
Novel Anti Double-Stranded Nucleic Acids Full-Length Recombinant Camelid Heavy-Chain Antibody for the Detection of miRNA


Abstract
:
1. Introduction
2. Results
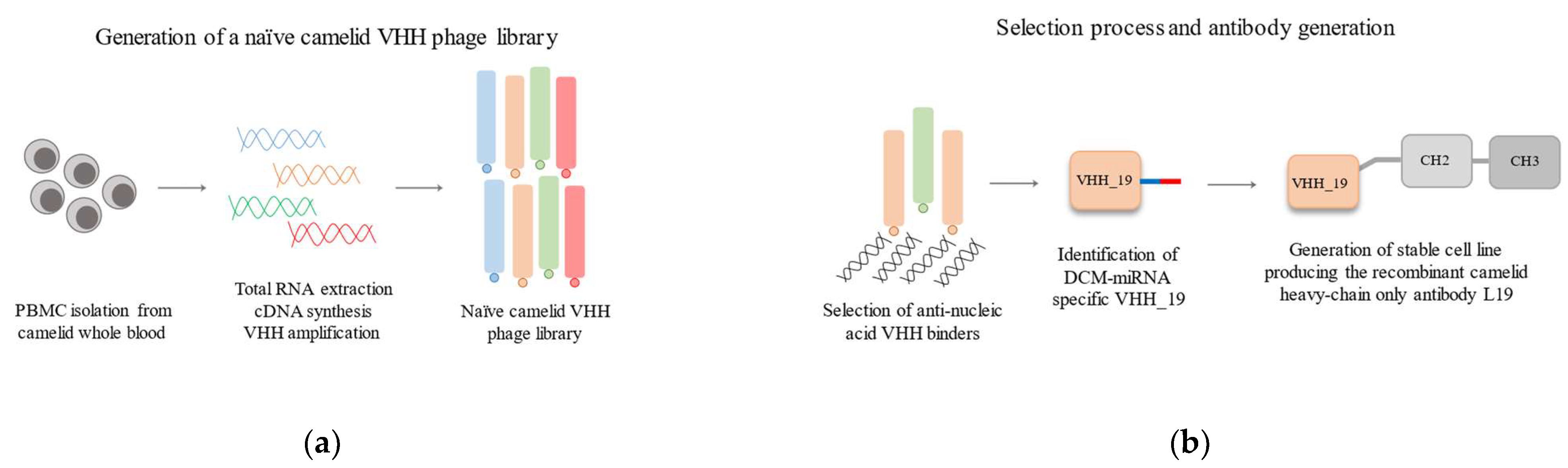
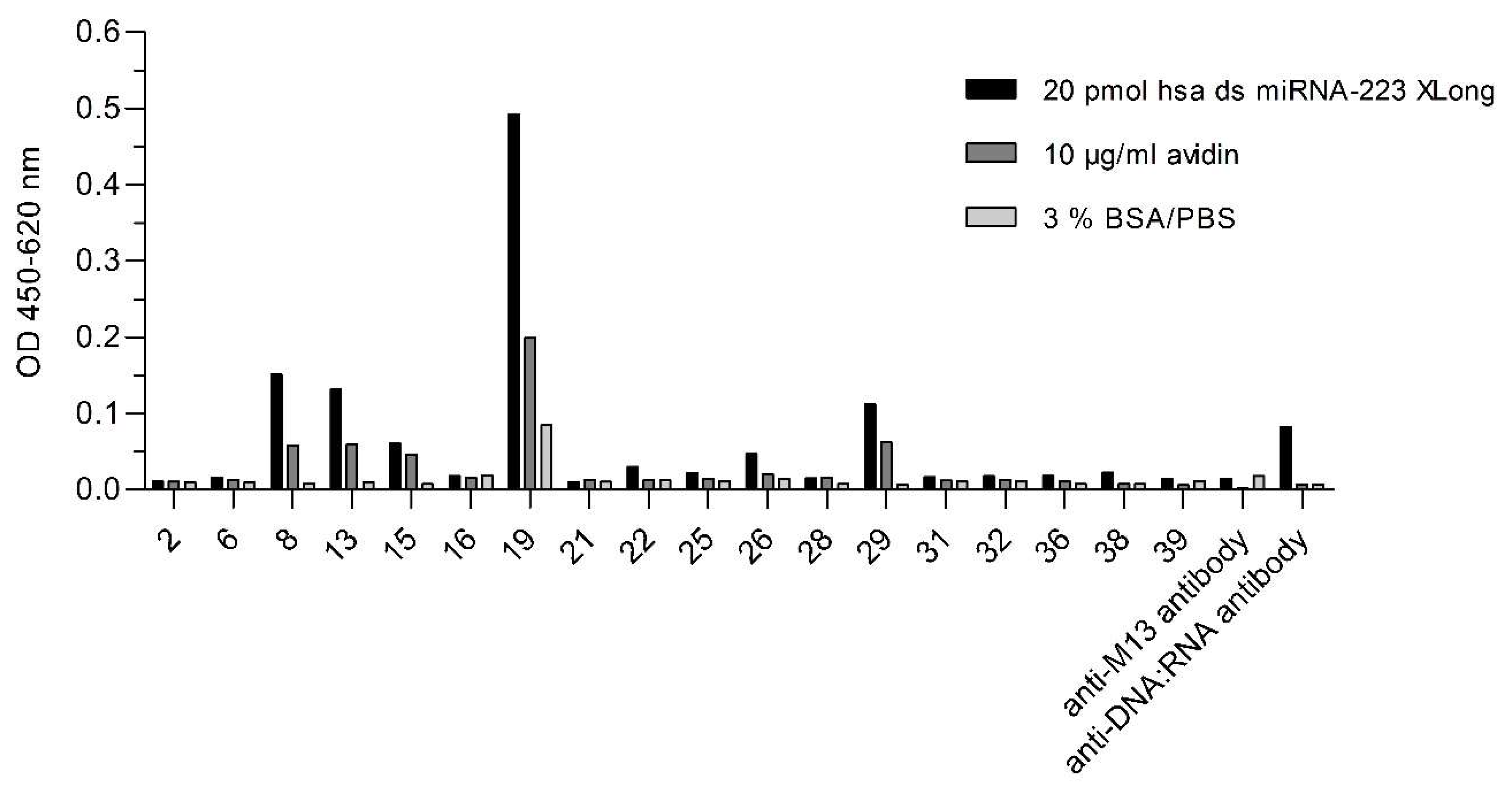
2.1. Selection of ds miRNA VHH Binder
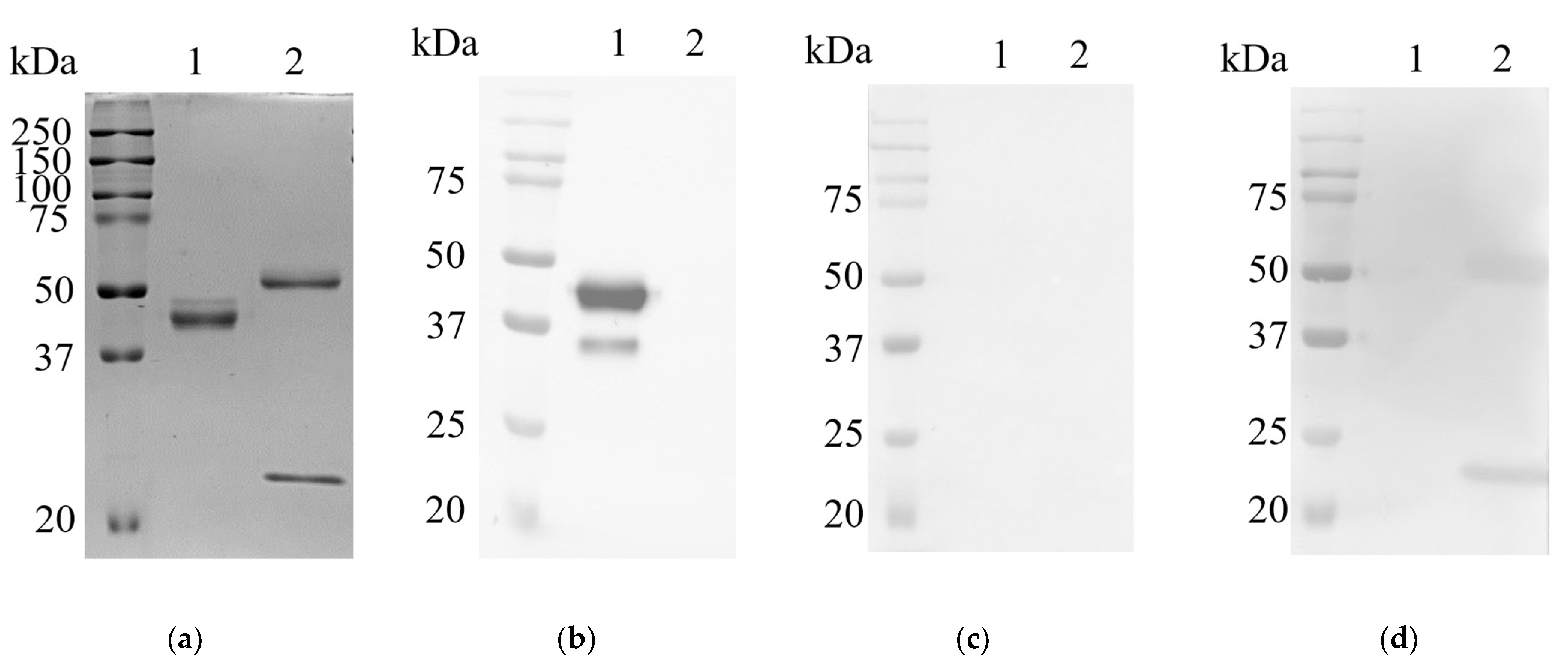
2.2. Expression and Purification of Recombinant VHH_19
2.3. Generation and Purification of Recombinant Full-Length Camelid Heavy-Chain Only Antibody L19
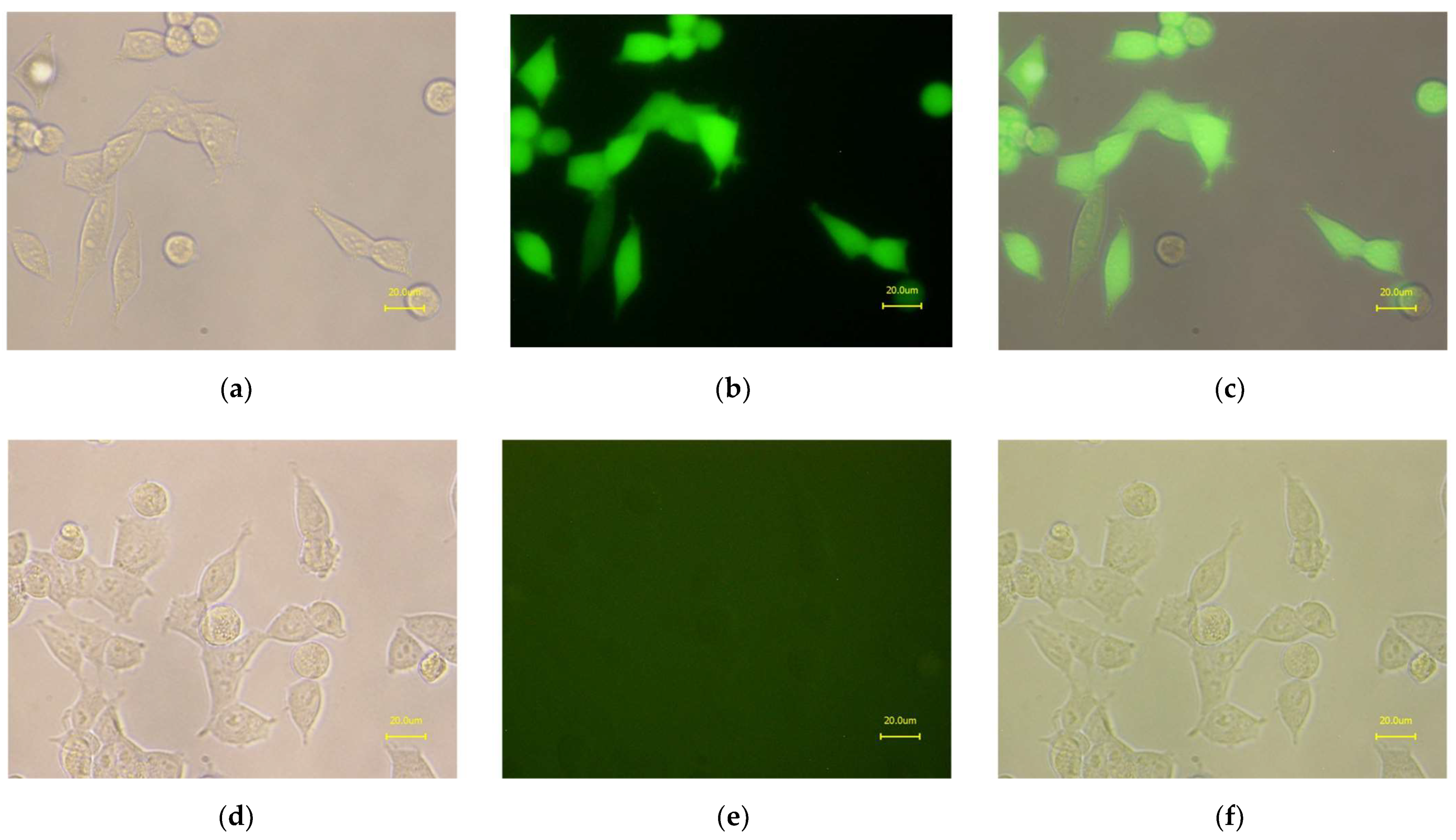
2.4. Detection of L19 with Western Blot Analysis
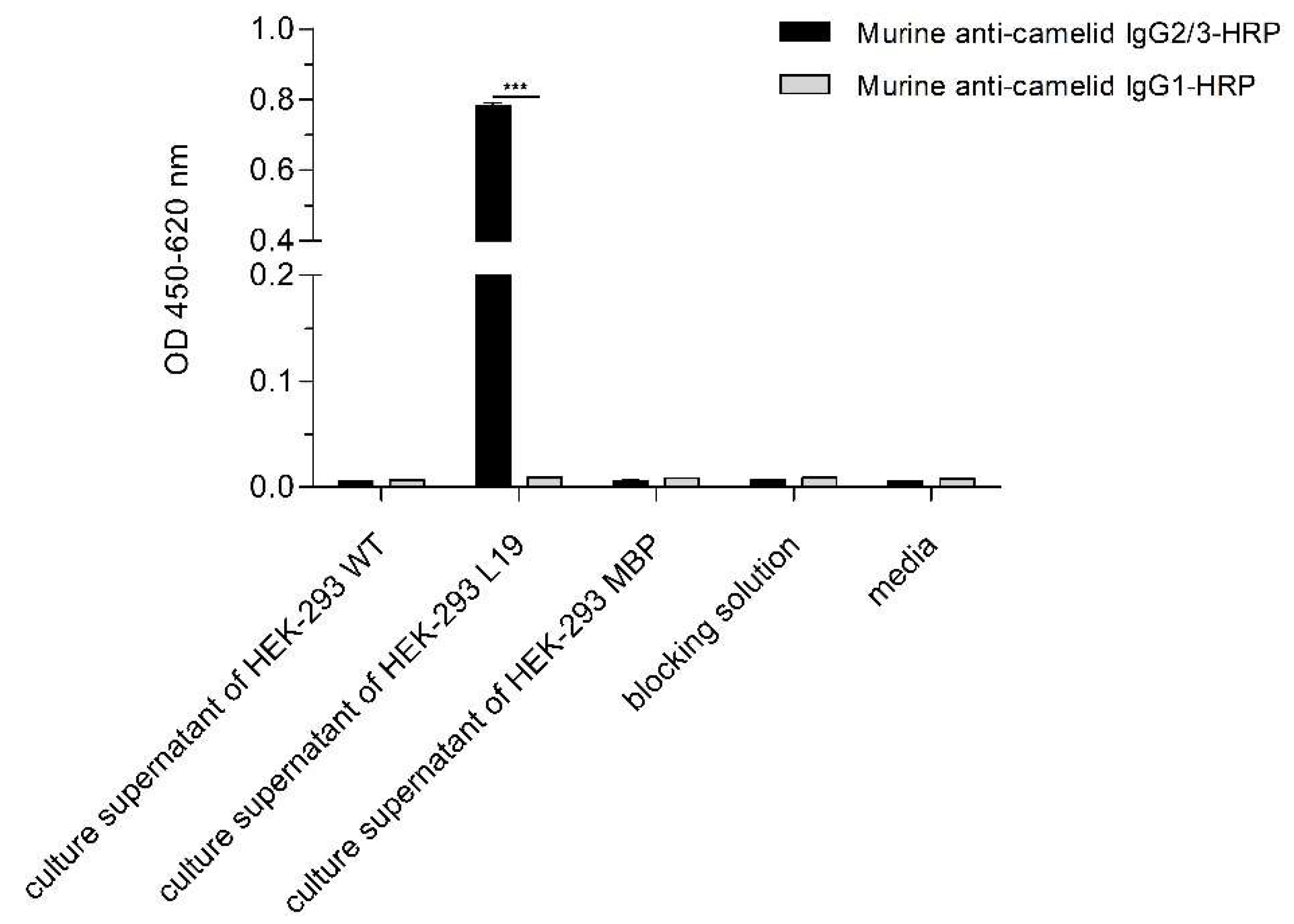
2.5. ELISA with the Purified L19
3. Discussion
4. Materials and Methods
4.1. Used miRNAs
4.2. Extraction of Total RNA from Camelid Peripheral Blood Mononuclear Cells
4.3. Construction of Naïve Phage Library
4.4. Panning the Naïve VHH Phage Library
4.5. Monoclonal Phage Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Expression and Purification of Recombinant VHH_19
4.7. Generation of Full-Length Recombinant Camelid Heavy-Chain Only Antibody L19
4.8. ELISA to Detect Secreted L19 Antibodies
4.9. Purification of L19 from the Culture Supernatant
4.10. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
4.11. ELISA with the Purified L19
4.12. Software
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Synthetic RNA | hsa bio ds miRNA-93 XLong | hsa bio ds miRNA-223 XLong | |
|---|---|---|---|
| FI | FI | ||
| 0.1 fmol/µL | without VHH_19 | 17 | 2 |
| 0.01 fmol/µL | 0 | 0 | |
| 1 atom/L | 0 | 0 | |
| 0.1 fmol/µL | with VHH_19 | 137 | 3 |
| 0.01 fmol/µL | 0 | 0 | |
| 1 atom/L | 0 | 0 |
| miRNA Real Sample | miRNA-93 | miRNA-223 | |
|---|---|---|---|
| FI | FI | ||
| Diseased | Without VHH_19 | 20 | 10 |
| with VHH_19 | + (16) | − (0) | |
| Healthy | Without VHH_19 | 11 | 11 |
| with VHH_19 | − (0) | + (10) |
