous alteration. Proc. Natl. Acad. Sci. USA 2019, 116, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Staroszczyk, H.; Pielichowska, J.; Sztuka, K.; Stangret, J.; Kołodziejska, I. Molecular and structural characteristics of cod gelatin films modified with EDC and TGase. Food Chem. 2012, 130, 335–343. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, H.-J. Fourier Transform Infrared Spectroscopy (FT-IR) and Simple Algorithm Analysis for Rapid and Non-Destructive Assessment of Developmental Cotton Fibers. Sensors 2017, 17, 1469. [Google Scholar] [CrossRef]
- Zając, A.; Hanuza, J.; Wandas, M.; Dymińska, L. Determination of N-acetylation degree in chitosan using Raman spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 134, 114–120. [Google Scholar] [CrossRef]
- Pezzotti, G.; Zhu, W.; Chikaguchi, H.; Marin, E.; Boschetto, F.; Masumura, T.; Sato, Y.-I.; Nakazaki, T. Raman Molecular Fingerprints of Rice Nutritional Quality and the Concept of Raman Barcode. Front. Nutr. 2021, 8, 334. [Google Scholar] [CrossRef]
- Wiercigroch, E.; Szafraniec, E.; Czamara, K.; Pacia, M.Z.; Majzner, K.; Kochan, K.; Kaczor, A.; Baranska, M.; Malek, K. Raman and infrared spectroscopy of carbohydrates: A review. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 317–335. [Google Scholar] [CrossRef]
- Dhayal, M.; Ratner, D.M. XPS and SPR Analysis of Glycoarray Surface Density. Langmuir 2009, 25, 2181–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqaheem, Y.; Alomair, A.A. Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes. Membranes 2020, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, L.E.; Li, A.; Faulds, K.; Graham, D. Ratiometric analysis using Raman spectroscopy as a powerful predictor of structural properties of fatty acids. R. Soc. Open Sci. 2018, 5, 181483. [Google Scholar] [CrossRef]
- Garidel, P.; Schott, H. Fourier-transform midinfrared spectroscopy for analysis and screening of liquid protein formulations. Part 2: Detailed analysis and applications. Bioprocess Int. 2006, 4, 48–55. [Google Scholar]
- Horstman, R.; Peters, K.; Gebremedhin, S.; Meltzer, R.; Bruce Vieth, M.; Cameron, D.; Moffatt, D. Deconvolution, Derivation, and Smoothing of Spectra Using Fourier Transforms. J. Test. Eval. 1984, 12, 78. [Google Scholar] [CrossRef]
- Haris, P.I.; Chapman, D. Does Fourier-transform infrared spectroscopy provide useful information on protein structures? Trends Biochem. Sci. 1992, 17, 328–333. [Google Scholar] [CrossRef]
- Rahmelow, K.; Hübner, W. Secondary structure determination of proteins in aqueous solution by infrared spectroscopy: A comparison of multivariate data analysis methods. Anal. Biochem. 1996, 241, 5–13. [Google Scholar] [CrossRef]
- Kauppinen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier Self-Deconvolution: A Method for Resolving Intrinsically Overlapped Bands. Appl. Spectrosc. 1981, 35, 271–276. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Li, Y.; Lv, M.; Li, P.; Xu, H.; Wang, L. Preparation and characterization of novel curdlan/chitosan blending membranes for antibacterial applications. Carbohydr. Polym. 2011, 84, 952–959. [Google Scholar] [CrossRef]
- Przekora, A.; Ginalska, G. Addition of 1,3-β-d-glucan to chitosan-based composites enhances osteoblast adhesion, growth, and proliferation. Int. J. Biol. Macromol. 2014, 70, 474–481. [Google Scholar] [CrossRef]
