
Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma

 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Patient Population
2.2. Sample Collection and Processing
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. High Performance Liquid Chromatography and Tandem Mass Spectrometry (MS)
2.4.1. Sample Preparation and Digestion
2.4.2. Data Acquisition
2.4.3. Protein Quantitation
2.5. Statistical Analysis
2.5.1. Clinical Variables and ELISA
2.5.2. MS Protein Analysis
2.5.3. Identification of Differentially Expressed Proteins
2.5.4. Identification of Protein Predictors of EoT
2.5.5. Identification of Intracellular Proteins Associated with End-Organ Damage
3. Results
3.1. Clinical Characteristics
3.2. ELISA
3.3. MS Proteomics
4. Discussion
4.1. Damage Associated Molecular Patterns
4.2. End-Organ Damage
4.3. Coagulation
4.4. Complement
4.5. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kochanek, K.D.; Murphy, S.L.; Xu, J.; Arias, E. Deaths: Final Data for 2017. Natl. Vital Stat. Rep. 2019, 68, 9. [Google Scholar]
- Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Injuries and Violence Are Leading Causes of Death. 2020. Available online: https://www.cdc.gov/injury/wisqars/animated-leading-causes.html (accessed on 20 October 2020).
- Frydrych, L.M.; Keeney-Bonthrone, T.P.; Gwinn, E.; Wakam, G.K.; Anderson, M.S.; Delano, M.J. Short-term versus long-term trauma mortality: A systematic review. J. Trauma Acute Care Surg. 2019, 87, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Ostrowski, S.R. Acute coagulopathy of trauma: Balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med. Hypotheses 2010, 75, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.B. A novel and potentially unifying mechanism for shock induced early coagulopathy. Ann. Surg. 2011, 254, 201–202. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Ostrowski, S.R. Shock induced endotheliopathy (SHINE) in acute critical illness—A unifying pathophysiologic mechanism. Crit. Care. 2017, 21, 25. [Google Scholar] [CrossRef] [Green Version]
- White, N.J.; Ward, K.R.; Pati, S.; Strandenes, G.; Cap, A.P. Hemorrhagic blood failure: Oxygen debt, coagulopathy, and endothelial damage. J. Trauma Acute Care Surg. 2017, 82 (Suppl. 1), S41–S49. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Reine, T.M.; Lanzalaco, F.; Kristiansen, O.; Enget, A.R.; Satchell, S.; Jenssen, T.G.; Kolset, S.O. Matrix metalloproteinase-9 mediated shedding of syndecan-4 in glomerular endothelial cells. Microcirculation 2019, 26, e12534. [Google Scholar] [CrossRef]
- Stepp, M.A.; Pal-Ghosh, S.; Tadvalkar, G.; Pajoohesh-Ganji, A. Syndecan-1 and Its Expanding List of Contacts. Adv. Wound Care 2015, 4, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Bjerkvig, C.K.; Strandenes, G.; Eliassen, H.S.; Spinella, P.C.; Fosse, T.K.; Cap, A.P.; Ward, K.R. ‘Blood failure‘ time to view blood as an organ: How oxygen debt contributes to blood failure and its implications for remote damage control resuscitation. Transfusion 2016, 56 (Suppl. 2), S182–S189. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, E.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E. Plasma colloid osmotic pressure is an early indicator of injury and hemorrhagic shock. Shock 2014, 41, 181–187. [Google Scholar] [CrossRef]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S.; Ostrowski, S.R.; Johansson, P.I.; Holcomb, J.B.; Wade, C.E. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Rodriguez, E.; Cardenas, J.C.; Lopez, E.; Cotton, B.A.; Tomasek, J.S.; Ostrowski, S.R.; Baer, L.A.; Stensballe, J.; Holcomb, J.B.; Johansson, P.I.; et al. Early Identification of the Patient with Endotheliopathy of Trauma by Arrival Serum Albumin. Shock 2018, 50, 31–37. [Google Scholar] [CrossRef]
- Hofmann, N.; Zipperle, J.; Jafarmadar, M.; Ashmwe, M.; Keibl, C.; Penzenstadler, C.; Ponschab, M.; Jafarmadar, B.; Redl, H.; Bahrami, S.; et al. Experimental Models of Endotheliopathy: Impact of Shock Severity. Shock 2018, 49, 564–571. [Google Scholar] [CrossRef]
- Di Battista, A.P.; Rizoli, S.B.; Lejnieks, B.; Min, A.; Shiu, M.Y.; Peng, H.T.; Baker, A.J.; Hutchison, M.G.; Churchill, N.; Inaba, K.; et al. Sympathoadrenal Activation is Associated with Acute Traumatic Coagulopathy and Endotheliopathy in Isolated Brain Injury. Shock 2016, 46 (Suppl. 1), 96–103. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, E.; Cardenas, J.C.; Cox, C.S.; Kitagawa, R.S.; Stensballe, J.; Holcomb, J.B.; Johansson, P.I.; Wade, C.E. Traumatic brain injury is associated with increased syndecan-1 shedding in severely injured patients. Scand. J. Trauma Resusc. Emerg. Med. 2018, 26, 102. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Ostrowski, S.R. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann. Surg. 2017, 265, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. High circulating adrenaline levels at admission predict increased mortality after trauma. J. Trauma Acute Care Surg. 2012, 72, 428–436. [Google Scholar] [CrossRef]
- Albert, V.; Subramanian, A.; Agrawal, D.; Pati, H.P.; Gupta, S.D.; Mukhopadhyay, A.K. Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome. Med. Sci. 2018, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, S.R.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Johansson, P.I. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A prospective observational study of 404 severely injured patients. J. Trauma Acute Care Surg. 2017, 82, 293–301. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60–66. [Google Scholar] [CrossRef]
- Johansson, P.I.; Sørensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit. Care 2011, 15, R272. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Sørensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. High sCD40L levels early after trauma are associated with enhanced shock, sympathoadrenal activation, tissue and endothelial damage, coagulopathy and mortality. J. Thromb Haemost. 2012, 10, 207–216. [Google Scholar] [CrossRef]
- Naumann, D.N.; Hazeldine, J.; Davies, D.J.; Bishop, J.; Midwinter, M.J.; Belli, A.; Harrison, P.; Lord, J.M. Endotheliopathy of Trauma is an on-Scene Phenomenon, and is Associated with Multiple Organ Dysfunction Syndrome: A Prospective Observational Study. Shock 2018, 49, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011, 254, 194–200. [Google Scholar] [CrossRef]
- Cobb, J.P.; O’Keefe, G.E. Injury research in the genomic era. Lancet 2004, 363, 2076–2083. [Google Scholar] [CrossRef]
- Alpantaki, K.; Tsiridis, E.; Pape, H.C.; Giannoudis, P.V. Application of clinical proteomics in diagnosis and management of trauma patients. Injury 2007, 38, 263–271. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Wade, C.E.; Matijevic, N.; Wang, Y.W.; Rodriguez, E.G.; Lopez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Chen, T.A.; Tomasek, J.S.; et al. Absences of Endothelial Microvesicle Changes in the Presence of the Endotheliopathy of Trauma. Shock 2019, 51, 180–184. [Google Scholar] [CrossRef]
- Wei, S.; Gonzalez Rodriguez, E.; Chang, R.; Holcomb, J.B.; Kao, L.S.; Wade, C.E. Elevated Syndecan-1 after Trauma and Risk of Sepsis: A Secondary Analysis of Patients from the Pragmatic, Randomized Optimal Platelet and Plasma Ratios (PROPPR) Trial. J. Am. Coll. Surg. 2018, 227, 587–595. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hastie, T.; Qian, J. Glmnet vignette. Stanford 2014, 9, 1–30. Available online: https://hastie.su.domains/Papers/Glmnet_Vignette.pdf (accessed on 11 January 2021).
- Steyerberg, E.W.; Harrell, F.E., Jr.; Borsboom, G.J.; Eijkemans, M.J.; Vergouwe, Y.; Habbema, J.D. Internal validation of predictive models: Efficiency of some procedures for logistic regression analysis. J. Clin. Epidemiol. 2001, 54, 774–781. [Google Scholar] [CrossRef]
- Steyerberg, E.W. Clinical Prediction Models: A Practical Approach to Development, Validation, and Updating; Springer International Publishing AG: Cham, Switzerland, 2019. [Google Scholar]
- Hatton, G.E.; Isbell, K.D.; Henriksen, H.H.; Stensballe, J.; Brummerstedt, M.; Johansson, P.I.; Kao, L.S.; Wade, C.E. Endothelial Dysfunction is Associated with Increased Incidence, Worsened Severity, and Prolonged Duration of Acute Kidney Injury after Severe Trauma. Shock 2021, 55, 311–315. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Sroussi, H.Y.; Berline, J.; Dazin, P.; Green, P.; Palefsky, J.M. S100A8 triggers oxidation-sensitive repulsion of neutrophils. J. Dent. Res. 2006, 85, 829–833. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Hsu, K.; Tedla, N.; Chung, Y.M.; Chow, S.; Herbert, C.; Geczy, C.L. S100A8 induces IL-10 and protects against acute lung injury. J. Immunol. 2014, 192, 2800–2811. [Google Scholar] [CrossRef]