References
- Wienholds, E.; Plasterk, R.H.A. MicroRNA Function in Animal Development. FEBS Lett. 2005, 579, 5911–5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cressatti, M.; Juwara, L.; Galindez, J.M.; Velly, A.M.; Nkurunziza, E.S.; Marier, S.; Canie, O.; Gornistky, M.; Schipper, H.M. Salivary MicroR-153 and MicroR-223 Levels as Potential Diagnostic Biomarkers of Idiopathic Parkinson’s Disease. Mov. Disord. 2020, 35, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Wang, Y.; Hu, L.; Xue, S.; Wang, Y.; Zhang, L.; Zhang, Y.; Qi, H.; Yu, H.; Aung, L.H.H.; et al. Combined Detection of MiR-21-5p, MiR-30a-3p, MiR-30a-5p, MiR-155-5p, MiR-216a and MiR-217 for Screening of Early Heart Failure Diseases. Biosci. Rep. 2020, 40, BSR20191653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauth, M.; Hegewald, A.B.; Schmitz, L.; Krone, D.J.; Saul, M.J. Validation of Extracellular MiRNA Quantification in Blood Samples Using RT-qPCR. FASEB BioAdvances 2019, 1, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, B.; Peplow, P. MicroRNAs in Blood and Cerebrospinal Fluid as Diagnostic Biomarkers of Multiple Sclerosis and to Monitor Disease Progression. Neural. Regen. Res. 2020, 15, 606. [Google Scholar] [CrossRef]
- Raeisi, F.; Mahmoudi, E.; Dehghani-Samani, M.; Hosseini, S.S.E.; Ghahfarrokhi, A.M.; Arshi, A.; Forghanparast, K.; Ghazanfari, S. Differential Expression Profile of MiR-27b, MiR-29a, and MiR-155 in Chronic Lymphocytic Leukemia and Breast Cancer Patients. Mol. Ther.-Oncolytics 2020, 16, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Siegismund, C.S.; Rohde, M.; Kühl, U.; Escher, F.; Schultheiss, H.P.; Lassner, D. Absent MicroRNAs in Different Tissues of Patients with Acquired Cardiomyopathy. Genom. Proteom. Bioinform. 