- Permyakova, E.S.; Konopatsky, A.S.; Ershov, K.I.; Bakhareva, K.I.; Sitnikova, N.A.; Shtansky, D.V.; Solovieva, A.O.; Manakhov, A.M. Ag-Contained Superabsorbent Curdlan–Chitosan Foams for Healing Wounds in a Type-2 Diabetic Mice Model. Pharmaceutics 2022, 14, 724. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liao, D.; Zou, Y.; Chi, H. The effects of chitosan supplementation on body weight and body composition: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2020, 60, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Elieh-Ali-Komi, D.; Hamblin, M.R. Chitin and Chitosan: Production and Application of Versatile Biomedical Nanomaterials. Int. J. Adv. Res. 2016, 4, 411–427. [Google Scholar]
- Yen, M.-T.; Yang, J.-H.; Mau, J.-L. Physicochemical characterization of chitin and chitosan from crab shells. Carbohydr. Polym. 2009, 75, 15–21. [Google Scholar] [CrossRef]
- Maver, U.; Maver, T.; Znidarsic, A.; Persin, Z.; Gaberscek, M.; Stana-Kleinschek, K. Use of Afm Force Spectroscopy for Assessment of Polymer Response to Conditions Similar to the Wound, during Healing. Mater. Tehnol. 2011, 45, 259–263. [Google Scholar]
- Cai, S.; Wu, C.; Yang, W.; Liang, W.; Yu, H.; Liu, L. Recent advance in surface modification for regulating cell adhesion and behaviors. Nanotechnol. Rev. 2020, 9, 971–989. [Google Scholar] [CrossRef]
- Hsissou, R.; Bekhta, A.; Dagdag, O.; El Bachiri, A.; Rafik, M.; Elharfi, A. Rheological properties of composite polymers and hybrid nanocomposites. Heliyon 2020, 6, e04187. [Google Scholar] [CrossRef]
- Abdel-Mohsen, A.M.; Abdel-Rahman, R.M.; Kubena, I.; Kobera, L.; Spotz, Z.; Zboncak, M.; Prikryl, R.; Brus, J.; Jancar, J. Chitosan-glucan complex hollow fibers reinforced collagen wound dressing embedded with aloe vera. Part I: Preparation and characterization. Carbohydr. Polym. 2020, 230, 115708. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber/cm−1 | The Type of Vibration and Assignment * | ||
|---|---|---|---|
| Chitosan | 1,3-β-d-Glucan | Chitosan/ 1,3-β-d-Glucan 90 °C | |
| 3353 | - | - | ν (OH) of glucopyranose units |
| 3289 | 3300 | 3291 | ν (OH), water |
| 3085 | - | - | ν (N–H) |
| 2914 | - | - | ν (CH2) |
| 2871 | 2884 | 2881 | ν (C–H) |
| 1650 | 1641 | 1646 | 80% ν (C=O), 20% ν (C–N) in chit and chit/glu, τ (HOH), amide I, water |
| 1566 | - | 1584 | 60% τ (N–H), 30% ν (C–N), 10% ν (C–C), amide II |
| - | - | 1454 | δs (CH3), δs (CH2) |
| 1419 | 1421 | 1421 | δs (CH3), δs (CH2), δs (CH), νs (C=O) |
| 1374 | 1368 | 1371 | δs (CH3) |
| 1313 | 1312 | 1312 | δ (C–H), δ (CH2) δ (OH), N-acetylglucosamine (chitosan), amide III |
| 1259 | 1252 | 1259 | ν (C–O), ν (N–H) in chit and chit/glu, ν (C–O–C), ν (C–OH) |
| - | - | 1241 | ν (C–H) in rings |
| 1198 | 1203 | 1202 | ν CH2OH |
| 1150 | 1156 | 1155 | τ (C–H), νas (C–O), ν (C–N) in chit and chit/glu, νas (C–O–C) in β-glycosidic linked rings |
| - | 1106 | 1108 | ν (C–C), ν (C–O) |
| 1061 | 1066 | 1066 | ν (C–O) |
| 1026 | 1029 | 1027 | ν skeletal (C=O) |
| 992 | 992 | 992 | δ (C=O), τ (C–C) |
| 949 | - | - | trans ν (C=C) |
| 894 | 887 | 889 | δ (C–H), β-glycosidic bonds |
| Raman Shift/cm−1 | The Type of Vibration and Assignment * | ||
|---|---|---|---|
| Chitosan | 1,3-β-d-Glucan | Chitosan/ 1,3-β-d-Glucan 90 °C | |
| 289 | 302 | 293 | δ (C–C–C) |
| 329 | 319 | 324 | δ (C–C–C) |
| - | - | 337 | τ (O–C–O) |
| 376 | 350 | 357 | δ (C–C–C) |
| - | 399 | 397 | δ C–C(=O)C |
| 423 | 427 | 425 | τ out of plane (H–C–C=O) |
| 440 | - | - | νs (NH2) in rings |
| 485 | 472 | 467 | τ (N–C=O) in chit and chit/glu, τ in plane (C–O–C) |
| - | - | 511 | τ in plane (C–O–C) |
| - | 527 | 527 | δ (C–C–N) in chit/glu, δ (C–C–C) |
| 576 | 570 | 579 | δ (C–C–C) |
| - | 611 | 608 | τ out of plane (C–H) |
| - | - | 562 | τ (C–C=O) |
| - | - | 829 | δ aromatic (C–N=C) |
| 896 | 890 | 892 | δ out of plane (C–H), β-glycosidic bond |
| - | - | 919 | νs (C–O–C) |
| - | - | 938 | δ out of plane (C–H) |
| 948 | - | 954 | ν (C–H) in rings |