- Raftery, M.J.; Yang, Z.; Valenzuela, S.M.; Geczy, C.L. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J. Biol. Chem. 2001, 276, 33393–33401. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.A.; Raftery, M.J.; Walsh, J.; Alewood, P.; Iismaa, S.E.; Thliveris, S.; Geczy, C.L. Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J. Biol. Chem. 1999, 274, 8561–8569. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Raftery, M.J.; Geczy, C.L. Oxidative modifications of DAMPs suppress inflammation: The case for S100A8 and S100A9. Antioxid Redox Signal. 2011, 15, 2235–2248. [Google Scholar] [CrossRef]
- Goyette, J.; Geczy, C.L. Inflammation-associated S100 proteins: New mechanisms that regulate function. Amino Acids. 2011, 41, 821–842. [Google Scholar] [CrossRef]
- Lim, S.Y.; Raftery, M.J.; Goyette, J.; Hsu, K.; Geczy, C.L. Oxidative modifications of S100 proteins: Functional regulation by redox. J. Leukoc. Biol. 2009, 86, 577–587. [Google Scholar] [CrossRef]
- Sroussi, H.Y.; Berline, J.; Palefsky, J.M. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J. Leukoc. Biol. 2007, 81, 818–824. [Google Scholar] [CrossRef]
- Smith, M.M. Histone structure and function. Curr. Opin. Cell Biol. 1991, 3, 429–437. [Google Scholar] [CrossRef]
- Huang, H.; Evankovich, J.; Yan, W.; Nace, G.; Zhang, L.; Ross, M.; Liao, X.; Billiar, T.; Xu, J.; Esmon, C.T.; et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 2011, 54, 999–1008. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Dinarvand, P.; Hassanian, S.M.; Qureshi, S.H.; Manithody, C.; Eissenberg, J.C.; Yang, L.; Rezaie, A.R. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood 2014, 123, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Chaaban, H.; Keshari, R.S.; Silasi-Mansat, R.; Popescu, N.I.; Mehta-D’Souza, P.; Lim, Y.P.; Lupu, F. Inter-α inhibitor protein and its associated glycosaminoglycans protect against histone-induced injury. Blood 2015, 125, 2286–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildhagen, K.C.; García de Frutos, P.; Reutelingsperger, C.P.; Schrijver, R.; Aresté, C.; Ortega-Gómez, A.; Deckers, N.M.; Hemker, H.C.; Soehnlein, O.; Nicolaes, G.A. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 2014, 123, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemberton, A.D.; Brown, J.K.; Inglis, N.F. Proteomic identification of interactions between histones and plasma proteins: Implications for cytoprotection. Proteomics 2010, 10, 1484–1493. [Google Scholar] [CrossRef]
- Johansson, P.I.; Windeløv, N.A.; Rasmussen, L.S.; Sørensen, A.M.; Ostrowski, S.R. Blood levels of histone-complexed DNA fragments are associated with coagulopathy, inflammation and endothelial damage early after trauma. J. Emerg. Trauma Shock 2013, 6, 171–175. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Esmon, C.T. Molecular circuits in thrombosis and inflammation. Thromb. Haemost. 2013, 109, 416–420. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Semeraro, N.; Colucci, M. Extracellular histones promote fibrinolysis by single-chain urokinase-type plasminogen activator in a factor seven activating protease-dependent way. Thromb. Res. 2020, 196, 193–199. [Google Scholar] [CrossRef]
- Jin, Z.G.; Melaragno, M.G.; Liao, D.F.; Yan, C.; Haendeler, J.; Suh, Y.A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 2000, 87, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.W.; Baldi, R.F.; Soni, S.; Handslip, R.; Tan, Y.Y.; O’Dea, K.P.; Malesevic, M.; McAuley, D.F.; O’Kane, C.M.; Patel, B.V.; et al. Secreted Extracellular Cyclophilin A Is a Novel Mediator of Ventilator-induced Lung Injury. Am. J. Respir. Crit. Care Med. 2021, 204, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, B.; Yarlett, N.; Strupp, A.; Cerami, A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc. Natl. Acad. Sci. USA 1992, 89, 3511–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawar, F.U.; Xiong, Y.; Khattak, M.N.K.; Li, J.; Lin, L.; Mei, J. Potential role of cyclophilin A in regulating cytokine secretion. J. Leukoc. Biol. 2017, 102, 989–992. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Fontaine, M.; Lepape, A.; Piriou, V.; Venet, F.; Friggeri, A. Innate danger signals in acute injury: From bench to bedside. Anaesth. Crit. Care Pain Med. 2016, 35, 283–292. [Google Scholar] [CrossRef]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. 2020, 46, 751–775. [Google Scholar] [CrossRef] [Green Version]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Matzinger, P. Essay 1: The Danger model in its historical context. Scand. J Immunol. 2001, 54, 4–9. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Marinković, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Araki, K.; Kinoshita, R.; Tomonobu, N.; Gohara, Y.; Tomida, S.; Takahashi, Y.; Senoo, S.; Taniguchi, A.; Itano, J.; Yamamoto, K.I.; et al. The heterodimer S100A8/A9 is a potent therapeutic target for idiopathic pulmonary fibrosis. J. Mol. Med. 2021, 99, 131–145. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; He, Y.; Hwang, S.; Bertola, A.; Mackowiak, B.; Ahmed, Y.A.; Seo, W.; Ma, J.; Wang, X.; Park, S.H.; et al. E-Selectin-Dependent Inflammation and Lipolysis in Adipose Tissue Exacerbate Steatosis-to-NASH Progression via S100A8/9. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 151–171. [Google Scholar] [CrossRef]
- Lu, G.; Jia, Z.; Zu, Q.; Zhang, J.; Zhao, L.; Shi, H. Inhibition of the cyclophilin A-CD147 interaction attenuates right ventricular injury and dysfunction after acute pulmonary embolism in rats. J. Biol. Chem. 2018, 293, 12199–12208. [Google Scholar] [CrossRef]
- Seizer, P.; Schönberger, T.; Schött, M.; Lang, M.R.; Langer, H.F.; Bigalke, B.; Krämer, B.F.; Borst, O.; Daub, K.; Heidenreich, O.; et al. EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 2010, 209, 51–57. [Google Scholar] [CrossRef]
- Balsley, M.A.; Malesevic, M.; Stemmy, E.J.; Gigley, J.; Jurjus, R.A.; Herzog, D.; Bukrinsky, M.I.; Fischer, G.; Constant, S.L. A cell-impermeable cyclosporine A derivative reduces pathology in a mouse model of allergic lung inflammation. J. Immunol. 2010, 185, 7663–7670. [Google Scholar] [CrossRef]
- Von Ungern-Sternberg, S.N.I.; Vogel, S.; Walker-Allgaier, B.; Geue, S.; Maurer, A.; Wild, A.M.; Münzer, P.; Chatterjee, M.; Heinzmann, D.; Kremmer, E.; et al. Extracellular Cyclophilin A Augments Platelet-Dependent Thrombosis and Thromboinflammation. Thromb. Haemost. 2017, 117, 2063–2078. [Google Scholar] [CrossRef]
- Heinzmann, D.; Bangert, A.; Müller, A.M.; von Ungern-Sternberg, S.N.; Emschermann, F.; Schönberger, T.; Chatterjee, M.; Mack, A.F.; Klingel, K.; Kandolf, R.; et al. The Novel Extracellular Cyclophilin A (CyPA)—Inhibitor MM284 Reduces Myocardial Inflammation and Remodeling in a Mouse Model of Troponin I -Induced Myocarditis. PLoS ONE 2015, 10, e0124606. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Zhang, Q.Z.; Hu, M.Q.; Li, F.X.; Fu, J.M.; Zhu, Z.D.; Li, Q.K.; Yang, Z.; Quan, J.M. Targeting Extracellular Cyclophilin A via an Albumin-Binding Cyclosporine A Analogue. ChemMedChem 2021, 16, 3649–3652. [Google Scholar] [CrossRef]
- Hernandez, I.; Tesoro, L.; Ramirez-Carracedo, R.; Diez-Mata, J.; Sanchez, S.; Saura, M.; Zamorano, J.L.; Zaragoza, C.; Botana, L. Ivabradine Induces Cardiac Protection against Myocardial Infarction by Preventing Cyclophilin-A Secretion in Pigs under Coronary Ischemia/Reperfusion. Int. J. Mol. Sci. 2021, 22, 2902. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, K.; Feng, C.; Peng, Z.; Luo, W. Therapeutic effect of SP-8356 on pulmonary embolism-associated cardiac injury is mediated by its ability to suppress apoptosis and inflammation. J. Cell Mol. Med. 2021, 25, 5260–5268. [Google Scholar] [CrossRef]
- Nakahara, M.; Ito, T.; Kawahara, K.; Yamamoto, M.; Nagasato, T.; Shrestha, B.; Yamada, S.; Miyauchi, T.; Higuchi, K.; Takenaka, T.; et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS ONE 2013, 8, e75961. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Gandhi, A.A.; Smith, S.A.; Wang, Q.; Chiang, D.; Yalavarthi, S.; Ali, R.A.; Liu, C.; Sule, G.; Tsou, P.S.; et al. Endothelium-protective, histone-neutralizing properties of the polyanionic agent defibrotide. JCI Insight 2021, 6, e149149. [Google Scholar] [CrossRef]
- Viemann, D.; Strey, A.; Janning, A.; Jurk, K.; Klimmek, K.; Vogl, T.; Hirono, K.; Ichida, F.; Foell, D.; Kehrel, B.; et al. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood 2005, 105, 2955–2962. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P.A. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Vogl, T.; Ludwig, S.; Goebeler, M.; Strey, A.; Thorey, I.S.; Reichelt, R.; Foell, D.; Gerke, V.; Manitz, M.P.; Nacken, W.; et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 2004, 104, 4260–4268. [Google Scholar] [CrossRef] [Green Version]
- Simard, J.C.; Girard, D.; Tessier, P.A. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J. Leukoc. Biol. 2010, 87, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Riva, M.; Källberg, E.; Björk, P.; Hancz, D.; Vogl, T.; Roth, J.; Ivars, F.; Leanderson, T. Induction of nuclear factor-κB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 2012, 137, 172–182. [Google Scholar] [CrossRef]