2016, 14, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Han, J.; Chen, J.; Dong, J.; Xia, Y.; Liu, J.; Jiang, Y.; Dai, J.; Lu, J.; Jin, G.; et al. Plasma MiRNAs as Early Biomarkers for Detecting Hepatocellular Carcinoma: Plasma MiRNAs and Hepatocellular Carcinoma. Int. J. Cancer 2015, 137, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Zajdel, M.; Rymkiewicz, G.; Sromek, M.; Cieslikowska, M.; Swoboda, P.; Kulinczak, M.; Goryca, K.; Bystydzienski, Z.; Blachnio, K.; Ostrowska, B.; et al. Tumor and Cerebrospinal Fluid MicroRNAs in Primary Central Nervous System Lymphomas. Cancers 2019, 11, 1647. [Google Scholar] [CrossRef] [Green Version]
- Aleshcheva, G.; Pietsch, H.; Escher, F.; Schultheiss, H. MicroRNA Profiling as a Novel Diagnostic Tool for Identification of Patients with Inflammatory and/or Virally Induced Cardiomyopathies. ESC Heart Fail. 2021, 8, 408–422. [Google Scholar] [CrossRef]
- Winfield, J.B.; Faiferman, I.; Koffler, D. Avidity of Anti-DNA Antibodies in Serum and IgG Glomerular Eluates from Patients with Systemic Lupus Erythematosus. Association of High Avidity Antinative DNA Antibody with Glomerulonephritis. J. Clin. Investig. 1977, 59, 90–96. [Google Scholar] [CrossRef]
- Bizzaro, N.; Villalta, D.; Giavarina, D.; Tozzoli, R. Are Anti-Nucleosome Antibodies a Better Diagnostic Marker than Anti-DsDNA Antibodies for Systemic Lupus Erythematosus? A Systematic Review and a Study of Metanalysis. Autoimmun. Rev. 2012, 12, 97–106. [Google Scholar] [CrossRef]
- Gualtierotti, R.; Biggioggero, M.; Penatti, A.E.; Meroni, P.L. Updating on the Pathogenesis of Systemic Lupus Erythematosus. Autoimmun. Rev. 2010, 10, 3–7. [Google Scholar] [CrossRef]
- Mehra, S.; Fritzler, M.J. The Spectrum of Anti-Chromatin/Nucleosome Autoantibodies: Independent and Interdependent Biomarkers of Disease. J. Immunol. Res. 2014, 2014, 368274. [Google Scholar] [CrossRef] [Green Version]