| - | 984 | 976 | ring skeleton stretching vibrations sensitive to anomeric structure of glucose |
| - | - | 997 | ν (C–O–C) |
| 1043 | 1037 | 1034 | δ (C–H), (C–C), (C–OH) |
| - | - | 1063 | ν (C–O–C) in rings |
| 1099 | 1095 | 1092 | νs (C–O–C) in rings |
| 1115 | 1118 | 1116 | νas (C–O–C), ether |
| 1149 | 1142 | 1146 | ν (C–N) in chit and chit/glu, ν (C–C) |
| 1200 | 1205 | 1202 | ν (C–CH) |
| 1229 | 1238 | 1228 | ν (C–H) in rings |
| 1266 | 1266 | 1253 | δ in plane (C–H), CH2OH |
| - | 1319 | 1315 | δ in plane (C–H) |
| 1346 | - | - | τ (C–H) |
| 1376 | 1364 | 1362 | ν (C–N) in chit and chit/glu, ν (C–H), ν (C–OH) |
| 1413 | 1412 | 1410 | δas (CH3) |
| - | - | 1422 | ν (C–C) |
| 1460 | 1459 | 1461 | δas (C–H), τ in plane (CH2) |
| 1661 | - | - | ν (C=O), amide I |
| 1856 | - | - | ν (C–C), τ (C=O) |
| The FT-IR Absorbance Ratio | Sample | ||
|---|---|---|---|
| Chitosan * | 1,3-β-d-Glucan ** | Chitosan/ 1,3-β-d-Glucan 90 °C *** | |
| Aamide I/Aamide II | 1.200 ± 0.068 | – | 1.490 ± 0.048 |
| ACH2/ACH3 | 0.838 ± 0.015 | 0.737 ± 0.035 | 0.834 ± 0.025 |
| AC=O/AC–O | 0.997 ± 0.026 | 1.228 ± 0.036 | 1.131 ± 0.016 |
| The Raman intensity ratio | |||
| ICH and/or C–OH/ICH3 | 1.819 ± 0.156 | 2.008 ± 0.039 | 1.844 ± 0.067 |
| IC–O–C/IC–C | 1.944 ± 0.129 | 1.844 ± 0.067 | 1.451 ± 0.058 |
| Binding Energy/eV High Resolution Spectra | Chemical Composition | |||||
|---|---|---|---|---|---|---|
| Glucan | C1s | O1s | N1s | C:O At. % ratio (C:N) | species | |
| 284.6 | 2.15 | C-C/C-H | C—64.3 | |||
| 286.3 | 532.8 | 399.7 | −13.39 | C-OH, C-N | O—29.9 | |
| 287.6 | 531.3 | O-C-O, C=O | N—4.8 | |||
| 282.4 | Si-C-O | Na—0.0 | ||||
| 535.8 | H2O | Si—0.3 | ||||
| 401.6 | C-NHx | Ca—0.0 | ||||
| S—0.8 | ||||||
| P—0.0 | ||||||
| Pb—0.0 | ||||||
| Cl—0.0 | ||||||
| Chitosan | C1s | O1s | N1s | C:O At. % ratio (C:N) | species | |
| 284.6 | 2.58 | C-C/C-H | C—67.2 | |||
| 286.1 | 532.7 | 399.9 | −12.24 | C-OH, C-N | O—26.0 | |
| 287.9 | 531.1 | O-C-O, C=O | N—4.9 | |||
| 282.3 | Si-C-O | Na—0.8 | ||||
| 535.5 | H2O | Si—0.0 | ||||
| 401.9 | C-NHx | Ca—0.0 | ||||
| S—0.4 | ||||||
| P—0.0 | ||||||
| Pb—0.0 | ||||||
| Cl—0.4 | ||||||
| Chitosan/β-1,3-glucan | C1s | O1s | N1s | C:O At. % ratio (C:N) | species | |
| 284.6 | 2.32 | C-C/C-H | C—62.9 | |||
| 286.1 | 532.7 | 399.9 | −13.81 | C-OH, C-N | O—26.1 | |
| 287.9 | 531.1 | O-C-O, C=O | N—4.5 | |||
| 282.3 | Si-C-O | Na—3.5 | ||||
| 535.5 | H2O | Si—0.8 | ||||
| 401.9 | C-NHx | Ca—0.7 | ||||
| S—0.8 | ||||||
| P—0.4 | ||||||
| Pb—0.0 | ||||||
| Cl—0.2 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gieroba, B.; Sroka-Bartnicka, A.; Kazimierczak, P.; Kalisz, G.; Lewalska-Graczyk, A.; Vivcharenko, V.; Nowakowski, R.; Pieta, I.S.; Przekora, A. Surface Chemical and Morphological Analysis of Chitosan/1,3-β-d-Glucan Polysaccharide Films Cross-Linked at 90 °C. Int. J. Mol. Sci. 2022, 23, 5953. https://doi.org/10.3390/ijms23115953
Gieroba B, Sroka-Bartnicka A, Kazimierczak P, Kalisz G, Lewalska-Graczyk A, Vivcharenko V, Nowakowski R, Pieta IS, Przekora A. Surface Chemical and Morphological Analysis of Chitosan/1,3-β-d-Glucan Polysaccharide Films Cross-Linked at 90 °C. International Journal of Molecular Sciences. 2022; 23(11):5953. https://doi.org/10.3390/ijms23115953
Chicago/Turabian StyleGieroba, Barbara, Anna Sroka-Bartnicka, Paulina Kazimierczak, Grzegorz Kalisz, Agnieszka Lewalska-Graczyk, Vladyslav Vivcharenko, Robert Nowakowski, Izabela S. Pieta, and Agata Przekora. 2022. "Surface Chemical and Morphological Analysis of Chitosan/1,3-β-d-Glucan Polysaccharide Films Cross-Linked at 90 °C" International Journal of Molecular Sciences 23, no. 11: 5953. https://doi.org/10.3390/ijms23115953