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit. Care 2014, 18, 543. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Leiva, M.C.; Fischkoff, S.A.; Handschumacher, R.E.; Lyttle, C.R. Leukocyte chemotactic activity of cyclophilin. J. Biol. Chem. 1992, 267, 11968–11971. [Google Scholar] [CrossRef]
- Jin, Z.G.; Lungu, A.O.; Xie, L.; Wang, M.; Wong, C.; Berk, B.C. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1186–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Raftery, M.; Cai, H.; Hsu, K.; Yan, W.X.; Hseih, H.L.; Watts, R.N.; Richardson, D.; Thomas, S.; Perry, M.; et al. S-nitrosylated S100A8: Novel anti-inflammatory properties. J. Immunol. 2008, 181, 5627–5636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, A.; Arai, S.; Yamada, S.; Nagae, A.; Saita, N.; Itoh, H.; Uemoto, S.; Totani, M.; Ikemoto, M. Dynamic mobility of immunological cells expressing S100A8 and S100A9 in vivo: A variety of functional roles of the two proteins as regulators in acute inflammatory reaction. Inflammation 2012, 35, 409–419. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Y.; Engeland, C.G.; Gordon, S.C.; Sroussi, H.Y. The anti-oxidative, anti-inflammatory, and protective effect of S100A8 in endotoxemic mice. Mol. Immunol. 2013, 53, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Sui, J.; Lu, R.; Halkidis, K.; Kocher, N.K.; Cao, W.; Marques, M.B.; Zheng, X.L. Plasma levels of S100A8/A9, histone/DNA complexes, and cell-free DNA predict adverse outcomes of immune thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2021, 19, 370–379. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Kremer Hovinga, J.A.; Schatzberg, D.; Wagner, D.D.; Lämmle, B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood 2012, 120, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453. [Google Scholar] [CrossRef]
- Wang, X.; Guan, M.; Zhang, X.; Ma, T.; Wu, M.; Li, Y.; Chen, X.; Zheng, Y. The Association Between S100A8/A9 and the Development of Very Late Stent Thrombosis in Patients with Acute Myocardial Infarction. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943295. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Lam, F.W.; Cruz, M.A.; Leung, H.C.; Parikh, K.S.; Smith, C.W.; Rumbaut, R.E. Histone induced platelet aggregation is inhibited by normal albumin. Thromb. Res. 2013, 132, 69–76. [Google Scholar] [CrossRef]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Vulliamy, P.; Gillespie, S.; Armstrong, P.C.; Allan, H.E.; Warner, T.D.; Brohi, K. Histone H4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc. Natl. Acad. Sci. USA 2019, 116, 17444–17449. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, N.; Di Cera, E. Dual effect of histone H4 on prothrombin activation. J. Thromb. Haemost. 2016, 14, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Seizer, P.; Ungern-Sternberg, S.N.; Schönberger, T.; Borst, O.; Münzer, P.; Schmidt, E.M.; Mack, A.F.; Heinzmann, D.; Chatterjee, M.; Langer, H.; et al. Extracellular cyclophilin A activates platelets via EMMPRIN (CD147) and PI3K/Akt signaling, which promotes platelet adhesion and thrombus formation in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Ohara, S.; Abe, Y.; Watanabe, M.; Suyama, K.; Sato, M.; Hashimoto, K.; Hosoya, M. The role of serum myeloid-related protein 8/14 complex in Henoch-Schönlein purpura nephritis. Pediatr. Nephrol. 2012, 27, 65–71. [Google Scholar] [CrossRef]
- Chen, X.; Tao, T.; Wang, H.; Zhao, H.; Lu, L.; Wu, F. Arterial Thrombosis Is Accompanied by Elevated Mitogen-Activated Protein Kinase (MAPK) and Cyclooxygenase-2 (COX-2) Expression via Toll-Like Receptor 4 (TLR-4) Activation by S100A8/A9. Med. Sci. Monit. 2018, 24, 7673–7681. [Google Scholar] [CrossRef]
- Stocca, A.; O’Toole, D.; Hynes, N.; Hynes, S.O.; Mashayekhi, K.; McGinley, L.; O’Connell, E.; Coleman, C.; Sultan, S.; Duffy, A.; et al. A role for MRP8 in in stent restenosis in diabetes. Atherosclerosis 2012, 221, 325–332. [Google Scholar] [CrossRef]
- Collier, D.M.; Villalba, N.; Sackheim, A.; Bonev, A.D.; Miller, Z.D.; Moore, J.S.; Shui, B.; Lee, J.C.; Lee, F.K.; Reining, S.; et al. Extracellular histones induce calcium signals in the endothelium of resistance-sized mesenteric arteries and cause loss of endothelium-dependent dilation. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1309–H1322. [Google Scholar] [CrossRef]
- Xie, Y.; Li, X.; Ge, J. Cyclophilin A-FoxO1 signaling pathway in endothelial cell apoptosis. Cell Signal. 2019, 61, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lessner, S.M.; Sakurai, Y.; Galis, Z.S. Cyclophilin A as a novel biphasic mediator of endothelial activation and dysfunction. Am. J. Pathol. 2004, 164, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Kim, Y.; Nigro, P.; Smolock, E.M.; Berk, B.C. Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc. Res. 2014, 101, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, H.; Chen, X.; Jiang, Y.; Huang, Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLoS ONE 2014, 9, e90472. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Guo, B.; Jia, Q.; Chen, Y.U.; Gao, X.; Xu, S. S100A9-containing serum exosomes of burn injury patients promote permeability of pulmonary microvascular endothelial cells. J. Biosci. 2021, 46, 33. [Google Scholar] [CrossRef]
- Rasmuson, J.; Kenne, E.; Wahlgren, M.; Soehnlein, O.; Lindbom, L. Heparinoid sevuparin inhibits Streptococcus-induced vascular leak through neutralizing neutrophil-derived proteins. Faseb. J. 2019, 33, 10443–10452. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.T.; Zhang, N.; Dart, C.; Wang, S.S.; Thachil, J.; Guan, Y.; Wang, G.; Toh, C. Human CRP defends against the toxicity of circulating histones. J. Immunol. 2013, 191, 2495–2502. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Bond Lau, W.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Bruhn, L.V.; Lauridsen, K.G.; Schmidt, A.S.; Rickers, H.; Bach, L.F.; Løfgren, B.; Hornung, N. Elevated calprotectin in patients with atrial fibrillation with and without heart failure. Scand. J. Clin. Lab. Investig. 2017, 77, 210–215. [Google Scholar] [CrossRef]
- Imbalzano, E.; Mandraffino, G.; Casciaro, M.; Quartuccio, S.; Saitta, A.; Gangemi, S. Pathophysiological mechanism and therapeutic role of S100 proteins in cardiac failure: A systematic review. Heart Fail. Rev. 2016, 21, 463–473. [Google Scholar] [CrossRef]
- Lu, N.F.; Jiang, L.; Zhu, B.; Yang, D.G.; Zheng, R.Q.; Shao, J.; Yuan, J.; Xi, X.M. Elevated Plasma Histone H4 Levels Are an Important Risk Factor in the Development of Septic Cardiomyopathy. Balk. Med. J. 2020, 37, 72–78. [Google Scholar] [CrossRef]
- Shah, M.; He, Z.; Rauf, A.; Kalkhoran, S.B.; Heiestad, C.M.; Stensløkken, K.O.; Parish, C.R.; Soehnlein, O.; Arjun, S.; Davidson, S.M.; et al. Extracellular histones are a target in myocardial ischaemia reperfusion injury. Cardiovasc. Res. 2021, 118, 1115–1125. [Google Scholar] [CrossRef]
- Lu, N.F.; Jiang, L.; Zhu, B.; Yang, D.G.; Zheng, R.Q.; Shao, J.; Xi, X.M. Elevated plasma histone H4 level predicts increased risk of mortality in patients with sepsis. Ann. Palliat. Med. 2020, 9, 1084–1091. [Google Scholar] [CrossRef]
- Yu, W.; Jianhong, L.; Weili, W.; Peng, S.; Zhongyang, S. Effects of CyPA signal pathway in myocardial tissue after cardiopulmonary resuscitation in rats. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2015, 27, 965–969. [Google Scholar]
- Seizer, P.; Ochmann, C.; Schönberger, T.; Zach, S.; Rose, M.; Borst, O.; Klingel, K.; Kandolf, R.; MacDonald, H.R.; Nowak, R.A.; et al. Disrupting the EMMPRIN (CD147)-cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1377–1386. [Google Scholar] [CrossRef] [Green Version]
- Seko, Y.; Fujimura, T.; Taka, H.; Mineki, R.; Murayama, K.; Nagai, R. Hypoxia followed by reoxygenation induces secretion of cyclophilin A from cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 2004, 317, 162–168. [Google Scholar] [CrossRef]
- Yan, J.; Zang, X.; Chen, R.; Yuan, W.; Gong, J.; Wang, C.; Li, Y. The clinical implications of increased cyclophilin A levels in patients with acute coronary syndromes. Clin. Chim. Acta 2012, 413, 691–695. [Google Scholar] [CrossRef]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2021, 109, 67–72. [Google Scholar] [CrossRef]
- Hasenauer, A.; Bédat, B.; Parapanov, R.; Lugrin, J.; Debonneville, A.; Abdelnour-Berchtold, E.; Gonzalez, M.; Perentes, J.Y.; Piquilloud, L.; Szabo, C.; et al. Effects of cold or warm ischemia and ex-vivo lung perfusion on the release of damage associated molecular patterns and inflammatory cytokines in experimental lung transplantation. J. Heart Lung Transplant. 2021, 40, 905–916. [Google Scholar] [CrossRef]