- Stollar, B.D. Double-Helical Polynucleotides: Immunochemical Recognition of Differing Conformations. Science 1970, 169, 609–611. [Google Scholar] [CrossRef]
- Cros, P.; Kurfiirst, R.; Allibert, P.; Battail, N.; Piga, N.; Roig, V.; Thuong, N.T.; Mandrand, B.; Hélène, C. Monoclonal Antibodies Targeted to A-Oligonucleotides. Characterisation and Application in Nucleic Acid Detection. Nucleic Acids Res. 1994, 22, 2951–2957. [Google Scholar] [CrossRef]
- Nakazato, H. Radioimmunoassay of an Antibody to ΦX174 DNA·RNA Hybrid. Anal. Biochem. 1979, 98, 74–80. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Matsumoto, T.; Okuhara, E.; Shikata, E. Immunogenicity of Rice Dwarf Virus-Ribonucleic Acid. Tohoku J. Exp. Med. 1977, 122, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Francki, R.I.B. Presence of Antibodies to Double-Stranded RNA in Sera of Rabbits Immunized with Rice Dwarf and Maize Rough Dwarf Viruses. Virology 1973, 56, 404–406. [Google Scholar] [CrossRef]
- Hu, Z.; Leppla, S.H.; Li, B.; Elkins, C.A. Antibodies Specific for Nucleic Acids and Applications in Genomic Detection and Clinical Diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 895–916. [Google Scholar] [CrossRef]
- Stollar, B.D.; Voss, E.W. Antibodies to DN. Crit. Rev. Biochem. 1986, 20, 1–36. [Google Scholar] [CrossRef] [PubMed]
- David Stollar, B. The Experimental Induction of Antibodies to Nucleic Acids. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1980; Volume 70, pp. 70–85. ISBN 978-0-12-181970-5. [Google Scholar]
- Erlanger, B.F.; Beiser, S.M. Antibodies specific for ribonucleosides and ribonucleotides and their reaction with dna. Proc. Natl. Acad. Sci. USA 1964, 52, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of Monoclonal Antibody to DNA · RNA and Its Application to Immunodetection of Hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-Based Products as Research and Diagnostic Tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef]