- Saito, T.; Liu, M.; Binnie, M.; Sato, M.; Hwang, D.; Azad, S.; Machuca, T.N.; Zamel, R.; Waddell, T.K.; Cypel, M.; et al. Distinct expression patterns of alveolar “alarmins” in subtypes of chronic lung allograft dysfunction. Am. J. Transplant. 2014, 14, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Zheng, Y.; Fan, B.; Zhang, L.; Zhu, F.; Wang, X.; Chen, Z.; Tan, X.; Wei, Q. Serum levels of interleukins and S100A8/A9 correlate with clinical severity in patients with dermatomyositis-associated interstitial lung disease. BMC Pulm. Med. 2020, 20, 196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Guan, L.; Mao, L.; Li, S.; Zhao, J. Histone H4 aggravates inflammatory injury through TLR4 in chlorine gas-induced acute respiratory distress syndrome. J. Occup. Med. Toxicol. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guan, L.; Yu, J.; Zhao, Z.; Mao, L.; Li, S.; Zhao, J. Pulmonary endothelial activation caused by extracellular histones contributes to neutrophil activation in acute respiratory distress syndrome. Respir. Res. 2016, 17, 155. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, W.M.; Damsker, J.M.; Falahati, R.; Okwumabua, I.; Kelly-Welch, A.; Keegan, A.D.; Vanpouille, C.; Lee, J.J.; Dent, L.A.; Leitenberg, D.; et al. Novel approach to inhibit asthma-mediated lung inflammation using anti-CD147 intervention. J. Immunol. 2006, 177, 4870–4879. [Google Scholar] [CrossRef] [Green Version]
- Nikolakopoulou, Z.; Hector, L.R.; Creagh-Brown, B.C.; Evans, T.W.; Quinlan, G.J.; Burke-Gaffney, A. Plasma S100A8/A9 heterodimer is an early prognostic marker of acute kidney injury associated with cardiac surgery. Biomark. Med. 2019, 13, 205–218. [Google Scholar] [CrossRef]
- Fujiu, K.; Manabe, I.; Nagai, R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J. Clin. Investig. 2011, 121, 3425–3441. [Google Scholar] [CrossRef] [Green Version]
- Dessing, M.C.; Tammaro, A.; Pulskens, W.P.; Teske, G.J.; Butter, L.M.; Claessen, N.; van Eijk, M.; van der Poll, T.; Vogl, T.; Roth, J.; et al. The calcium-binding protein complex S100A8/A9 has a crucial role in controlling macrophage-mediated renal repair following ischemia/reperfusion. Kidney Int. 2015, 87, 85–94. [Google Scholar] [CrossRef]
- Tammaro, A.; Florquin, S.; Brok, M.; Claessen, N.; Butter, L.M.; Teske, G.J.D.; de Boer, O.J.; Vogl, T.; Leemans, J.C.; Dessing, M.C. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin. Exp. Immunol. 2018, 193, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Pepper, R.J.; Wang, H.H.; Rajakaruna, G.K.; Papakrivopoulou, E.; Vogl, T.; Pusey, C.D.; Cook, H.T.; Salama, A.D. S100A8/A9 (calprotectin) is critical for development of glomerulonephritis and promotes inflammatory leukocyte-renal cell interactions. Am. J. Pathol. 2015, 185, 1264–1274. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hägele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 2012, 23, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Ozols, E.; Kanellis, J.; Badal, S.S.; Liles, J.T.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin Inhibition Protects against Experimental Acute Kidney Injury and Renal Interstitial Fibrosis. Int. J. Mol. Sci. 2020, 22, 271. [Google Scholar] [CrossRef]
- Leong, K.G.; Ozols, E.; Kanellis, J.; Nikolic-Paterson, D.J.; Ma, F.Y. Cyclophilin A Promotes Inflammation in Acute Kidney Injury but Not in Renal Fibrosis. Int. J. Mol. Sci. 2020, 21, 3667. [Google Scholar] [CrossRef]
- Lee, C.C.; Chang, C.H.; Cheng, Y.L.; Kuo, G.; Chen, S.W.; Li, Y.J.; Chen, Y.T.; Tian, Y.C. Diagnostic Performance of Cyclophilin A in Cardiac Surgery-Associated Acute Kidney Injury. J. Clin. Med. 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, H.; Fujii, W.; Isokawa, S.; Ishizuka, K.; Wang, Y.; Watanabe, S.; Sanjoba, C.; Matsumoto, Y.; Goto, Y. Exacerbation of hepatic injury during rodent malaria by myeloid-related protein 14. PLoS ONE 2018, 13, e0199111. [Google Scholar] [CrossRef] [Green Version]
- Moles, A.; Murphy, L.; Wilson, C.L.; Chakraborty, J.B.; Fox, C.; Park, E.J.; Mann, J.; Oakley, F.; Howarth, R.; Brain, J.; et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 2014, 60, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Takai, S.; Higuchi, S.; Yano, A.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Involvement of immune- and inflammatory-related factors in flucloxacillin-induced liver injury in mice. J. Appl. Toxicol. 2015, 35, 142–151. [Google Scholar] [CrossRef]
- Wen, Z.; Liu, Y.; Li, F.; Ren, F.; Chen, D.; Li, X.; Wen, T. Circulating histones exacerbate inflammation in mice with acute liver failure. J. Cell Biochem. 2013, 114, 2384–2391. [Google Scholar] [CrossRef]
- Huang, H.; Chen, H.W.; Evankovich, J.; Yan, W.; Rosborough, B.R.; Nace, G.W.; Ding, Q.; Loughran, P.; Beer-Stolz, D.; Billiar, T.R.; et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J. Immunol. 2013, 191, 2665–2679. [Google Scholar] [CrossRef]
- Dear, J.W.; Simpson, K.J.; Nicolai, M.P.; Catterson, J.H.; Street, J.; Huizinga, T.; Craig, D.G.; Dhaliwal, K.; Webb, S.; Bateman, D.N.; et al. Cyclophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury. J. Immunol. 2011, 187, 3347–3352. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, H.H.; Caklili, O.T.; Coskunpinar, E. Serum concentrations of Cyclophilin A in patients with Nonalcoholic Fatty Liver Disease. Acta Gastroenterol. Belg. 2017, 80, 3–7. [Google Scholar]
- Tian, X.; Zhao, C.; Zhu, H.; She, W.; Zhang, J.; Liu, J.; Li, L.; Zheng, S.; Wen, Y.M.; Xie, Y. Hepatitis B virus (HBV) surface antigen interacts with and promotes cyclophilin a secretion: Possible link to pathogenesis of HBV infection. J. Virol. 2010, 84, 3373–3381. [Google Scholar] [CrossRef] [Green Version]
- Naoumov, N.V. Cyclophilin inhibition as potential therapy for liver diseases. J. Hepatol. 2014, 61, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Joly, P.; Marshall, J.C.; Tessier, P.A.; Massé, C.; Page, N.; Frenette, A.J.; Khazoom, F.; Le Guillan, S.; Berthiaume, Y.; Charbonney, E. S100A8/A9 and sRAGE kinetic after polytrauma; an explorative observational study. Scand. J. Trauma Resusc. Emerg. Med. 2017, 25, 114. [Google Scholar] [CrossRef] [Green Version]
- Kutcher, M.E.; Xu, J.; Vilardi, R.F.; Ho, C.; Esmon, C.T.; Cohen, M.J. Extracellular histone release in response to traumatic injury: Implications for a compensatory role of activated protein C. J. Trauma Acute Care Surg. 2012, 73, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- St John, A.; Wang, Y.; Chen, J.; Osborn, W.; Wang, X.; Lim, E.; Chung, D.; Stern, S.; White, N.; Fu, X.; et al. Plasma proteomic profile associated with platelet dysfunction after trauma. J. Thromb. Haemost. 2021, 19, 1666–1675. [Google Scholar] [CrossRef]
- Gao, Y.; Duan, J.; Ji, H.; Lu, W. Levels of S100 calcium binding protein B (S100B), neuron-specific enolase (NSE), and cyclophilin A (CypA) in the serum of patients with severe craniocerebral injury and multiple injuries combined with delirium transferred from the ICU and their prognostic value. Ann. Palliat. Med. 2021, 10, 3371–3378. [Google Scholar]
- Smith, J.G.W.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.H.; DiLullo, C.; Schultheiss, T.; Holtzer, S.; Murray, J.M.; Choi, J.; Fischman, D.A.; Holtzer, H. The vinculin/sarcomeric-alpha-actinin/alpha-actin nexus in cultured cardiac myocytes. J. Cell Biol. 1992, 117, 1007–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis-Owen, S.A.G.; Thompson, A.; Kemp, P.R.; Polkey, M.I.; Cookson, W.; Moffatt, M.F.; Natanek, S.A. COPD is accompanied by co-ordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix. Sci. Rep. 2018, 8, 12165. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Li, H.; Zhang, D.; Zhao, Y.; Jiang, N.; Zhao, X.; Zhang, Y.U.; Tan, J.; Fang, W.; Zhang, Y.; et al. Aortic wall proteomic analysis in spontaneously hypertensive rats with a blood pressure decrease induced by 6-week load-free swimming. Biomed. Rep. 2015, 3, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, Y.; Teng, Z.; Zhang, X.; Ding, M.; Zhang, Z.; Chen, J.; Xu, Y. DNA Microarray Analysis in Screening Features of Genes Involved in Spinal Cord Injury. Med. Sci. Monit. 2016, 22, 1571–1581. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Z.; Eralp Inan, O.; Kocaturk, M.; Baykal, A.T.; Hacariz, O.; Hatipoglu, I.; Tvarijonaviciute, A.; Cansev, M.; Ceron, J.; Ulus, I.H. Changes in serum proteins after endotoxin administration in healthy and choline-treated calves. BMC Vet. Res. 2016, 12, 210. [Google Scholar] [CrossRef] [Green Version]
- Aránega, A.E.; Reina, A.; Muros, M.A.; Alvarez, L.; Prados, J.; Aránega, A. Circulating alpha-actin protein in acute myocardial infarction. Int. J. Cardiol. 1993, 38, 49–55. [Google Scholar] [CrossRef]
- De’Ath, H.D.; Rourke, C.; Davenport, R.; Manson, J.; Renfrew, I.; Uppal, R.; Davies, L.C.; Brohi, K. Clinical and biomarker profile of trauma-induced secondary cardiac injury. Br. J. Surg. 2012, 99, 789–797. [Google Scholar] [CrossRef]
- Martin, M.; Mullenix, P.; Rhee, P.; Belzberg, H.; Demetriades, D.; Salim, A. Troponin increases in the critically injured patient: Mechanical trauma or physiologic stress? J. Trauma Acute Care Surg. 2005, 59, 1086–1091. [Google Scholar] [CrossRef]
- Baur, M.; Weber, B.; Lackner, I.; Gebhard, F.; Pfeifer, R.; Cinelli, P.; Halvachizadeh, S.; Teuben, M.; Lipiski, M.; Cesarovic, N.; et al. Structural alterations and inflammation in the heart after multiple trauma followed by reamed versus non-reamed femoral nailing. PLoS ONE 2020, 15, e0235220. [Google Scholar] [CrossRef]
- De’Ath, H.D.; Manson, J.; Davenport, R.; Glasgow, S.; Renfrew, I.; Davies, L.C.; Uppal, R.; Brohi, K. Trauma-induced secondary cardiac injury is associated with hyperacute elevations in inflammatory cytokines. Trauma-induced secondary cardiac injury is associated with hyperacute elevations in inflammatory cytokines. Shock 2013, 39, 415–420. [Google Scholar] [CrossRef]
- Wilson, N.M.; Wall, J.; Naganathar, V.; Brohi, K.; De’Ath, H.D. Mechanisms Involved in Secondary Cardiac Dysfunction in Animal Models of Trauma and Hemorrhagic Shock. Shock 2017, 48, 401–410. [Google Scholar] [CrossRef]
- Naganathar, S.; De’Ath, H.D.; Wall, J.; Brohi, K. Admission biomarkers of trauma-induced secondary cardiac injury predict adverse cardiac events and are associated with plasma catecholamine levels. J. Trauma Acute Care Surg. 2015, 79, 71–77. [Google Scholar] [CrossRef]
- Gawande, N.B.; Tumram, N.K.; Dongre, A.P. Cardiac changes in hospitalized patients of trauma. Shock 2014, 42, 211–217. [Google Scholar] [CrossRef]
- Cebelin, M.S.; Hirsch, C.S. Human stress cardiomyopathy. Myocardial lesions in victims of homicidal assaults without internal injuries. Hum. Pathol. 1980, 11, 123–132. [Google Scholar] [CrossRef]
- Wall, J.; Naganathar, S.; Praditsuktavorn, B.; Bugg, O.F.; McArthur, S.; Thiemermann, C.; Tremoleda, J.L.; Brohi, K. Modeling Cardiac Dysfunction Following Traumatic Hemorrhage Injury: Impact on Myocardial Integrity. Front. Immunol. 2019, 10, 2774. [Google Scholar] [CrossRef] [Green Version]
- Erukhimov, J.A.; Tang, Z.L.; Johnson, B.A.; Donahoe, M.P.; Razzack, J.A.; Gibson, K.F.; Lee, W.M.; Wasserloos, K.J.; Watkins, S.A.; Pitt, B.R. Actin-containing sera from patients with adult respiratory distress syndrome are toxic to sheep pulmonary endothelial cells. Am. J. Respir. Crit. Care Med. 2000, 162, 288–294. [Google Scholar] [CrossRef]
- Hazeldine, J.; Dinsdale, R.J.; Naumann, D.N.; Acharjee, A.; Bishop, J.R.B.; Lord, J.M.; Harrison, P. Traumatic injury is associated with reduced deoxyribonuclease activity and dysregulation of the actin scavenging system. Burns Trauma 2021, 9, tkab001. [Google Scholar] [CrossRef]
- Dinsdale, R.J.; Hazeldine, J.; Al Tarrah, K.; Hampson, P.; Devi, A.; Ermogenous, C.; Bamford, A.L.; Bishop, J.; Watts, S.; Kirkman, E.; et al. Dysregulation of the actin scavenging system and inhibition of DNase activity following severe thermal injury. Br. J. Surg. 2020, 107, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.R.; Moore, E.E.; Freeman, K.; Grubinger, N.D.; Hennig, G.W.; Cohen, M.J.; Samuels, J.M.; Hansen, K. Actin is associated with tissue injury in trauma patients and produces a hypercoagulable profile in vitro. J. Trauma Acute Care Surg. 2020, 89, 87–95. [Google Scholar] [CrossRef]
- Ordija, C.M.; Chiou, T.T.; Yang, Z.; Deloid, G.M.; de Oliveira Valdo, M.; Wang, Z.; Bedugnis, A.; Noah, T.L.; Jones, S.; Koziel, H.; et al. Free actin impairs macrophage bacterial defenses via scavenger receptor MARCO interaction with reversal by plasma gelsolin. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L1018–L1028. [Google Scholar] [CrossRef]
- Dahl, B.; Schiødt, F.V.; Rudolph, S.; Ott, P.; Kiaer, T.; Heslet, L. Trauma stimulates the synthesis of Gc-globulin. Intensive Care Med. 2001, 27, 394–399. [Google Scholar] [CrossRef]
- Dahl, B.; Schiødt, F.V.; Kiaer, T.; Ott, P.; Bondesen, S.; Tygstrup, N. Serum Gc-globulin in the early course of multiple trauma. Crit. Care Med. 1998, 26, 285–289. [Google Scholar] [CrossRef]
- Ley, E.J.; Brown, C.V.R.; Moore, E.E.; Sava, J.A.; Peck, K.; Ciesla, D.J.; Sperry, J.L.; Rizzo, A.G.; Rosen, N.G.; Brasel, K.J.; et al. Updated guidelines to reduce venous thromboembolism in trauma patients: A Western Trauma Association critical decisions algorithm. J. Trauma Acute Care Surg. 2020, 89, 971–981. [Google Scholar] [CrossRef]
- Shackford, S.R.; Cipolle, M.D.; Badiee, J.; Mosby, D.L.; Knudson, M.M.; Lewis, P.R.; McDonald, V.S.; Olson, E.J.; Thompson, K.A.; Van Gent, J.M.; et al. Determining the magnitude of surveillance bias in the assessment of lower extremity deep venous thrombosis: A prospective observational study of two centers. J. Trauma Acute Care Surg. 2016, 80, 734–739. [Google Scholar] [CrossRef]
- Thorson, C.M.; Ryan, M.L.; Van Haren, R.M.; Curia, E.; Barrera, J.M.; Guarch, G.A.; Busko, A.M.; Namias, N.; Livingstone, A.S.; Proctor, K.G. Venous thromboembolism after trauma: A never event?*. Crit. Care Med. 2012, 40, 2967–2973. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.R.; Espina, C.; Guedj, T.; Buaron, R.; Harrois, A.; Figueiredo, S.; Duranteau, J. High level of venous thromboembolism in critically ill trauma patients despite early and well-driven thromboprophylaxis protocol. Ann. Intensive Care 2017, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Velmahos, G.C.; Spaniolas, K.; Tabbara, M.; Abujudeh, H.H.; de Moya, M.; Gervasini, A.; Alam, H.B. Pulmonary embolism and deep venous thrombosis in trauma: Are they related? Arch. Surg. 2009, 144, 928–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gent, J.M.; Zander, A.L.; Olson, E.J.; Shackford, S.R.; Dunne, C.E.; Sise, C.B.; Badiee, J.; Schechter, M.S.; Sise, M.J. Pulmonary embolism without deep venous thrombosis: De novo or missed deep venous thrombosis? J. Trauma Acute Care Surg. 2014, 76, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Van Langevelde, K.; Srámek, A.; Vincken, P.W.; van Rooden, J.K.; Rosendaal, F.R.; Cannegieter, S.C. Finding the origin of pulmonary emboli with a total-body magnetic resonance direct thrombus imaging technique. Haematologica 2013, 98, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Pelsers, M.M.; Namiot, Z.; Kisielewski, W.; Namiot, A.; Januszkiewicz, M.; Hermens, W.T.; Glatz, J.F. Intestinal-type and liver-type fatty acid-binding protein in the intestine. Tissue distribution and clinical utility. Clin. Biochem. 2003, 36, 529–535. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Relja, B.; Szermutzky, M.; Henrich, D.; Maier, M.; de Haan, J.J.; Lubbers, T.; Buurman, W.A.; Marzi, I. Intestinal-FABP and liver-FABP: Novel markers for severe abdominal injury. Acad. Emerg. Med. 2010, 17, 729–735. [Google Scholar] [CrossRef]
- Voth, M.; Holzberger, S.; Auner, B.; Henrich, D.; Marzi, I.; Relja, B. I-FABP and L-FABP are early markers for abdominal injury with limited prognostic value for secondary organ failures in the post-traumatic course. Clin. Chem. Lab. Med. 2015, 53, 771–780. [Google Scholar] [CrossRef]
- Van de Poll, M.C.; Derikx, J.P.; Buurman, W.A.; Peters, W.H.; Roelofs, H.M.; Wigmore, S.J.; Dejong, C.H. Liver manipulation causes hepatocyte injury and precedes systemic inflammation in patients undergoing liver resection. World J. Surg. 2007, 31, 2033–2038. [Google Scholar] [CrossRef] [Green Version]
- Van den Broek, M.A.; Bloemen, J.G.; Dello, S.A.; van de Poll, M.C.; Olde Damink, S.W.; Dejong, C.H. Randomized controlled trial analyzing the effect of 15 or 30 min intermittent Pringle maneuver on hepatocellular damage during liver surgery. J. Hepatol. 2011, 55, 337–345. [Google Scholar] [CrossRef]
- Van den Broek, M.A.; Shiri-Sverdlov, R.; Schreurs, J.J.; Bloemen, J.G.; Bieghs, V.; Rensen, S.S.; Dejong, C.H.; Olde Damink, S.W. Liver manipulation during liver surgery in humans is associated with hepatocellular damage and hepatic inflammation. Liver Int. 2013, 33, 633–641. [Google Scholar] [CrossRef]
- Hanssen, S.J.; Derikx, J.P.; Vermeulen Windsant, I.C.; Heijmans, J.H.; Koeppel, T.A.; Schurink, G.W.; Buurman, W.A.; Jacobs, M.J. Visceral injury and systemic inflammation in patients undergoing extracorporeal circulation during aortic surgery. Ann. Surg. 2008, 248, 117–125. [Google Scholar] [CrossRef]
- Kunihara, T.; Kubota, S.; Shiiya, N.; Iizuka, K.; Sasaki, S.; Wakasa, S.; Matsuzaki, K.; Matsui, Y. Cytokine balance in hepatosplanchnic system during thoracoabdominal aortic aneurysm repair. J. Artif. Organs 2011, 14, 192–200. [Google Scholar] [CrossRef]