- Van der Linden, R.H.J.; Frenken, L.G.J.; de Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; de Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of Physical Chemical Properties of Llama VHH Antibody Fragments and Mouse Monoclonal Antibodies. Biochim. Et Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1999, 1431, 37–46. [Google Scholar] [CrossRef]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen Binding and Solubility Effects upon the Veneering of a Camel VHH in Framework-2 to Mimic a VH. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef]
- Dumoulin, M.; Conrath, K.; Van Meirhaeghe, A.; Meersman, F.; Heremans, K.; Frenken, L.G.J.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-Domain Antibody Fragments with High Conformational Stability. Protein Sci. 2009, 11, 500–515. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Kabat, E.A.; Wu, T.T.; Perry, H.M.; Gottesman, K.S.; Foeller, C. Sequences of Proteins of Immunological Interest; US Department of Health and Human Services; US Public Health Service NIH: Bethesda, MD, USA, 1991. [Google Scholar]
- Benes, V.; Castoldi, M. Expression Profiling of MicroRNA Using Real-Time Quantitative PCR, How to Use It and What Is Available. Methods 2010, 50, 244–249. [Google Scholar] [CrossRef]
- Creighton, C.J.; Reid, J.G.; Gunaratne, P.H. Expression Profiling of MicroRNAs by Deep Sequencing. Brief. Bioinform. 2009, 10, 490–497. [Google Scholar] [CrossRef]
- Davison, T.S.; Johnson, C.D.; Andruss, B.F. Analyzing Micro-RNA Expression Using Microarrays. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 411, pp. 14–34. ISBN 978-0-12-182816-5. [Google Scholar]