- Crapo, J.D.; Oury, T.; Rabouille, C.; Slot, J.W.; Chang, L.Y. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10405–10409. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Miguelsanz, J.; Vallecillo, N.; Garrido, F.; Reytor, E.; Pérez-Sala, D.; Pajares, M.A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1165–1182. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, H.; Niewiadomski, J.; Hirschberger, L.L.; Roman, H.B.; Mazor, K.M.; Liu, X.; Locasale, J.W.; Park, E.; Stipanuk, M.H. Downregulation of hepatic betaine:homocysteine methyltransferase (BHMT) expression in taurine-deficient mice is reversed by taurine supplementation in vivo. Amino Acids 2016, 48, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Lambert, I.H. Reactive oxygen species regulate swelling-induced taurine efflux in NIH3T3 mouse fibroblasts. J. Membr. Biol. 2003, 192, 19–32. [Google Scholar] [CrossRef]
- Abebe, W.; Mozaffari, M.S. Taurine depletion alters vascular reactivity in rats. Can. J. Physiol. Pharmacol. 2003, 81, 903–909. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, B.; Li, Y.; Sun, F.; Li, P.; Xia, W.; Zhou, X.; Li, Q.; Wang, X.; Chen, J.; et al. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension 2016, 67, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Ristori, M.T.; Verdetti, J. Effects of taurine on rat aorta in vitro. Fundam. Clin. Pharmacol. 1991, 5, 245–258. [Google Scholar] [CrossRef]
- Innes, D.; Sevitt, S. Coagulation and fibrinolysis in injured patients. J. Clin. Pathol. 1964, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Floccard, B.; Rugeri, L.; Faure, A.; Saint Denis, M.; Boyle, E.M.; Peguet, O.; Levrat, A.; Guillaume, C.; Marcotte, G.; Vulliez, A.; et al. Early coagulopathy in trauma patients: An on-scene and hospital admission study. Injury 2012, 43, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.C.; Craft, R.M.; Langdon, R.J.; Clanton, C.R.; Snider, C.C.; Wellons, D.D.; Dakin, P.A.; Lawson, C.M.; Enderson, B.L.; Kurek, S.J. Early evaluation of acute traumatic coagulopathy by thrombelastography. Transl. Res. 2009, 154, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Rizoli, S.B.; Scarpelini, S.; Callum, J.; Nascimento, B.; Mann, K.G.; Pinto, R.; Jansen, J.; Tien, H.C. Clotting factor deficiency in early trauma-associated coagulopathy. J. Trauma 2011, 71 (Suppl. 1), S427–S434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolley, T.; Gwyther, R.; Parmar, K.; Kirkman, E.; Watts, S.; Midwinter, M.; Lucca, J.D.; Hunt, B.J. A prospective observational study of acute traumatic coagulopathy in traumatic bleeding from the battlefield. Transfusion 2020, 60 (Suppl. 3), S52–S61. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.O.; Scarpelini, S.; Pinto, R.; Tien, H.C.; Callum, J.; Rizoli, S.B. Hypoperfusion in severely injured trauma patients is associated with reduced coagulation factor activity. J. Trauma 2011, 71 (Suppl. 1), S435–S440. [Google Scholar] [CrossRef]
- Theusinger, O.M.; Baulig, W.; Seifert, B.; Müller, S.M.; Mariotti, S.; Spahn, D.R. Changes in coagulation in standard laboratory tests and ROTEM in trauma patients between on-scene and arrival in the emergency department. Anesth. Analg. 2015, 120, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Camire, R.M.; Pollak, E.S.; Kaushansky, K.; Tracy, P.B. Secretable human platelet-derived factor V originates from the plasma pool. Blood 1998, 92, 3035–3041. [Google Scholar] [CrossRef]
- Tracy, P.B.; Eide, L.L.; Bowie, E.J.; Mann, K.G. Radioimmunoassay of factor V in human plasma and platelets. Blood 1982, 60, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Chesney, C.M.; Pifer, D.; Colman, R.W. Subcellular localization and secretion of factor V from human platelets. Proc. Natl. Acad. Sci. USA 1981, 78, 5180–5184. [Google Scholar] [CrossRef] [Green Version]
- Wencel-Drake, J.D.; Dahlback, B.; White, J.G.; Ginsberg, M.H. Ultrastructural localization of coagulation factor V in human platelets. Blood 1986, 68, 244–249. [Google Scholar] [CrossRef]
- Tracy, P.B.; Giles, A.R.; Mann, K.G.; Eide, L.L.; Hoogendoorn, H.; Rivard, G.E. Factor V (Quebec): A bleeding diathesis associated with a qualitative platelet Factor V deficiency. J. Clin. Investig. 1984, 74, 1221–1228. [Google Scholar] [CrossRef]
- Kahr, W.H.; Zheng, S.; Sheth, P.M.; Pai, M.; Cowie, A.; Bouchard, M.; Podor, T.J.; Rivard, G.E.; Hayward, C.P. Platelets from patients with the Quebec platelet disorder contain and secrete abnormal amounts of urokinase-type plasminogen activator. Blood 2001, 98, 257–265. [Google Scholar] [CrossRef]
- Bertina, R.M.; Koeleman, B.P.; Koster, T.; Rosendaal, F.R.; Dirven, R.J.; de Ronde, H.; van der Velden, P.A.; Reitsma, P.H. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994, 369, 64–67. [Google Scholar] [CrossRef]
- Ridker, P.M.; Miletich, J.P.; Hennekens, C.H.; Buring, J.E. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997, 277, 1305–1307. [Google Scholar] [CrossRef]
- Zivelin, A.; Griffin, J.H.; Xu, X.; Pabinger, I.; Samama, M.; Conard, J.; Brenner, B.; Eldor, A.; Seligsohn, U. A single genetic origin for a common Caucasian risk factor for venous thrombosis. Blood 1997, 89, 397–402. [Google Scholar] [CrossRef]
- Van Mens, T.E.; Levi, M.; Middeldorp, S. Evolution of Factor V Leiden. Thromb. Haemost. 2013, 110, 23–30. [Google Scholar]
- Lindqvist, P.G.; Dahlbäck, B. Carriership of Factor V Leiden and evolutionary selection advantage. Curr. Med. Chem. 2008, 15, 1541–1544. [Google Scholar] [CrossRef]
- Lindqvist, P.G.; Svensson, P.J.; Dahlbäck, B.; Marsál, K. Factor V Q506 mutation (activated protein C resistance) associated with reduced intrapartum blood loss—A possible evolutionary selection mechanism. Thromb. Haemost. 1998, 79, 69–73. [Google Scholar]
- Lindqvist, P.G.; Svensson, P.J.; Marsaál, K.; Grennert, L.; Luterkort, M.; Dahlbäck, B. Activated protein C resistance (FV:Q506) and pregnancy. Thromb. Haemost. 1999, 81, 532–537. [Google Scholar] [CrossRef]
- Lindqvist, P.G.; Zöller, B.; Dahlbäck, B. Improved hemoglobin status and reduced menstrual blood loss among female carriers of factor V Leiden--an evolutionary advantage? Thromb. Haemost. 2001, 86, 1122–1123. [Google Scholar] [PubMed]
- Gando, S.; Shiraishi, A.; Wada, T.; Yamakawa, K.; Fujishima, S.; Saitoh, D.; Kushimoto, S.; Ogura, H.; Abe, T.; Otomo, Y. A multicenter prospective validation study on disseminated intravascular coagulation in trauma-induced coagulopathy. J. Thromb. Haemost. 2020, 18, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Raza, I.; Davenport, R.; Rourke, C.; Platton, S.; Manson, J.; Spoors, C.; Khan, S.; De’Ath, H.D.; Allard, S.; Hart, D.P.; et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J. Thromb. Haemost. 2013, 11, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Blombäck, M.; Eklund, J.; Hellgren, M.; Lagerkranser, M.; Swedenborg, J. Blood coagulation and fibrinolytic factors as well as their inhibitors in trauma. Scand. J. Clin. Lab. Investig. Suppl. 1985, 178, 15–23. [Google Scholar]
- Cardenas, J.C.; Matijevic, N.; Baer, L.A.; Holcomb, J.B.; Cotton, B.A.; Wade, C.E. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock 2014, 41, 514–521. [Google Scholar] [CrossRef]
- Cardenas, J.C.; Wade, C.E.; Cotton, B.A.; George, M.J.; Holcomb, J.B.; Schreiber, M.A.; White, N.J. TEG Lysis Shutdown Represents Coagulopathy in Bleeding Trauma Patients: Analysis of the PROPPR Cohort. Shock 2019, 51, 273–283. [Google Scholar] [CrossRef]
- Chapman, M.P.; Moore, E.E.; Moore, H.B.; Gonzalez, E.; Gamboni, F.; Chandler, J.G.; Mitra, S.; Ghasabyan, A.; Chin, T.L.; Sauaia, A.; et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 2016, 80, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Hasan, A.A.; Cines, D.B.; Ngaiza, J.R.; Jaffe, E.A.; Schmaier, A.H. High-molecular-weight kininogen is exclusively membrane bound on endothelial cells to influence activation of vascular endothelium. Blood 1995, 85, 3134–3143. [Google Scholar] [CrossRef]
- Kolte, D.; Osman, N.; Yang, J.; Shariat-Madar, Z. High molecular weight kininogen activates B2 receptor signaling pathway in human vascular endothelial cells. J. Biol. Chem. 2011, 286, 24561–24571. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.P.; Ghebrehiwet, B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol. Immunol. 2010, 47, 2161–2169. [Google Scholar] [CrossRef]
- Reddigari, S.; Silverberg, M.; Kaplan, A.P. Assembly of the human plasma kinin-forming cascade along the surface of vascular endothelial cells. Int. Arch. Allergy Immunol. 1995, 107, 93–94. [Google Scholar] [CrossRef]
- Murray, M.A.; Heistad, D.D.; Mayhan, W.G. Role of protein kinase C in bradykinin-induced increases in microvascular permeability. Circ. Res. 1991, 68, 1340–1348. [Google Scholar] [CrossRef] [Green Version]
- Oh-ishi, S.; Hayashi, I.; Yamaki, K.; Utsunomiya, I.; Hayashi, M.; Yamasu, A.; Nakano, T. Role of high molecular weight (HMW)-kininogen in inflammatory exudation: Evidence with the studies of the HMW-kininogen deficient rat. Adv. Exp. Med. Biol. 1989, 247a, 145–152. [Google Scholar]
- Kaplan, A.P.; Ghebrehiwet, B.; Silverberg, M.; Sealey, J.E. The intrinsic coagulation-kinin pathway, complement cascades, plasma renin-angiotensin system, and their interrelationships. Crit. Rev. Immunol. 1981, 3, 75–93. [Google Scholar]