- Ponsel, D.; Neugebauer, J.; Ladetzki-Baehs, K.; Tissot, K. High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation. Molecules 2011, 16, 3675–3700. [Google Scholar] [CrossRef] [Green Version]
- Sabir, J.S.M.; Atef, A.; El-Domyati, F.M.; Edris, S.; Hajrah, N.; Alzohairy, A.M.; Bahieldin, A. Construction of Naïve Camelids VHH Repertoire in Phage Display-Based Library. Comptes Rendus Biol. 2014, 337, 244–249. [Google Scholar] [CrossRef]
- Saerens, D.; Frederix, F.; Reekmans, G.; Conrath, K.; Jans, K.; Brys, L.; Huang, L.; Bosmans, E.; Maes, G.; Borghs, G.; et al. Engineering Camel Single-Domain Antibodies and Immobilization Chemistry for Human Prostate-Specific Antigen Sensing. Anal. Chem. 2005, 77, 7547–7555. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Fijten, H.P.D. Improved functional immobilization of llama single-domain antibody fragments to polystyrene surfaces using small peptides. J. Immunoass. Immunochem. 2012, 33, 234–251. [Google Scholar] [CrossRef]
- Orenes-Piñero, E.; Marín, F.; Lip, G.Y.H. MiRNA-197 and MiRNA-223 and Cardiovascular Death in Coronary Artery Disease Patients. Ann. Transl. Med. 2016, 4, 200. [Google Scholar] [CrossRef] [Green Version]
- Geithe, C.; Zeng, B.; Schmidt, C.; Dinter, F.; Roggenbuck, D.; Lehmann, W.; Dame, G.; Schierack, P.; Hanack, K.; Rödiger, S. A Multiplex Microchamber Diffusion Assay for the Antibody-Based Detection of MicroRNAs on Randomly Ordered Microbeads. Biochemistry 2021. [Google Scholar]
- Kappel, A.; Backes, C.; Huang, Y.; Zafari, S.; Leidinger, P.; Meder, B.; Schwarz, H.; Gumbrecht, W.; Meese, E.; Staehler, C.F.; et al. MicroRNA In Vitro Diagnostics Using Immunoassay Analyzers. Clin. Chem. 2015, 61, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.V.; Piro, B.; Reisberg, S.; Duc, H.T.; Pham, M.C. Antibodies Directed to RNA/DNA Hybrids: An Electrochemical Immunosensor for MicroRNAs Detection Using Graphene-Composite Electrodes. Anal. Chem. 2013, 85, 8469–8474. [Google Scholar] [CrossRef]