- Cap, A.P. Plasmin: A driver of hemovascular dysfunction. Blood 2016, 128, 2375–2376. [Google Scholar] [CrossRef] [Green Version]
- Renné, T.; Schuh, K.; Müller-Esterl, W. Local bradykinin formation is controlled by glycosaminoglycans. J. Immunol. 2005, 175, 3377–3385. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Ledford, B.E. Expression of the haemopexin-transport system in cultured mouse hepatoma cells. Links between haemopexin and iron metabolism. Biochem. J. 1988, 256, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Montecinos, L.; Eskew, J.D.; Smith, A. What Is Next in This “Age” of Heme-Driven Pathology and Protection by Hemopexin? An Update and Links with Iron. Pharmaceuticals 2019, 12, 144. [Google Scholar] [CrossRef] [Green Version]
- Wagener, B.M.; Hu, P.J.; Oh, J.Y.; Evans, C.A.; Richter, J.R.; Honavar, J.; Brandon, A.P.; Creighton, J.; Stephens, S.W.; Morgan, C.; et al. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: A preclinical experimental study. PLoS Med. 2018, 15, e1002522. [Google Scholar] [CrossRef] [Green Version]
- Morgan, H.P.; Schmidt, C.Q.; Guariento, M.; Blaum, B.S.; Gillespie, D.; Herbert, A.P.; Kavanagh, D.; Mertens, H.D.; Svergun, D.I.; Johansson, C.M.; et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol. 2011, 18, 463–470. [Google Scholar]
- Jokiranta, T.S.; Cheng, Z.Z.; Seeberger, H.; Jòzsi, M.; Heinen, S.; Noris, M.; Remuzzi, G.; Ormsby, R.; Gordon, D.L.; Meri, S.; et al. Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am. J. Pathol. 2005, 167, 1173–1181. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Noris, M.; Remuzzi, G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001, 60, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Dopler, A.; Guntau, L.; Harder, M.J.; Palmer, A.; Höchsmann, B.; Schrezenmeier, H.; Simmet, T.; Huber-Lang, M.; Schmidt, C.Q. Self versus Nonself Discrimination by the Soluble Complement Regulators Factor H and FHL-1. J. Immunol. 2019, 202, 2082–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Kuraya, M.; Hamasaki, N.; Tsujimura, M.; Shiraki, H.; Fujita, T. Activation of the lectin complement pathway by H-ficolin (Hakata antigen). J. Immunol. 2002, 168, 3502–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorski, J.P.; Hugli, T.E.; Müller-Eberhard, H.J. C4a: The third anaphylatoxin of the human complement system. Proc. Natl. Acad. Sci. USA 1979, 76, 5299–5302. [Google Scholar] [CrossRef] [Green Version]
- Saadi, S.; Holzknecht, R.A.; Patte, C.P.; Stern, D.M.; Platt, J.L. Complement-mediated regulation of tissue factor activity in endothelium. J. Exp. Med. 1995, 182, 1807–1814. [Google Scholar] [CrossRef]
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989, 264, 9053–9060. [Google Scholar] [CrossRef]
- Kilgore, K.S.; Shen, J.P.; Miller, B.F.; Ward, P.A.; Warren, J.S. Enhancement by the complement membrane attack complex of tumor necrosis factor-alpha-induced endothelial cell expression of E-selectin and ICAM-1. J. Immunol. 1995, 155, 1434–1441. [Google Scholar]
- Kilgore, K.S.; Flory, C.M.; Miller, B.F.; Evans, V.M.; Warren, J.S. The membrane attack complex of complement induces interleukin-8 and monocyte chemoattractant protein-1 secretion from human umbilical vein endothelial cells. Am. J. Pathol. 1996, 149, 953–961. [Google Scholar]
- Kilgore, K.S.; Schmid, E.; Shanley, T.P.; Flory, C.M.; Maheswari, V.; Tramontini, N.L.; Cohen, H.; Ward, P.A.; Friedl, H.P.; Warren, J.S. Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappa B activation. Am. J. Pathol. 1997, 150, 2019–2031. [Google Scholar]
- Dobrina, A.; Pausa, M.; Fischetti, F.; Bulla, R.; Vecile, E.; Ferrero, E.; Mantovani, A.; Tedesco, F. Cytolytically inactive terminal complement complex causes transendothelial migration of polymorphonuclear leukocytes in vitro and in vivo. Blood 2002, 99, 185–192. [Google Scholar] [CrossRef]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997, 185, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Atefi, G.; Aisiku, O.; Shapiro, N.; Hauser, C.; Dalle Lucca, J.; Flaumenhaft, R.; Tsokos, G.C. Complement Activation in Trauma Patients Alters Platelet Function. Shock 2016, 46 (Suppl. 1), 83–88. [Google Scholar] [CrossRef]
- Del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Muroya, T.; Kannan, L.; Ghiran, I.C.; Shevkoplyas, S.S.; Paz, Z.; Tsokos, M.; Dalle Lucca, J.J.; Shapiro, N.I.; Tsokos, G.C. C4d deposits on the surface of RBCs in trauma patients and interferes with their function. Crit. Care Med. 2014, 42, e364–e372. [Google Scholar] [CrossRef] [PubMed]
- Calfee, C.S.; Delucchi, K.; Parsons, P.E.; Thompson, B.T.; Ware, L.B.; Matthay, M.A. Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir. Med. 2014, 2, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Calfee, C.S.; Delucchi, K.L.; Sinha, P.; Matthay, M.A.; Hackett, J.; Shankar-Hari, M.; McDowell, C.; Laffey, J.G.; O’Kane, C.M.; McAuley, D.F. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: Secondary analysis of a randomised controlled trial. Lancet Respir. Med. 2018, 6, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Wiggers, H.C.; Ingraham, R.C.; Dille, J. Hemorrhagic-hypotension shock in locally anesthetized dogs. Am. J. Physiol. Leg. Content 1945, 143, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.; Seligman, A.M. Traumatic shock: Iv. a study of the problem of the “lost plasma” in hemorrhagic shock by the use of radioactive plasma protein. J. Clin. Investig. 1943, 22, 285–303. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| n | All Trauma Subjects (n = 99) | n | Non-EoT (n = 37) | n | EoT (n = 62) | p-Value | |
|---|---|---|---|---|---|---|---|
| Demographics | |||||||
| Age (years) | 99 | 44 (34, 54) | 37 | 46 (36, 58) | 62 | 44 (32, 51) | 0.265 |
| Body mass index | 96 | 27.95 (25, 31) | 37 | 28.3 (25, 31) | 59 | 27.2 (25, 31) | 0.698 |
| Male | 99 | 73 (74%) | 37 | 25 (68%) | 62 | 48 (77%) | 0.347 |
| Race | 99 | 37 | 62 | 0.616 | |||
| White | 70 (71%) | 42 (67.74%) | 28 (75.68%) | ||||
| African American | 27 (27%) | 9 (24%) | 18 (29%) | ||||
| Asian/Pacific Islander | 2 (2%) | 1 (2.7%) | 1 (1.6%) | ||||
| Ethnicity | 99 | 37 | 62 | 1 | |||
| Hispanic | 23 (24%) | 8 (22%) | 15 (25%) | ||||
| Injury Characteristics | |||||||
| Injury severity score | 99 | 25 (16, 29) | 37 | 22 (9, 25) | 62 | 26 (22, 34) | <0.001 * |
| Head AIS | 0 (0, 0) | 0 (0,0) | 0 (0,0) | 0.907 | |||
| Face AIS | 0 (0, 0) | 0 (0,0) | 0 (0,0) | 0.154 | |||
| Chest AIS | 3 (0, 4) | 0 (0, 3) | 3 (0,4) | 0.026 * | |||
| Abdomen AIS | 0 (0, 4) | 0 (0, 2) | 2 (0, 4) | 0.001 * | |||
| Extremity AIS | 0 (0, 3) | 0 (0, 3) | 0 (0, 3) | 0.177 | |||
| External AIS | 1 (0, 2) | 1 (0, 3) | 1 (1, 2) | 0.188 | |||
| Mechanism of injury | 99 | 37 | 62 | 0.005 * | |||
| Blunt | 55 (56%) | 16 (43%) | 39 (63%) | ||||
| Penetrating | 25 (25%) | 8 (22%) | 17 (27%) | ||||
| Burn | 19 (19%) | 13 (35%) | 6 (10%) | ||||
| Admission Vital Signs and Labs | |||||||
| Glasgow Coma Scale | 99 | 37 | 62 | 0.018 * | |||
| 3 | 37 (37.4%) | 10 (27.0%) | 27 (43.5%) | ||||
| 7 | 2 (2.0%) | 0 (0.0%) | 2 (3.2%) | ||||
| 8 | 1 (1.0%) | 0 (0.0%) | 1 (1.6%) | ||||
| 9 | 2 (2.0%) | 0 (0.0%) | 2 (3.2%) | ||||
| 11 | 1 (1.0%) | 0 (0.0%) | 1 (1.6%) | ||||
| 13 | 2 (2.0%) | 1 (2.7%) | 1 (1.6%) | ||||
| 14 | 5 (5.1%) | 1 (2.7%) | 4 (6.5%) | ||||
| 15 | 49 (49.5%) | 25 (67.6%) | 24 (38.7%) | ||||
| Systolic BP (mm Hg) | 97 | 118 (99, 137) | 35 | 125 (113, 139) | 62 | 110 (97, 130) | 0.018 |
| Heart rate (bpm) | 97 | 100 (83, 116) | 35 | 96 (79, 110) | 62 | 101 (84, 121) | 0.252 |
| Creatinine (mg/dL) | 97 | 1.21 (1.00, 1.47) | 37 | 1.08 (0.95, 1.30) | 60 | 1.32 (1.01, 1.57) | 0.003 * |
| Glucose (mg/dL) | 97 | 163 (121, 213) | 37 | 153 (117, 180) | 60 | 180 (133, 230) | 0.020 * |
| Albumin (g/dL) | 34 | 3.10 (2.73, 3.40) | 13 | 3.00 (2.90, 3.50) | 21 | 3.10 (2.70, 3.40) | 0.845 |
| Base excess (mmol/L) | 98 | −6 (−9, −2) | 37 | −2 (−6, −2) | 61 | −8 (−12, −4) | <0.001 * |
| pH | 96 | 7.26 (7.18, 7.35) | 36 | 7.34 (7.28, 7.35) | 60 | 7.20 (7.10, 7.28) | <0.001 * |
| Platelet count (K/uL) | 95 | 230 (187, 281) | 37 | 242 (199, 314) | 58 | 215 (164, 277) | 0.127 |
| ACT (s) | 91 | 113 (105, 121) | 34 | 105 (105, 113) | 57 | 113 (105, 121) | 0.047 * |
| TEG R-time (min) | 91 | 0.7 (0.6, 0.8) | 34 | 0.6 (0.6, 0.7) | 57 | 0.70 (0.6, 0.8) | 0.091 |
| TEG K-time (min) | 90 | 1.30 (1.10, 1.80) | 34 | 1.20 (1.02, 1.50) | 56 | 1.45 (1.20, 1.80) | 0.023 * |
| TEG α-angle (degrees) | 91 | 74 (70, 77) | 34 | 76 (72,78) | 57 | 73 (69, 76) | 0.016 * |
| TEG maximum amplitude (mm) | 91 | 64 (59, 68) | 34 | 66 (62, 69) | 57 | 62 (58, 66) | 0.002 * |
| TEG G-value (Kdynes/cm2) | 91 | 8.70 (7.15, 10.50) | 34 | 9.75 (8.03, 11.30) | 57 | 8.20 (6.80, 9.50) | 0.002 * |
| TEG estimated lysis (%) | 91 | 1 (0.1, 2.4) | 34 | 1.2 (0.2, 2.5) | 57 | 0.9 (0.1, 2.5) | 0.961 |