- Ye, J.-D.; Tereshko, V.; Frederiksen, J.K.; Koide, A.; Fellouse, F.A.; Sidhu, S.S.; Koide, S.; Kossiakoff, A.A.; Piccirilli, J.A. Synthetic Antibodies for Specific Recognition and Crystallization of Structured RNA. Proc. Natl. Acad. Sci. USA 2008, 105, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.; Lee, J.; Hong, H.J.; Park, I.; Lee, Y. RNA Recognition by a Human Antibody against Brain Cytoplasmic 200 RNA. RNA 2014, 20, 805–814. [Google Scholar] [CrossRef] [Green Version]
- Schlör, A.; Holzlöhner, P.; Listek, M.; Grieß, C.; Butze, M.; Micheel, B.; Hentschel, C.; Sowa, M.; Roggenbuck, D.; Schierack, P.; et al. Generation and Validation of Murine Monoclonal and Camelid Recombinant Single Domain Antibodies Specific for Human Pancreatic Glycoprotein 2. New Biotechnol. 2018, 45, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zola, H.; Fusco, M.; Macardle, P.J.; Flego, L.; Roberton, D. Expression of Cytokine Receptors by Human Cord Blood Lymphocytes: Comparison with Adult Blood Lymphocytes. Pediatr. Res. 1995, 38, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Thompson, A.A.; Fan, Y.; Lou, J.; Conrad, F.; Ho, M.; Pires-Alves, M.; Wilson, B.A.; Stevens, R.C.; Marks, J.D. A Single-Domain Llama Antibody Potently Inhibits the Enzymatic Activity of Botulinum Neurotoxin by Binding to the Non-Catalytic α-Exosite Binding Region. J. Mol. Biol. 2010, 397, 1106–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A General Protocol for the Generation of Nanobodies for Structural Biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Phage Display; Cold Spring Harbor Laboratory Pr: 2004; The Scripps Research Institute: San Diego, CA, USA, 2004; ISBN 978-0-87969-740-2.