| Transfusions | |||||||
| Any blood product prehospital to 24 h | 99 | 71 (72%) | 37 | 21 (57%) | 62 | 50 (81%) | 0.020 * |
| Outcomes | |||||||
| ICU-free days (30 days) | 99 | 19 (0, 28) | 37 | 26 (0, 30) | 62 | 9 (0, 27) | 0.058 |
| ICU days | 99 | 3, (0, 11) | 37 | 2 (0, 10) | 62 | 4 (0, 13) | 0.450 |
| 12-h mortality | 99 | 13 (13%) | 37 | 1 (3%) | 62 | 12 (19%) | 0.028 * |
| 24-h mortality | 99 | 14 (14%) | 37 | 1 (3%) | 62 | 13 (21%) | 0.015 * |
| 72-h mortality | 99 | 15 (15%) | 37 | 1 (3%) | 62 | 14 (23%) | 0.008 * |
| In-hospital mortality | 99 | 37 (37%) | 37 | 10 (27%) | 62 | 27 (44%) | 0.133 |
| Complications | |||||||
| Acute renal failure | 99 | 27 (27%) | 37 | 10 (27%) | 62 | 17 (27%) | 1 |
| Deep vein thrombosis | 99 | 1 (1%) | 37 | 1 (3%) | 62 | 0 (0%) | 0.374 |
| Pulmonary embolism | 99 | 7 (7%) | 37 | 0 (0%) | 62 | 7 (11%) | 0.043 * |
| Pneumonia | 99 | 16 (16%) | 37 | 2 (5%) | 62 | 14 (23%) | 0.026 * |
| Sepsis | 99 | 19 (19%) | 37 | 7 (19%) | 62 | 12 (20%) | 1 |
| Urinary tract infection | 99 | 6 (6%) | 37 | 4 (11%) | 62 | 2 (3%) | 0.193 |
| ARDS | 99 | 6 (6%) | 37 | 3 (8%) | 62 | 3 (5%) | 0.668 |
| Respiratory failure | 99 | 40 (40%) | 37 | 14 (38%) | 62 | 26 (42%) | 0.833 |
| SIRS | 99 | 7 (7%) | 37 | 4 (11%) | 62 | 3 (5%) | 0.419 |
| Decubitus ulcer | 99 | 3 (3%) | 37 | 0 (0%) | 62 | 3 (5%) | 0.291 |
| Multiple organ failure | 99 | 17 (17%) | 37 | 7 (19%) | 62 | 10 (16%) | 0.786 |
| n | All Trauma Subjects (n = 99) | Non-EoT (n = 62) | EoT (n = 37) | p-Value | |
|---|---|---|---|---|---|
| Epinephrine | 99 | 315 (140, 906) | 241 (89, 392) | 458 (201, 2136) | 0.004 * |
| Norepinephrine | 99 | 1295 (595, 2432) | 780 (388, 1308) | 1799 (803, 3642) | <0.001 * |
| Soluble Thrombomodulin | 99 | 6.6 (5.2, 9.5) | 5.2 (4.7, 6.5) | 7.5 (6.1, 11.4) | <0.001 * |
| Syndecan-1 | 99 | 67 (28, 184) | 25 (19, 31) | 165 (79, 195) | <0.001 * |
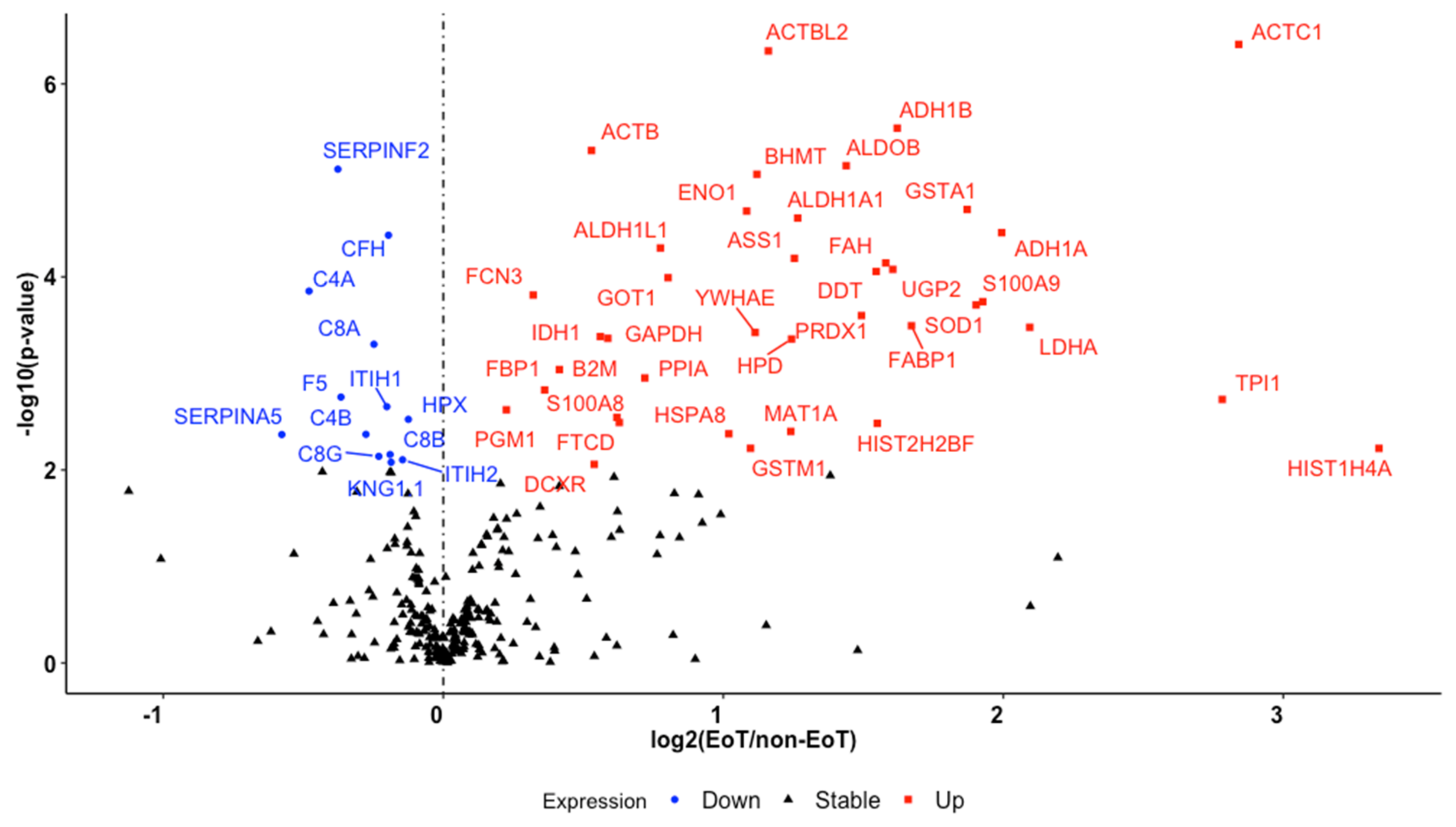
| Gene Symbol | Protein Description | Raw p-Value ¥ | Adjusted p-Value £ | Fold Change ± |
|---|---|---|---|---|
| HIST1H4A | Histone H4 | 5.97 × 10−3 | 3.69 × 10−2 | 3.34 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 3.90 × 10−7 | 6.61 × 10−5 | 2.84 |
| TPI1 | Isoform 2 of triosephosphate isomerase | 1.86 × 10−3 | 1.54 × 10−2 | 2.78 |
| LDHA | L-lactate dehydrogenase A chain | 3.33 × 10−4 | 3.86 × 10−3 | 2.09 |
| ADH1A | Alcohol dehydrogenase 1A | 3.48 × 10−5 | 8.94 × 10−4 | 1.99 |
| S100A9 | Protein S100-A9 | 1.80 × 10−4 | 2.49 × 10−3 | 1.93 |
| SOD1 | Superoxide dismutase | 1.95 × 10−4 | 2.58 × 10−3 | 1.90 |
| GSTA1 | Glutathione S-transferase | 2.00 × 10−5 | 6.71 × 10−4 | 1.87 |
| FABP1 | Fatty acid-binding protein, liver | 3.20 × 10−4 | 3.86 × 10−3 | 1.67 |
| ADH1B | Alcohol dehydrogenase 1B | 2.87 × 10−6 | 2.78 × 10−4 | 1.62 |
| UGP2 | UTP--glucose-1-phosphate uridylyltransferase | 8.35 × 10−5 | 1.50 × 10−3 | 1.61 |
| FAH | Fumarylacetoacetase | 7.17 × 10−5 | 1.39 × 10−3 | 1.58 |
| HIST2H2BF | Histone H2B type 2-F | 3.29 × 10−3 | 2.33 × 10−2 | 1.55 |
| DDT | D-dopachrome decarboxylase | 8.78 × 10−5 | 1.50 × 10−3 | 1.55 |
| PRDX1 | Peroxiredoxin-1 | 2.51 × 10−4 | 3.17 × 10−3 | 1.49 |
| ALDOB | Fructose-bisphosphate aldolase B | 7.04 × 10−6 | 3.58 × 10−4 | 1.44 |
| ALDH1A1 | Retinal dehydrogenase 1 | 2.46 × 10−5 | 7.12 × 10−4 | 1.27 |
| ASS1 | Argininosuccinate synthase | 6.42 × 10−5 | 1.33 × 10−3 | 1.25 |
| HPD | 4-hydroxyphenylpyruvate dioxygenase | 4.40 × 10−4 | 4.40 × 10−3 | 1.24 |
| MAT1A | S-adenosylmethionine synthase isoform type-1 | 3.99 × 10−3 | 2.75 × 10−2 | 1.24 |
| ACTBL2 | Beta-actin-like protein 2 O | 4.56 × 10−7 | 6.61 × 10−5 | 1.16 |
| BHMT | Betaine--homocysteine S-methyltransferase 1 | 8.64 × 10−6 | 3.58 × 10−4 | 1.12 |
| YWHAE | 14-3-3 protein epsilon | 3.77 × 10−4 | 4.20 × 10−3 | 1.11 |
| GSTM1 | Glutathione S-transferase Mu 1 | 5.97 × 10−3 | 3.69 × 10−2 | 1.10 |
| ENO1 | Alpha-enolase | 2.08 × 10−5 | 6.71 × 10−4 | 1.08 |
| HSPA8 | Heat shock cognate 71 kDa protein | 4.22 × 10−3 | 2.77 × 10−2 | 1.02 |
| GOT1 | Aspartate aminotransferase, cytoplasmic | 1.02 × 10−4 | 1.64 × 10−3 | 0.80 |
| ALDH1L1 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 5.02 × 10−5 | 1.12 × 10−3 | 0.78 |
| PPIA | Peptidyl-prolyl cis-trans isomerase A | 1.11 × 10−3 | 1.01 × 10−2 | 0.72 |
| FTCD | Formimidoyltransferase-cyclodeaminase | 3.22 × 10−3 | 2.33 × 10−2 | 0.63 |
| S100A8 | Protein S100-A8 | 2.85 × 10−3 | 2.17 × 10−2 | 0.62 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 4.33 × 10−4 | 4.40 × 10−3 | 0.59 |
| IDH1 | Isocitrate dehydrogenase [NADP] cytoplasmic | 4.15 × 10−4 | 4.40 × 10−3 | 0.56 |
| DCXR | L-xylulose reductase | 8.75 × 10−3 | 4.88 × 10−2 | 0.54 |
| ACTB | Actin, cytoplasmic 1 | 4.90 × 10−6 | 3.55 × 10−4 | 0.53 |
| B2M | Beta-2-microglobulin | 9.11 × 10−4 | 8.52 × 10−3 | 0.41 |
| FBP1 | Fructose-1,6-bisphosphatase 1 | 1.48 × 10−3 | 1.30 × 10−2 | 0.36 |
| FCN3 | Ficolin-3 OS = Homo sapiens | 1.54 × 10−4 | 2.24 × 10−3 | 0.32 |
| PGM1 | Phosphoglucomutase-1 | 2.38 × 10−3 | 1.87 × 10−2 | 0.23 |
| HPX | Hemopexin | 2.99 × 10−3 | 2.22 × 10−2 | −0.12 |
| ITIH2 | Inter-alpha-trypsin inhibitor heavy chain H2 | 7.85 × 10−3 | 4.55 × 10−2 | −0.15 |
| KNG1 | Kininogen-1 | 8.33 × 10−3 | 4.73 × 10−2 | −0.19 |
| C8B | Complement component C8 beta chain | 6.92 × 10−3 | 4.18 × 10−2 | −0.19 |
| CFH | Complement factor H | 3.70 × 10−5 | 8.94 × 10−4 | −0.20 |
| ITIH1 | Inter-alpha-trypsin inhibitor heavy chain H1 | 2.21 × 10−3 | 1.78 × 10−2 | −0.20 |
| C8G | Complement component C8 gamma chain | 7.23 × 10−3 | 4.28 × 10−2 | −0.23 |
| C8A | Complement component C8 alpha chain | 4.99 × 10−4 | 4.82 × 10−3 | −0.25 |
| C4B | Complement C4-B | 4.27 × 10−3 | 2.77 × 10−2 | −0.28 |
| F5 | Coagulation factor V | 1.76 × 10−3 | 1.50 × 10−2 | −0.37 |
| SERPINF2 | Alpha-2-antiplasmin | 7.66 × 10−6 | 3.58 × 10−4 | −0.38 |
| C4A; C4B | Complement C4-A | 1.40 × 10−4 | 2.14 × 10−3 | −0.48 |
| SERPINA5 | Plasma serine protease inhibitor | 4.30 × 10−3 | 2.77 × 10−2 | −0.58 |
| AST | ALT | |||
|---|---|---|---|---|
| r | p-Value | r | p-Value | |
| BHMT | 0.837 (0. 0.729, 0.904) | <0.001 * | 0.894 (0.820, 0.938) | <0.001 * |
| ADH1A | 0.907 (0. 842, 0.946) | <0.001 * | 0.795 (0.666, 0.878) | <0.001 * |
| MAT1A | 0.441 (0.188, 0.639) | 0.001 * | 0.310 (0.038, 0.539) | 0.027 * |
| HPD | 0.631 (0.430, 0.772) | <0.001 * | 0.654 (0.461, 0.787) | <0.001 * |
| FABP1 | 0.828 (0.716, 0.899) | <0.001 * | 0.732 (0.572, 0.838) | <0.001 * |
| non-AKI n = 72 | AKI n = 27 | p-Value | |
|---|---|---|---|
| BHMT | 2,039,625 (1,498,128, 5,608,227) | 1,276,029 (1,285,297, 2,118,322) | 0.226 |
| HPD | 3,018,214 (2,053,260, 8,459,705) | 3,118,767 (1,450,326, 5,226,505) | 0.188 |
| FABP1 | 3,362,220 (1,877,758, 16,830,020) | 3,032,475 (2,430,624, 5,302,253) | 0.997 |
| S100-A8, S-100A9, S100-A8/A9 | Histones | PPIA | |
|---|---|---|---|
| Pro-inflammatory | [87,88,89,90,91] | [50,51,53,60,92] | [66,93,94] |
| Anti-inflammatory | [41,42,95,96,97] | ||
| Thrombosis | [87,98,99,100,101] | [52,85,102,103,104,105,106,107] | [65,80,108] |
| Endothelial impairment and activation | [109,110,111] | [52,53,54,57,60,92,112] | [94,113,114,115] |
| Endothelial permeability | [87,116,117,118] | [56,118,119] | |
| Cardiac injury | [74,120,121,122,123] | [124,125,126] | [127,128,129,130] |
| Pulmonary injury | [52,131,132,133,134] | [56,60,135,136] | [64,137] |
| Renal injury | [138,139,140,141,142] | [52,143,144] | [145,146,147] |
| Hepatic injury | [76,148,149,150] | [50,51,151,152] | [153,154,155,156] |
| Trauma populations |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krocker, J.D.; Lee, K.H.; Henriksen, H.H.; Wang, Y.-W.W.; Schoof, E.M.; Karvelsson, S.T.; Rolfsson, Ó.; Johansson, P.I.; Pedroza, C.; Wade, C.E. Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma. Int. J. Mol. Sci. 2022, 23, 6213. https://doi.org/10.3390/ijms23116213
Krocker JD, Lee KH, Henriksen HH, Wang Y-WW, Schoof EM, Karvelsson ST, Rolfsson Ó, Johansson PI, Pedroza C, Wade CE. Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma. International Journal of Molecular Sciences. 2022; 23(11):6213. https://doi.org/10.3390/ijms23116213
Chicago/Turabian StyleKrocker, Joseph D., Kyung Hyun Lee, Hanne H. Henriksen, Yao-Wei Willa Wang, Erwin M. Schoof, Sigurdur T. Karvelsson, Óttar Rolfsson, Pär I. Johansson, Claudia Pedroza, and Charles E. Wade. 2022. "Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma" International Journal of Molecular Sciences 23, no. 11: 6213. https://doi.org/10.3390/ijms23116213
APA StyleKrocker, J. D., Lee, K. H., Henriksen, H. H., Wang, Y.-W. W., Schoof, E. M., Karvelsson, S. T., Rolfsson, Ó., Johansson, P. I., Pedroza, C., & Wade, C. E. (2022). Exploratory Investigation of the Plasma Proteome Associated with the Endotheliopathy of Trauma. International Journal of Molecular Sciences, 23(11), 6213. https://doi.org/10.3390/ijms23116213