- Coomber, D.W.J. Panning of Antibody Phage-Display Libraries: Standard Protocols. In Antibody Phage Display; Humana Press: Totowa, NJ, USA, 2001; Volume 178, pp. 133–145. ISBN 978-1-59259-240-1. [Google Scholar]
- Holzlöhner, P.; Butze, M.; Maier, N.; Hebel, N.; Schliebs, E.; Micheel, B.; Füner, J.; Heidicke, G.; Hanack, K. Generation of Murine Monoclonal Antibodies with Specificity against Conventional Camelid IgG1 and Heavy-Chain Only IgG2/3. Vet. Immunol. Immunopathol. 2018, 197, 1–6. [Google Scholar] [CrossRef] [PubMed]






| Primer Name | Sequence (5′ → 3′) | Reference |
|---|---|---|
| P12 | GTCCTGGCTGCTCTTCTACAAGG | [46] |
| P13 | ATGGAGAGGACGTCCTTGGGT | [46] |
| mL93 | ACCGTGGCCCAGGCGGCCCAGGTGCAGCTGCAGGAGTCTGGRGGAGG | [47] modified |
| mL94 | GTGCTGGCCGGCCTGGCCGCTGGAGACGGTGACCTGGGT | [47] modified |
| mL95 | GTGCTGGCCGGCCTGGCCTGAGGAGACGGTGACCTGGGT | [47] modified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, M.; Weichelt, U.; Rödiger, S.; Hanack, K. Novel Anti Double-Stranded Nucleic Acids Full-Length Recombinant Camelid Heavy-Chain Antibody for the Detection of miRNA. Int. J. Mol. Sci. 2022, 23, 6275. https://doi.org/10.3390/ijms23116275
Czarnecka M, Weichelt U, Rödiger S, Hanack K. Novel Anti Double-Stranded Nucleic Acids Full-Length Recombinant Camelid Heavy-Chain Antibody for the Detection of miRNA. International Journal of Molecular Sciences. 2022; 23(11):6275. https://doi.org/10.3390/ijms23116275
Chicago/Turabian StyleCzarnecka, Malgorzata, Ulrike Weichelt, Stefan Rödiger, and Katja Hanack. 2022. "Novel Anti Double-Stranded Nucleic Acids Full-Length Recombinant Camelid Heavy-Chain Antibody for the Detection of miRNA" International Journal of Molecular Sciences 23, no. 11: 6275. https://doi.org/10.3390/ijms23116275